")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Amino Acid Conjugates of Aminothiazole and Aminopyridine as Potential Anticancer Agents: Synthesis, Molecular Docking and in vitro Evaluation
Authors Naz S, Shah FA , Nadeem H, sarwar S , Tan Z, Imran M , Ali T , Li JB, Li S
Received 11 December 2020
Accepted for publication 27 February 2021
Published 1 April 2021 Volume 2021:15 Pages 1459—1476
DOI https://doi.org/10.2147/DDDT.S297013
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Shagufta Naz,1,2 Fawad Ali Shah,1 Humaira Nadeem,1 Sadia Sarwar,1 Zhen Tan,3 Muhammad Imran,1 Tahir Ali,2 Jing Bo Li,2 Shupeng Li4
1Riphah Institute of Pharmaceutical Sciences, Riphah International University, Islamabad, 44000, Pakistan; 2Shenzhen University Clinical Research Center for Neurological Diseases, Health Management Center, Shenzhen University General Hospital, Shenzhen University Clinical Medical Academy, Shenzhen University, Shenzhen, People’s Republic of China; 3Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, People’s Republic of China; 4State Key Laboratory of Oncogenomics, School of Chemical Biology and Biotechnology, Shenzhen Graduate School, Peking University, Shenzhen, People’s Republic of China
Correspondence: Humaira Nadeem
Riphah Institute of Pharmaceutical Sciences, Riphah International University, G-7/4, 7th Avenue, Islamabad, 44000, Pakistan
Tel +92 51-2891835
Fax +92 51-8350180
Email [email protected]
Shupeng Li
State Key Laboratory of Oncogenomics, School of Chemical Biology and Biotechnology, Shenzhen Graduate School, Peking University, Shenzhen, People’s Republic of China
Email [email protected]
Purpose: The development of resistance to available anticancer drugs is increasingly becoming a major challenge and new chemical entities could be unveiled to compensate this therapeutic failure. The current study demonstrated the synthesis of 2-aminothiazole [S3(a-d) and S5(a-d)] and 2-aminopyridine [S4(a-d) and S6(a-d)] derivatives that can target multiple cellular networks implicated in cancer development.
Methods: Biological assays were performed to investigate the antioxidant and anticancer potential of synthesized compounds. Redox imbalance and oxidative stress are hallmarks of cancer, therefore, synthesized compounds were preliminarily screened for their antioxidant activity using DPPH assay, and further five derivatives S3b, S3c, S4c, S5b, and S6c, with significant antioxidant potential, were selected for investigation of in vitro anticancer potential. The cytotoxic activities were evaluated against the parent (A2780) and cisplatin-resistant (A2780CISR) ovarian cancer cell lines. Further, Molecular docking studies of active compounds were performed to determine binding affinities.
Results: Results revealed that S3c, S5b, and S6c displayed promising inhibition in cisplatin-resistant cell lines in comparison to parent cells in terms of both resistance factor (RF) and IC50 values. Moreover, S3c proved to be most active compound in both parent and resistant cell lines with IC50 values 15.57 μM and 11.52 μM respectively. Our docking studies demonstrated that compounds S3c, S5b, and S6c exhibited significant binding affinity with multiple protein targets of the signaling cascade.
Conclusion: Anticancer activities of compounds S3c, S5b, and S6c in cisplatin-resistant cell lines suggested that these ligands may contribute as lead compounds for the development of new anticancer drugs.
Keywords: thiazole, pyridine, antioxidant activity, anticancer activity, molecular docking
Introduction
Ovarian cancer is considered an immense human health hazard and it is the fifth leading cause of death worldwide. A woman’s risk of getting invasive ovarian cancer is about 1 in 71 during her lifetime.1 Currently, new drug development has focused on the identification of novel targets and/or for ovarian cancer and its inhibitors.2 Over the past decades, most of the chemotherapeutic substances were designed against a specific target in the signaling network. Although these strategies initially increased curative efficacies, but the chances of re-occurrence are prevailing due to the increased resistance rate. Resistance likely develops due to the “rewiring” of subnetworks, including pathway re-programming and cross-activation in response to external stimuli.3 Thus, single-target therapies using highly specific compounds would likely fail in cancer treatment unless the compounds can disrupt multiple targets in the network. To deal with this problem, drug strategies called “network medicine” has been recently emerged to combat chemotherapeutic drug resistance.3–5 Epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), protein kinase B (PKB/AKT), mitogen-activated protein kinases (MAPKs), anaplastic lymphoma kinase (ALK), cytotoxic T-lymphocyte associated antigen-4 (CTLA-4), phosphoinositide-3-Kinase (PI3K) are the key signaling networks involved in the development of various types of cancers and are inhibited by different anticancer drugs.4–8 Furthermore, cisplatin and other platinum compounds have been used for several types of human malignancies, however, their efficacy is often compromised by tumor resistance.9 Moreover, currently marketed non-metallic anticancer drugs are also associated with intolerable side effects.10 To overcome this, new suitable alternative chemical entities must be ruled out which could offer considerable safety and efficacy.11 Several mechanisms are involved in cisplatin resistance and result in severe limitations in clinical use.12–14
Heterocyclic compounds are of great interest in the field of synthetic organic chemistry and medicinal chemistry.15,16 Thiazoles and pyridine are heterocyclic compounds and have demonstrated beneficial biological activities including antidiabetic, antimicrobial, antiviral, anti-inflammatory, anticancer, anti-alzheimer, antihypertensive, antioxidant, and hepatoprotective activities.17–27 Anticancer activities of the thiazole scaffold are well established as multiple therapeutically active agents like bleomycin, vosaroxin, epothilones, and dasatinib belong to this class.28 The groove-binding anticancer agents’ such as dactinomycin, netropsin, and thia-netropsin do also share thiazole moiety in their structure.29 Other 2-aminothiazole analogues such as giroline30 and cantharidin31 exhibited increased apoptotic activity compared to their parent compounds. Moreover, pyridine derivatives such as imatinib mesylate (Gleevec) and aminopyridine derivatives have also been used as potential anticancer agents.32–35 Furthermore, free radicals have a significant detrimental propensity for DNA, proteins, and cellular membranes, and thus could exacerbate the pathogenesis of several diseases including cancer.36,37 Thus, by decreasing free radical generation and oxidative stress, antioxidants could ameliorate DNA damage, and consequently mutagenesis.38 Several studies reported the antioxidant activities of 2-aminothiazole and 2-aminopyridine derivatives.39,40 There is a great tendency to conjugate amino acid residues with various bioactive heterocyclic compounds in the field of biomedical research. Various products reported by conjugation of amino acid residue with heterocyclic motif resulted in enhanced potency, solubility, cell permeability, selectivity, in vivo stability, and decreased toxicity of heterocycles.41 Derivatives of aminothiazole with amino acids possess pronounced antibacterial and antifungal activities.41,42 Keeping in view the above literature and safety profile of amino acid conjugates, we presented in this study synthesis of new derivatives of ethyl-2-aminothiazole-4-carboxylate and 2-aminopyridine to explore their antioxidant and anticancer potential. Moreover, molecular docking studies against selected proteins from the cell signaling pathway also provided insight into the possible mechanism of active compounds at the molecular level to elaborate the concept of network medicine.
Materials and Methods
Chemistry
General Information
All the starting materials were purchased from Sigma-Aldrich (St. Louis, MO, USA), Daejung (South Korea), and Alfa-Aesar (Germany). Digital Gallenkamp (Sanyo) apparatus was used to record the melting points of final compounds and was uncorrected. Proton NMR (1H NMR) and carbon-13 (13C NMR) spectra were measured on a Bruker AV400 spectrophotometer in CD3OD, CDCl3, and DMSO-d6 at 400 MHz using TMS (Tetramethyl silane) as an internal standard. Alpha Bruker FTIR spectrophotometer (ATR eco ZnSe, νmax in cm−1) was used to record FTIR spectra and elemental analysis was conducted using a LECO-183 CHNS analyzer. All reactions were monitored by thin-layer chromatography (TLC). Merck silica gel HF-254 was used for column chromatographic purification of products using (pet.ether: ethyl acetate, 4:1) as eluent. All chemicals used were of high analytical grade (99% HPLC).
General Procedure for the Preparation of Ethyl-2-Aminothiazole-4-Carboxylate (3)
A mixture of ethyl bromopyruvate (0.05 mol, 9.75 g) and thiourea (0.10 mol, 7.61 g) in absolute ethanol (53mL) was refluxed for 24 h. The completion of the reaction was checked by thin layer chromatography (TLC). After completion, the reaction mixture was allowed to cool to room temperature and concentrated in vacuo to half the original volume. The remaining ethanol solution was poured into water and made alkaline (pH 10) with 2N NaOH. Light brown solid precipitated immediately. The mixture was stirred for about 10 min and then the solid was removed by vacuum filtration and dried to give the desired product.47
Light brown crystals; yield: 68%; m.p. found: 176 °C; m.p. reported: 175–180 °C; Rf = 0.18 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3290 (NH2, str.), 1733 (C=O, ester, str.), 1615 (C=N, str.), 1540 (C=C, str.); 1H NMR (400 MHz, CD3OD) δ ppm 7.4 (s, 1H, H-1), 5.8 (brs, 2H, NH2), 4.32 (q, J=7.2Hz, 2H, H-2), 1.35 (t, J=7.2Hz, 3H, H-3); 13C NMR (400 MHz, CD3OD) δ ppm 167.8, 161.5, 148.2, 116.7, 61.3, 15.2; Anal. calcd for C6H8O2N2S (172.20): C, 41.86; N, 16.27; H, 4.68. Found: C, 41.79; N, 16.19; H, 4.61%.
General Procedure for the Preparation of N,N-Phthaloyl Protected Amino Acids (S1a-S1d)
A mixture of respective amino acids (0.03 mol) and finely ground phthalic anhydride (0.03 mol, 4.44 g) was heated with stirring in an oil bath at 185–190°C for 30 min. After cooling, the solid material was dissolved in hot methanol (20 mL) and filtered. The product was allowed to crystallize out slowly in appropriate solvent (methanol/water, 1:1).48
White crystals; yield: 90%; m.p. found: 197 °C; m.p. reported: 197–198 °C; Rf = 0.64 (ethyl acetate: pet.ether 3:1); FTIR (νmax cm−1) 1716 (C=O, acid, str.), 1632, 1536 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CD3OD) δ ppm 7.91–7.79 (m, 4H, Ar-H), 4.52 (s, 2H, H-4); 13C NMR (400 MHz, CD3OD) δ ppm 170, 167.8, 167.2, 134.7, 133.9, 131.9, 131.3, 122.6, 122.1, 38.7; Anal. calcd for C10H7O4N (205.16): C, 58.53; N, 6.82; H, 3.41. Found: C, 58.51; N, 6.79; H, 3.38%.
White crystals; yield: 91%; m.p. found: 177 °C; m.p. reported: 178 °C; Rf = 0.71 (ethyl acetate: pet.ether 3:1); IR (νmax cm−1) 1714 (C=O, acid, str.), 1636,1534 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CD3OD) δ ppm 7.90–7.77 (m, 8H, Ar-H), 5.41 (d, J=4.8Hz, 1H, H-4), 3.67 (m, 2H, H-5); 13C NMR (400 MHz, CD3OD) δ ppm 176.5, 168.6, 168.1, 138.4, 135.8, 134.7, 131.6, 131.2, 128.2, 127.7, 125.5, 121.8, 121.1, 117.5, 117.2, 62.5, 35.3; Anal. calcd for C17H13O4N (295.28): C, 69.15; N, 47.45; H, 4.41. Found: C, 69.05; N, 47.40; H, 4.38%.
White crystals; yield: 83%; m.p. found: 110 °C; m.p. reported: 110 °C; Rf = 0.73 (ethyl acetate: pet.ether 3:1); FTIR (νmax cm−1) 1718 (C=O, acid, str.), 1633,1532 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CD3OD) δ ppm 7.91 (d, 2H, J=8Hz, H-8,8ʹ, Ar-H), 7.78 (d, J=8Hz, 2H, H-9,9ʹ, Ar-H), 5.06 (dd, J1,J2=4.8Hz, 1H, H-4), 2.40 (m, 2H, H-5), 1.54 (m, 1H, H-6), 1.0 (d, J=6.4Hz, 6H, H-7a,7b); 13C NMR (400 MHz, CD3OD) δ ppm 180.3, 169.9, 169.3, 137.4, 136.3, 128.7, 128.4, 120.5, 120.1, 63.2, 37.5, 26.8, 20.9, 19.3; Anal. calcd for C14H15O4N (261.27): C, 64.36; N, 5.36; H, 5.75. Found: C, 64.30; N, 5.33; H, 5.71%.
White crystals; yield: 75%; m.p. found: 112 °C; m.p. reported: 110–112 °C; Rf = 0.8 (ethyl acetate: pet.ether 3:1); FTIR (νmax cm−1) 1716 (C=O, acid, str.), 1637,1530 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CD3OD) δ ppm 7.87–7.79 (m, 4H, Ar-H), 4.60 (d, J=8Hz, 1H, H-4), 2.87 (m, 1H, H-5), 1.57 (m, 2H, H-6), 1.11 (d, J=8Hz, 3H, H-10), 0.91 (t, J=8Hz, 3H, H-7); 13C NMR (400 MHz, CD3OD) δ ppm 174.6, 165.7, 165.3, 135.5, 134.4, 127.8, 126.3, 118.7, 118.4, 65.3, 36.8, 23.2, 16.6, 10.5; Anal. calcd for C14H15O4N (261.27): C, 64.36; N, 5.36; H, 5.75. Found: C, 64.29; N, 5.30; H, 5.67%.
General Procedure for the Preparation of N,N-Phthaloyl Amino Acid Chlorides (S2a-S2d)
To a solution of respective N,N-phthaloyl amino acids (0.005 mol) in dichloromethane (25 mL), thionyl chloride (0.015 mol, 1.08 mL) was added and refluxed for 8 h. The progress of the reaction was monitored by TLC and after the completion, the excess of thionyl chloride was evaporated. The resulting product was used without further purification.49
General Procedure for the Preparation of Amides (S3a-S3d and S4a-S4d)
To a solution of respective N,N-phthaloyl amino acid chlorides (0.01 mol) in 20 mL of dichloromethane and 5 mL DMF (dimethylformamide), (0.01 mol, 2.06 g) of DCC (dicyclohexyl carbodiimide) was added. The reaction mixture was stirred for 0.5 h followed by the dropwise addition of ethyl-2-aminothiazole-4-carboxylate (0.01 mol, 1.72 g) or 2-aminopyridine (0.01 mol, 0.94 g) previously dissolved in 20 mL of dichloromethane. Further, the reaction mixture was stirred at 45°C for 2 h followed by overnight stirring at room temperature. The completion of the reaction was checked by TLC (pet.ether: ethyl acetate, 3:1). After completion, the mixture was allowed to cool, resulted in white dicyclohexylurea precipitates which were further removed by filtration. The excess of solvent was removed under reduced pressure and the corresponding product was dried and recrystallized from chloroform.50
Brown solid; yield: 72%; m.p. 171 °C; Rf = 0.37 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3298 (N-H, str.), 1726 (C=O, ester, str.), 1698 (C=O, amide, str.), 1631,1539 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CDCl3) δ ppm 9.25 (brs,1H, NH), 7.90–7.78 (m, 4H, aromatic), 7.28 (s, 1H, H-1), 4.52 (s, 2H, H-4), 3.74 (q, J=7.2Hz, 2H, H-2), 1.3 (t, J=7.2Hz, 3H, H-3); 13C NMR (400 MHz, CDCl3) δ ppm 190.1, 170.4, 167.5, 167.1, 158.2, 139.4, 133.6, 133.1, 130.4, 129.5, 123.8, 123.1, 113.5, 48.4, 39.6, 6.9; Anal. calcd for C16H13O5N3S (343.35): C, 53.48; N, 11.69; H, 3.62. Found: C, 52.70; N, 10.45; H, 3.10%.
White crystals; yield: 88%; m.p. 153 °C; Rf = 0.29 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3322 (N-H, str.), 1730 (C=O, ester, str.), 1695 (C=O, amide, str.), 1662,1546 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CDCl3) δ ppm 10.11 (brs,1H, NH), 7.85–7.77 (m, 8H, Ar-H), 7.17 (s, 1H, H-1), 5.39 (d, J=4.8Hz, 1H, H-4), 4.42 (q, J=7.2Hz, 2H, H-2), 3.67 (m, 2H, H-5), 1.40 (t, J=7.2Hz, 3H, H-3); 13C NMR (400 MHz, CDCl3) δ ppm 191.2, 170.5, 167.7, 167.3, 156.4, 138.2, 137.4, 133.8, 132.1, 131.4, 131.1, 130.2, 129.8, 129.1, 128.7, 128.2, 123.5, 123.3, 115.4, 58.2, 37.9, 35.2, 8.3; Anal. calcd for C23H19O5N3S (433.47): C, 61.46; N, 9.35; H, 4.23. Found: C, 60.39; N, 8.51; H, 3.95%.
White crystals; yield: 86%; m.p. 79 °C; Rf = 0.36 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3359 (N-H, str.), 1733 (C=O, ester, str.), 1696 (C=O, amide, str.), 1650,1563 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CDCl3) δ ppm 9.78 (brs,1H, NH), 7.91–7.83 (m, 4H, Ar-H), 7.29 (s, 1H, H-1), 5.19 (dd, J1,J2=4.8Hz, 1H, H-4), 4.42 (q, J=7.2Hz, 2H, H-2), 2.20 (m, 1H, H-6), 1.60 (m, 2H, H-5), 1.41 (t, J=7.2Hz, 3H, H-3), 1.0 (d, J=6.4Hz, 6H, H-7a,7b); 13C NMR (400 MHz, CDCl3) δ ppm 189.4, 171.2, 168.5, 167.8, 157.1, 138.4, 134.4, 133.1, 132.4, 131.3, 124.6, 123.1, 117.5, 61.8, 36.9, 33.5, 26.2, 23.4, 22.5, 7.4; Anal. calcd for C20H21O5N3S (399.46): C, 57.83; N, 10.12; H, 5.06. Found: C, 56.45; N, 9.89; H, 4.82%.
White crystals; yield: 78%; m.p. 174 °C; Rf = 0.34 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3320 (N-H, str.), 1728 (C=O, ester, str.), 1692 (C=O, amide, str.), 1642,1521 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CDCl3) δ ppm 10.32 (brs,1H, NH), 7.87–7.79 (m, 4H, Ar-H), 7.28 (s, 1H, H-1), 4.78 (q, J=7.2Hz, 2H, H-2), 4.43 (d, J=8Hz, 1H, H-4), 2.87 (m, 1H, H-5), 1.57 (m, 2H, H-6), 1.42 (t, J=7.2Hz, 3H, H-3), 1.11 (d, J=8Hz 3H, H-10), 0.91 (t, J=8Hz, 3H, H-7); 13C NMR (400 MHz, CDCl3) δ ppm 190.4, 171.5, 167.5, 167.1, 156.3, 139.4, 133.6, 133.1, 132.8, 131.7, 123.6, 123.1, 115.3, 58.5, 34.2, 31.5, 27.2, 19.2, 12.6, 6.8; Anal. calcd for C20H21O5N3S (399.46): C, 57.83; N, 10.12; H, 5.06. Found: C, 56.58; N, 9.91; H, 4.87%.
Off-white solid; yield: 64%; m.p. 134 °C; Rf = 0.36 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3296 (N-H, str.), 1690 (C=O, amide, str.), 1647,1552 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CDCl3) δ ppm 9.11 (brs,1H, NH), 8.67 (d, J=4.1 Hz, 1H, pyridine), 8.29 (d, J=7.9Hz, 1H, pyridine), 7.83 (t, J=7.8Hz,1H, pyridine), 7.70 (dd, J1,J2= 7.2Hz, 1H, pyridine), 7.24–7.04 (m,4H, Ar-H), 4.52 (s, 2H, glycine); 13C NMR (400 MHz, CDCl3) δ ppm 170.5, 168.1, 167.8, 155.4, 149.2, 138.4, 134.4, 133.2, 131.8, 130.4, 124.8, 122.3, 119.2, 114.5, 43.7; Anal. calcd for C16H13O5N3S (343.35): C, 53.48; N, 11.69; H, 3.62. Found: C, 53.42; N, 11.60; H, 3.59%.
White crystals; yield: 85%; m.p. 174 °C; Rf = 0.27 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3313 (N-H, str.), 1698 (C=O, amide, str.), 1644,1523 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CDCl3) δ ppm 9.85 (brs,1H, NH), 8.63 (d, J=4.3 Hz, 1H, pyridine), 8.30 (d, J=8Hz, 1H, pyridine), 7.81 (t, J=7.7Hz,1H, pyridine), 7.70 (dd, J1,J2=7.1Hz, 1H, pyridine), 7.20–7.06 (m, 9H, Ar-H), 5.31 (d, J=4.8 Hz 1H, CH-α-carbon), 3.69 (m, 2H, CH2-phenyl); 13C NMR (400 MHz, CDCl3) δ ppm 171.2, 167.1, 166.9, 152.3, 148.2, 139.6, 137.2, 134.2, 133.5, 131.5, 131.1, 129.9, 129.1, 127.5, 126.9, 125.8, 124.8, 123.7, 118.2, 113.7, 62.6, 39.2; Anal. calcd for C23H19O5N3S (433.47): C, 61.46; N, 9.35; H, 4.23. Found: C, 61.39; N, 9.31; H, 4.20%.
White crystals; yield: 60%; m.p. 109 °C; Rf = 0.325 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3343 (N-H, str.), 1697 (C=O, amide, str.), 1635,1542 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CDCl3) δ ppm 9.68 (brs,1H, NH), 8.65 (d, J=4Hz, 1H, pyridine), 8.29 (d, J=7.9Hz, 1H, pyridine), 7.92 (t, J=7.8Hz, 1H, pyridine), 7.79 (dd, J1,J2=7Hz, 1H, pyridine), 7.28–7.06 (m, 4H, Ar-H), 5.11 (t, J=5Hz, 1H, CH-α-carbon), 2.01 (m, 1H, -CH(CH3)2-leucine), 1.56 (dd, 2H, -CH2-leucine), 1.0 (d, 6H, J=6.4Hz, -CH(CH3)2-leucine); 13C NMR (400 MHz, CDCl3) δ ppm 171.6, 167.9, 167.1, 157.4, 148.4, 136.5, 134.8, 134.2, 132.9, 131.4, 125.8, 124.2, 118.4, 115.5, 69.5, 38.7, 30.4, 29.5, 23.5; Anal. calcd for C20H21O5N3S (399.46): C, 57.83; N, 10.12; H, 5.06. Found: C, 57.80; N, 10.02; H, 5.02%.
White crystals; yield: 54%; m.p. 107 °C; Rf = 0.35 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3310 (N-H, str.), 1695 (C=O, amide, str.), 1640,1531 (C=O, phthaloyl amide, str.); 1H NMR (400 MHz, CDCl3) δ ppm 9.96 (brs,1H, NH), 9.49 (d, J=4Hz, 1H, pyridine), 8.32 (d, J=8Hz, 1H, pyridine), 8.20 (t, J=7.8Hz, 1H, pyridine), 7.90 (dd, J1,J2=7.2Hz, 1H, pyridine), 7.28–7.03 (m, 4H, Ar-H), 4.66 (d, J=8Hz, 1H, α-carbon), 2.87 (m, 1H, -CHCH3-isoleucine), 1.57 (m, 2H, -CH2CH3-isoleucine), 1.11 (d, J=8Hz, 3H, -CHCH3-isoleucine), 0.91 (t, J=8Hz, 3H, -CH2CH3-isoleucine); 13C NMR (400 MHz, CDCl3) δ ppm 170.4, 168.5, 167.9, 154.3, 149.2, 137.6, 133.8, 132.2, 131.9, 131.1, 124.8, 123.2, 117.5, 116.2, 58.5, 32.7, 29.3, 17.6, 12.3; Anal. calcd for C20H21O5N3S (399.46): C, 57.83; N, 10.12; H, 5.06. Found: C, 57.78; N, 10.10; H, 5.01%.
General Procedure for N-Deprotection of Amino Acids (S5a-S5d and S6a-S6d)
To a solution of respective phthalimides (0.01 mol) in boiling ethanol (22 mL), hydrazine hydrate 50% (0.01 mol, 0.32 g) was added. The completion of the reaction was checked by TLC. On cooling, the precipitates were collected, and recrystallized from ethanol.51
Brown solid; yield: 62%; m.p. 195 °C; Rf = 0.37 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3298 (N-H, str.), 3276 (N-H, str.), 3268 (NH2, str.), 3263 (NH2, str.), 1698 (C=O, amide, str.), 1646 (C=O, amide, str.); 1H NMR (400 MHz, DMSO-d6) δ ppm 9.25 (brs, 1H, NH), 8.79 (s, 1H, NHCO), 7.28 (s, 1H, H-1), 4.52 (s, 2H, H-4), 4.38 (s, 2H, NH2NH), 4.25 (s, 2H, NH2); 13C NMR (400 MHz, DMSO-d6) δ ppm 169.8, 162.4, 159.4, 150.4, 114.5, 45.2; Anal. calcd for C6H9O2N5S (215.23): C, 33.48; N, 32.54; H, 4.21. Found: C, 33.42; N, 32.40; H, 4.10%.
Light brown solid; yield: 88%; m.p. 206 °C; Rf = 0.29 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3299 (N-H, str.), 3273 (N-H, str.), 3269 (NH2, str.), 3261 (NH2, str.), 1696 (C=0, amide, str.), 1646 (C=O, amide, str.); 1H NMR (400 MHz, DMSO-d6) δ ppm 10.11 (brs,1H, NH), 8.89 (s, 1H, NHCO), 7.85–7.77 (m, 5H, Ar-H), 7.17 (s, 1H, H-1), 5.39 (d, J=4.8Hz, 1H, H-4), 4.39 (s, 2H, NH2NH), 4.28 (s, 2H, NH2), 3.67 (m, 2H, H-5); 13C NMR (400 MHz, DMSO-d6) δ ppm 174.2, 163.7, 160.1, 150.2, 137.4, 129.8, 128.5, 128.1, 127.9, 126.3, 120.5, 49.8, 34.2; Anal. calcd for C13H15O2N5S (305.35): C, 51.13; N, 22.94; H, 4.95. Found: C, 51.10; N, 22.89; H, 4.92%.
Orange solid; yield: 70%; m.p. 198 °C; Rf = 0.36 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3298 (N-H, str.), 3275 (N-H, str.), 3266 (NH2, str.), 3260 (NH2, str.), 1695 (C=0, amide, str.), 1647 (C=O, amide, str.); 1H NMR (400 MHz, DMSO-d6) δ ppm 9.78 (brs,1H, NH), 8.77 (s, 1H, NHCO), 7.29 (s, 1H, H-1), 5.19 (dd, J1,J2=4.8Hz, 1H, H-4), 4.38 (s, 2H, NH2NH), 4.26 (s, 2H, NH2), 2.20 (m, 1H, H-6), 1.60 (m, 2H, H-5), 1.0 (d, J=6.4Hz, 6H, H-7a,7b); 13C NMR (400 MHz, DMSO-d6) δ ppm 170.4, 161.5, 158.2, 147.8, 114.5, 53.1, 45.1, 20.4, 19.2, 17.3; Anal. calcd for C10H17O2N5S (271.33): C, 44.26; N, 25.81; H, 6.31. Found: C, 44.21; N, 25.79; H, 6.28%.
Brown solid; yield: 78%; m.p. 161 °C; Rf = 0.34 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3301 (N-H, str.), 3278 (N-H, str.), 3265 (NH2, str.), 3262 (NH2, str.), 1692 (C=O, amide, str.), 1646 (C=O, amide, str.); 1H NMR (400 MHz, DMSO-d6) δ ppm 10.32 (brs,1H, NH), 8.79 (s, 1H, NHCO), 7.28 (s, 1H, H-1), 4.43 (d, J=8Hz, 1H, H-4), 4.37 (s, 2H, NH2NH), 4.28 (s, 2H, NH2), 2.87 (m, 1H, H-5), 1.57 (m, 2H, H-6), 1.11 (d, J=8Hz 3H, H-10), 0.91 (t, J=8Hz, 3H, H-7); 13C NMR (400 MHz, DMSO-d6) δ ppm 172.4, 164.4, 158.9, 146.1, 116.5, 58.5, 39.4, 24.8, 18.4, 10.1; Anal. calcd for C10H17O2N5S (271.33): C, 44.26; N, 25.81; H, 6.31. Found: C, 44.20; N, 25.80; H, 6.29%.
Off-white solid; yield: 65%; m.p. 204 °C; Rf = 0.36 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3296 (N-H, str.), 3263 (NH2, str.), 1690 (C=O, amide, str.); 1H NMR (400 MHz, DMSO-d6) δ ppm 9.11 (brs,1H, NH), 8.67 (d, J=4.1 Hz, 1H, H-3ʹ), 8.29 (d, J=7.9Hz, 1H, H-6ʹ), 7.83 (t, J=7.8Hz,1H, H-4ʹ), 7.70 (dd, J1,J2= 7.2Hz, 1H, H-5ʹ), 4.52 (s, 2H, H-4), 4.26 (s, 2H, NH2); 13C NMR (400 MHz, DMSO-d6) δ ppm 168.4, 153.4, 148.2, 136.9, 117.6, 114.5, 44.5; Anal. calcd for C7H9ON3 (151.16): C, 55.62; N, 27.80; H, 6.00. Found: C, 55.60; N, 27.78; H, 5.98%.
Brown solid; yield: 85%; m.p. 247 °C, Rf = 0.27 (pet.ether:ethyl acetate 3:1), FTIR (νmax cm−1) 3313 (N-H, str.), 3268 (NH2, str.), 1698 (C=O, amide, str.); 1H NMR (400 MHz, DMSO-d6) δ ppm 9.85 (brs,1H, NH), 8.63 (d, J=4.3 Hz, 1H, H-3ʹ), 8.30 (d, J=8Hz, 1H, H-6ʹ), 7.81 (t, J=7.7Hz,1H, H-4ʹ), 7.70 (dd, J1,J2=7.1Hz, 1H, H-5ʹ), 7.20–7.06 (m, 5H, Ar-H), 5.31 (d, J=4.8 Hz 1H, H-4), 4.28 (s, 2H, NH2), 3.69 (m, 2H, H-5); 13C NMR (400 MHz, DMSO-d6) δ ppm 170.6, 151.9, 147.4, 138.6, 136.4, 130.7, 129.8, 128.5, 127.4, 126.7, 119.2, 114.6, 57.6, 36.4; Anal. calcd for C14H15ON3 (241.28): C, 69.69; N, 17.41; H, 6.27. Found: C, 69.58; N, 17.39; H, 6.20%.
Brown solid; yield: 60%; m.p. 231 °C; Rf = 0.35 (pet.ether:ethyl acetate 3:1); FTIR (νmax cm−1) 3343 (N-H, str.), 3269 (NH2, str.), 1697 (C=O, amide, str.), 1H NMR (400 MHz, DMSO-d6) δ ppm 9.68 (brs,1H, NH), 8.65 (d, J=4Hz, 1H, H-3ʹ), 8.29 (d, J=7.9Hz, 1H, H-6ʹ), 7.92 (t, J=7.8Hz, 1H, H-4ʹ), 7.79 (dd, J1,J2=7Hz, 1H, H-5ʹ), 4.27 (s, 2H, NH2), 5.11 (t, J=5Hz, 1H, H-4), 2.01 (m, 1H, H-6), 1.56 (dd, 2H, H-5), 1.0 (d, 6H, J=6.4Hz, H-7a,7b); 13C NMR (400 MHz, DMSO-d6) δ ppm 169.4, 153.8, 148.2, 137.6, 118.4, 115.7, 55.5, 43.7, 29.4, 25.3, 22.5; Anal. calcd for C11H17ON3 (207.27): C, 63.74; N, 20.27; H, 8.27. Found: C, 63.69; N, 20.25; H, 8.23%.
White crystals; yield: 75%; m.p. 236 °C; Rf = 0.35 (pet.ether: ethyl acetate 3:1); FTIR (νmax cm−1) 3310 (N-H, str.), 3270 (NH2, str.), 1695 (C=O, amide, str.); 1H NMR (400 MHz, DMSO-d6) δ ppm 9.96 (brs,1H, NH), 9.49 (d, J=4Hz, 1H, H-3ʹ), 8.32 (d, J=8Hz, 1H, H-6ʹ), 8.20 (t, J=7.8Hz, 1H, H-4ʹ), 7.90 (dd, J1,J2=7.2Hz, 1H, H-5ʹ), 4.66 (d, J=8Hz, 1H, H-4), 4.29 (s, 2H, NH2), 2.87 (m, 1H, H-5), 1.57 (m, 2H, H-6), 1.11 (d, J=8Hz, 3H, H-10), 0.91 (t, J=8Hz, 3H, H-7); 13C NMR (400 MHz, DMSO-d6) δ ppm 170.4, 152.4, 149.8, 138.6, 118.5, 114.3, 58.5, 35.7, 26.4, 18.3, 12.2; Anal. calcd for C11H17ON3 (207.27): C, 63.74; N, 20.27; H, 8.27. Found: C, 63.70; N, 20.25; H, 8.23%.
In vitro Antioxidant Activity
To preliminary evaluate the free radical scavenging potential of synthesized derivatives; an in vitro antioxidant activity assay was performed using DPPH.52 For the relative assay, a solution containing DPPH (3 mL) and methanol (1 mL) was designated as a negative control. Ascorbic acid was used as a reference standard. Different concentrations of test compounds were taken up to 3 mL and, 1 mM DPPH solution in methanol was added. These mixtures were kept for 30 min under dark. UV spectrophotometer was used to assess the free radical scavenging potential of test compounds through measuring absorbance at 517 nm. The inverse relationship between absorbance and test compound concentrations shows antioxidant potential. Furthermore, the color change also presents a potential indication that is from blue color to yellowish-orange color. Percent scavenging efficacy was determined using the formula:

In vitro Anticancer Activity
Preparation of Cells
Human ovarian cancer cell lines A2780 and A2780CISR were purchased from ECACC (93112519, 93112517 for A2780 and A2780CISR respectively). The ovarian cancer cell lines A2780 (parent) and A2780CISR (cisplatin-resistant) were grown in tissue culture flasks in an incubator at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. 4500 to 5500 cells per well in 10% FCS/RPMI 1640 culture medium were seeded into flat-bottomed 96-well culture plates and allowed to attach overnight. Cells were counted by Bio-Rad TC10 automated cell counter. 10µL of cell suspension was loaded onto the slide and inserted into the counter to count automatically. After counting cells, cell lines were seeded in two separate 96 well plates. 100 µL of cell mixture (cells in the medium) were added to each well. Prepared drug concentrations are shown in Table 1. The plates were incubated for 24 h at 37 °C in a humidified atmosphere to allow the cells to attach.
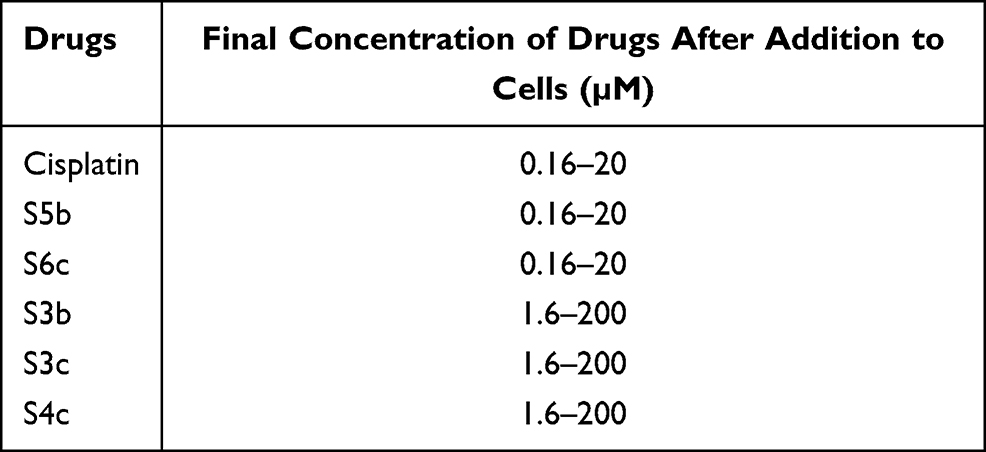 |
Table 1 The Concentration Range of Drugs is Prepared for Adding to Cells and the Final Concentration of Drugs After Addition to Cells |
Drug Addition to Cells
After the preparation of fivefold serial dilutions of the drugs in 10% FCS/RPMI 1640 medium, 100 μL of each drug were added to equal volumes of cell culture in triplicate wells. The plates were then left to incubate in the carbon dioxide incubator (5% carbon dioxide in the air, pH 7.4) for 72 h at 37 °C in a humidified atmosphere.
MTT Reduction Assay
MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] powder was dissolved in serum-free-RPMI-1640 medium at 1 mg/mL and filtered using DISMIC-25cs ADVANTEC cellulose acetate filter to remove any unwanted particles. The medium was removed and 50 μL of the MTT solution was added to each well of 96-well plate and incubated for 4 h in the same carbon dioxide incubator. Cisplatin was used as a standard drug. After incubation, the MTT solution was removed and 150 μL of DMSO was added to each well. The living cells if still present remained attached at the bottom. The yellow formazan crystals produced from the reduction of MTT was dissolved in DMSO and the optical density (OD) of the resulting solution was read with the Bio-Rad, Microplate Reader, Benchmark at 570 nm. The survival cell fractions were calculated according to the following equation of Mosmann (1983).
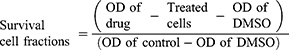
Dosage response curves have been constructed by plotting the percentage of viable cells against drug concentration. The drug concentration required to cause 50% cell death (IC50 value) was then calculated from the curve. For each drug concentration, at least three independent experiments were performed.53 RF (Resistance factor) was also calculated using the following formula.46
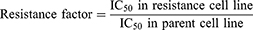
Molecular Docking
Molecular docking studies of the compounds S3c, S5b and S6c with better in vitro anticancer activities were performed against eleven protein targets, selected from cell signaling pathways (ie, EGFR, VEGFR, PDGFR, p110a/PI3K, RET, BCR-ABL, c-KIT, c-Raf, B-Raf, CTLA-4 and ALK) (PDB Codes: 1E90, 4LL0, 4AG8, 2L6W, 1ZZP, 2IVU, 3G0E, 3OMV, 2YJR, 3PPJ, 3OSK, respectively).
Structures of selected ligands ie S3c, S5b and S6c and standard inhibitors were prepared in ChemSketch and saved as mol files. Open Bable software was used to convert 2 dimensional (2D) structures to 3D.54 Accelrys Discovery Studio Visualizer (version 4.1) was used to convert the structures in PDB file format for use in AutoDock Vina.55 The 3-dimensional crystal structures of cancer targets (proteins and DNA) were retrieved from the RCSB Protein Data Bank (http://www.rcsb.org/pdb). The proteins were set up for docking by removing attached ligand and crystallographic water molecules using Accelrys Discovery Studio Visualizer (version 4.1). The optimized proteins were then saved as a PDB file format for docking.
Molecular docking studies were performed using AutoDock Vina software (version 1.5.6)56 to determine binding affinities of ligands with respective protein targets and their binding energy values were also compared with anticancer standard drugs against respective targets. The AutoDock tool was used to add polar hydrogens in a protein molecule. The binding site for docking was designed such that the entire receptor molecule was included within the selection grid. Default parameters were used for docking. Binding affinities of synthesized and standard ligands were analyzed based on minimum binding energy values. Ligand protein binding interactions of complexes with minimum docking scores were analyzed using Accelrys Discovery Studio Visualizer (version 4.1).
Statistical Analysis
Graph Pad Prism 6.0 (Graph-Pad, San Diego, CA, United States) was used to analyze the data. For biochemical data, one way-ANOVA followed by Tukey’s post hoc test was applied. Data were demonstrated as mean ± SEM. Significant statistical differences were at *p < 0.05.
Results
Chemistry
New derivatives of Aminothiazole and aminopyridine derivatives were synthesized using the scheme given in Figure 1. Initially, the nitrogen group of amino acids was protected using phthalic anhydride S1(a-d). These were further reacted with thionyl chloride to yield corresponding acid chlorides and condensed with ethyl-2-aminothiazole-4-carboxylate and 2-aminopyridine to afford corresponding amide derivatives S3(a-d) and S4(a-d) respectively. Finally, the N-deprotection of compounds S3(a-d) and S4(a-d) was carried out in the presence of hydrazine hydrate to yield compounds S5(a-d) and S6(a-d). The respective reaction mechanisms for N-deprotection of compounds S3 and S4 are shown in Figures 2 and 3 respectively.
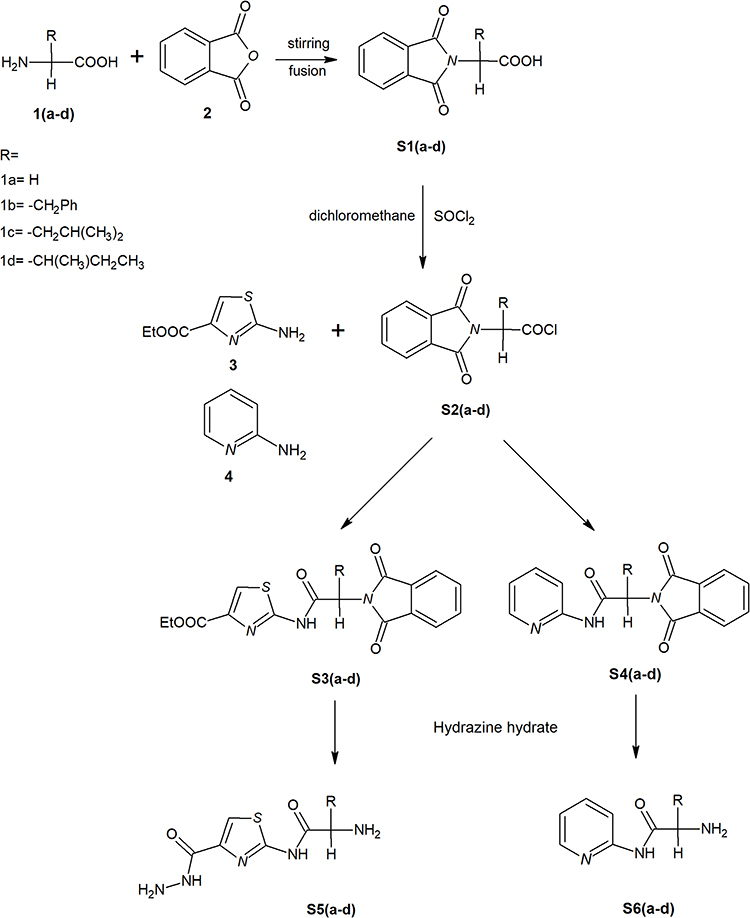 |
Figure 1 Synthesis of 2-aminothiazole and 2-aminopyridine derivatives. |
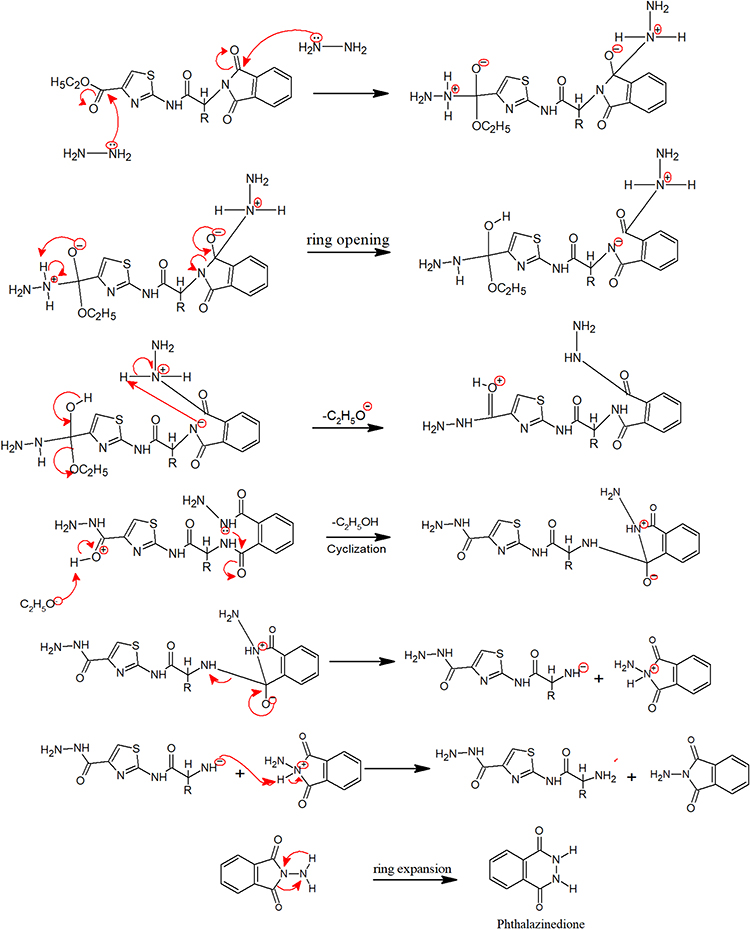 |
Figure 2 Proposed reaction mechanism to synthesize compound S5. |
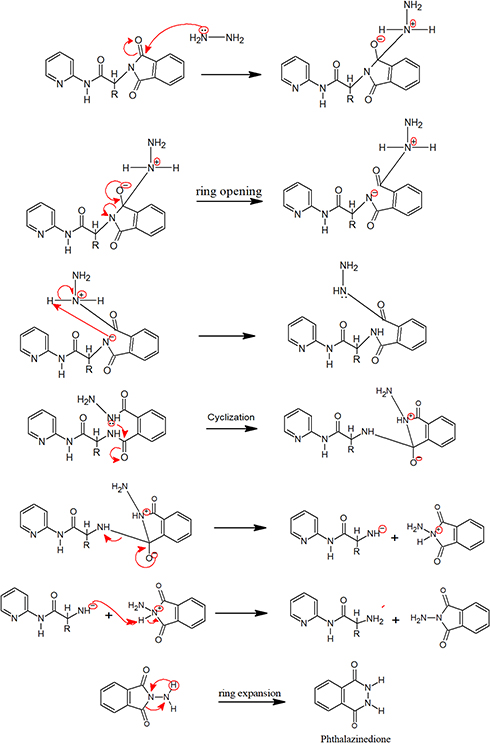 |
Figure 3 Proposed reaction mechanism to synthesize compound S6. |
In the FTIR spectra, the presence of amide stretch in compounds S3(a-d) and S4(a-d) ranging 1698–1690 cm−1 confirmed the formation of an amide linkage. Further, the 1H-NMR data of synthesized analogues presented a singlet of -NH proton ranging δ = 10.32–9.11 ppm, indicated the formation of coupled products. Similarly, in the case of S5(a-d) and S6(a-d) derivatives, the disappearance of carbonyl stretch for phthaloyl moiety in the range of 1635–1552 cm−1 confirmed the de-protection of amino acids.
In vitro Antioxidant Activity
2.2-diphenyl-1-picrylhydrazyl (DPPH) method was used to evaluate the antioxidant potential of newly synthesized compounds [S3(a-d), S4(a-d), S5(a-d), S6(a-d)] and the results are summarized in Figure 4. The antioxidant potential was calculated as the change in absorbance relative to the reference control. Probit regression was used to calculate EC50 values due to the non-linearity in regression.43 Compounds S3b, S3c, S4c, S5b, and S6c showed significant activities with EC50 values, 16.25 μg/mL, 14.16 μg/mL, 17.03 μg/mL, 12.97 μg/mL, and 15.19 μg/mL respectively to the reference ascorbic acid which was found as 10.41 μg/mL. Based on this preliminary assay, we selected five compounds (S3b, S3c, S4c, S5b, and S6c) for evaluation of their in vitro anticancer potential.
 |
Figure 4 DPPH radical scavenging activity of synthesized derivatives. |
In vitro Anticancer Activity
Synthesized derivatives S3b, S3c, S4c, S5b, and S6c exhibited relative significant antioxidant activities were further evaluated for anticancer potential in parent and cisplatin-resistant ovarian cancer cell lines, A2780 and A2780CISR respectively, while cisplatin was used as a standard drug. IC50 and the resistance factor (RF) values of standard drug cisplatin and tested compounds are given in Table 2. All the tested compounds exhibited a dose-dependent anti-proliferative effect against parent and cisplatin-resistant ovarian cancer cell lines. We demonstrated that compounds S3c, S5b, and S6c reversed the cisplatin resistance of cancer cells with IC50 values 11.52 µM, 14.32 µM, and 15.41 µM respectively relative to the standard drug cisplatin with IC50 values 16.43 µM. Furthermore, RF values for all the tested compounds S3b, S3c, S4c, S5b, and S6c were respectively ie 1.42, 0.74, 1.31, 0.56, and 0.49, while for cisplatin was 26.93. Moreover, compounds S3c, S5b, and S6c exhibited more potency in cisplatin-resistant cell lines than the parent cells by displaying less IC50 values, associated with RF values of 0.74, 0.56, and 0.49 respectively. Furthermore, derivative S3c demonstrated inhibitory effects both in parent and cisplatin-resistant ovarian cancer cell lines with an IC50 value of 15.57 µM, 11.52 µM respectively. This may be due to their variable effects on multiple proteins which are differentially expressed in sensitive and resistant cells (Figure 5).
 |
Table 2 Anticancer Activity of the Synthesized Thiazole and Pyridine Derivatives Against A2780 (Parent) and A2780CISR (Cisplatin-Resistant) Ovarian Cancer Cell Lines, Expressed as IC50 Values and Compared to Cisplatin, the Standard Drug |
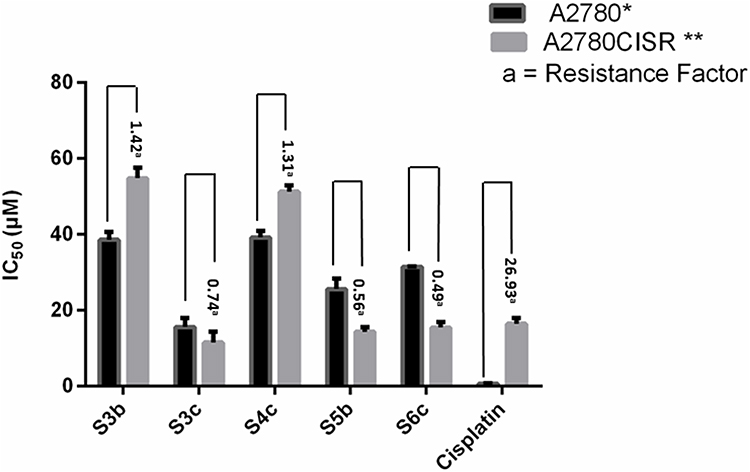 |
Figure 5 IC50 values of compounds and cisplatin (standard) as observed in the parent (A2780*) and cisplatin-resistant (A2780CISR**) ovarian cancer cell lines. Data are presented as mean ± SEM. One way ANOVA was used for the analysis of these results (n=5). |
Docking Evaluation
Synthesized compounds S3c, S5b, and S6c that exhibited significant in vitro anticancer activities were selected as ligands and docked against multiple protein targets such as EGFR, VEGFR, PDGFR, p110a/PI3K, RET, BCR-ABL, c-KIT, c-Raf, B-Raf, CTLA-4, and ALK. These targets were selected based upon their relative role in tumor development and are implicated in various cell signaling pathways. Further, the results of the docking study are summarized in Table 3. These results indicated that selected ligands demonstrated favorable interactions with multiple protein targets, which coincides with our experimental findings. Furthermore, compound S3c formed several hydrogen bonds with PDGFR, c-KIT, and ALK with the lowest binding energy values −5.2 Kcal/mol, −8.1 Kcal/mol, and −7.5 Kcal/mol respectively. Likely, compound S5b showed favorable docking against PDGFR, VEGFR, c-Raf, and CTLA-4 with binding energy values i.e −5.3 Kcal/mol, −8.6 Kcal/mol, −8.1 Kcal/mol, −5.7 Kcal/mol respectively that is comparable to standard inhibitors. It was analyzed that profound binding affinity was shown by compound S6c against BCR-Abl with the minimum free energy of binding −5.8 Kcal/mol. It was observed that these three derivatives possessed comparative binding affinity (possibly inhibitory) against EGFR and CTLA-4. As the p110a isoform of PI3K is mainly involved in the development of ovarian cancer, all three ligands were found to be active against this target. Compounds S3c, S5b, and S6c exhibited marked inhibition against p110a isoform of PI3K with binding energy values of −9.0 Kcal/mol, −8.5 Kcal/mol, and −7.9 Kcal/mol that is comparable to the reference compound ie −9.9 Kcal/mol.
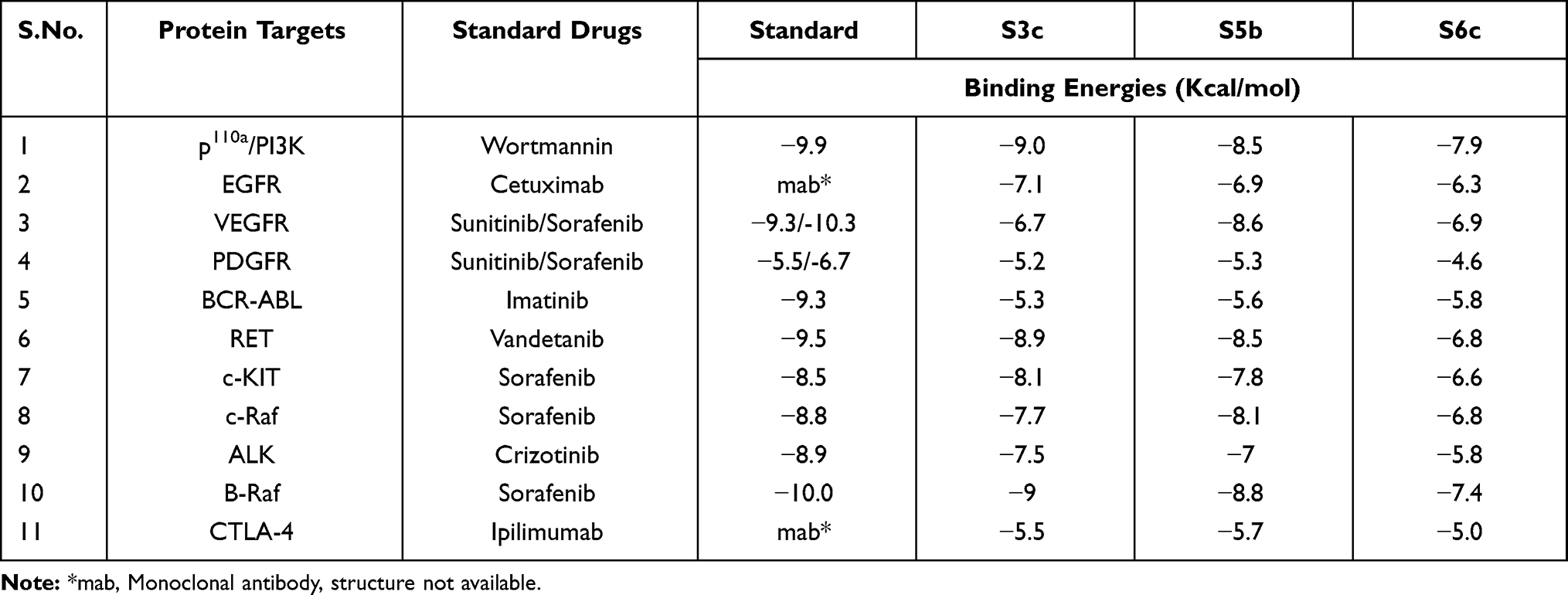 |
Table 3 Binding Energies of Ligand Molecules (S3c, S5b and S6c) and Standard Drugs with Target Proteins |
Binding mode and 2D interactions of compounds S3c & S5b with p110a isoform of PI3K, EGFR, VEGFR, and PDGFR are expressed in Figures 6–9 respectively, demonstrating that these derivatives can significantly inhibit multiple targets in cellular pathways.
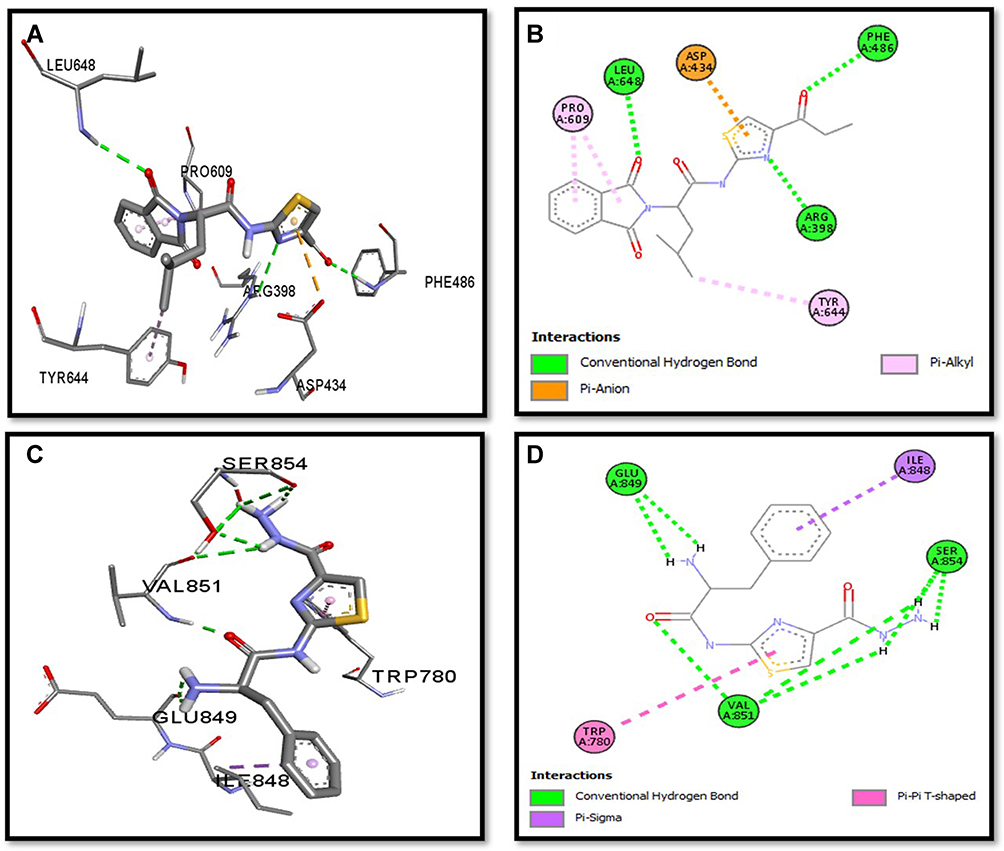 |
Figure 6 Post docking analysis visualized by Discovery Studio Visualizer in both 2D and 3D poses in the protein structures of p110a isoform of PI3K. Interaction between S3c and p110a isoform of PI3K (A and B), S5b and p110a isoform of PI3K (C and D). 3D poses (A and C) and 2D (B and D). |
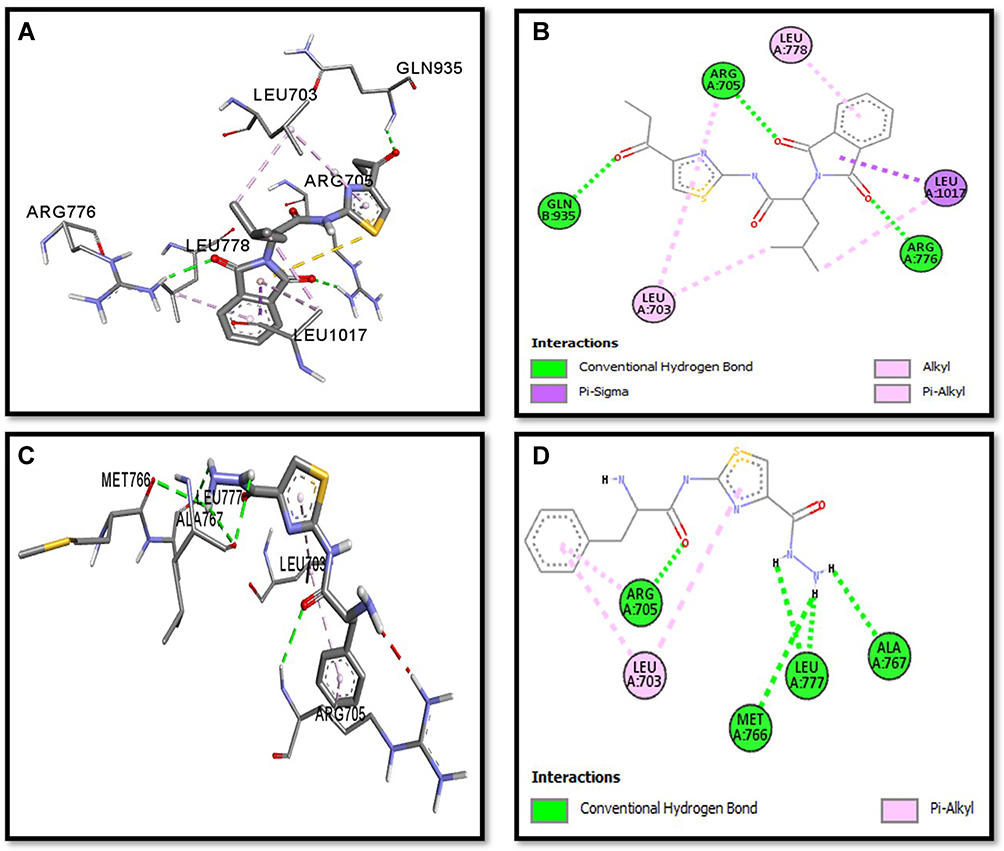 |
Figure 7 Post docking analysis visualized by Discovery Studio Visualizer in both 2D and 3D styles with EGFR. Interaction between S3c and EGFR (A and B), S5b and EGFR (C and D). 3D poses (A and C) and 2D (B and D). |
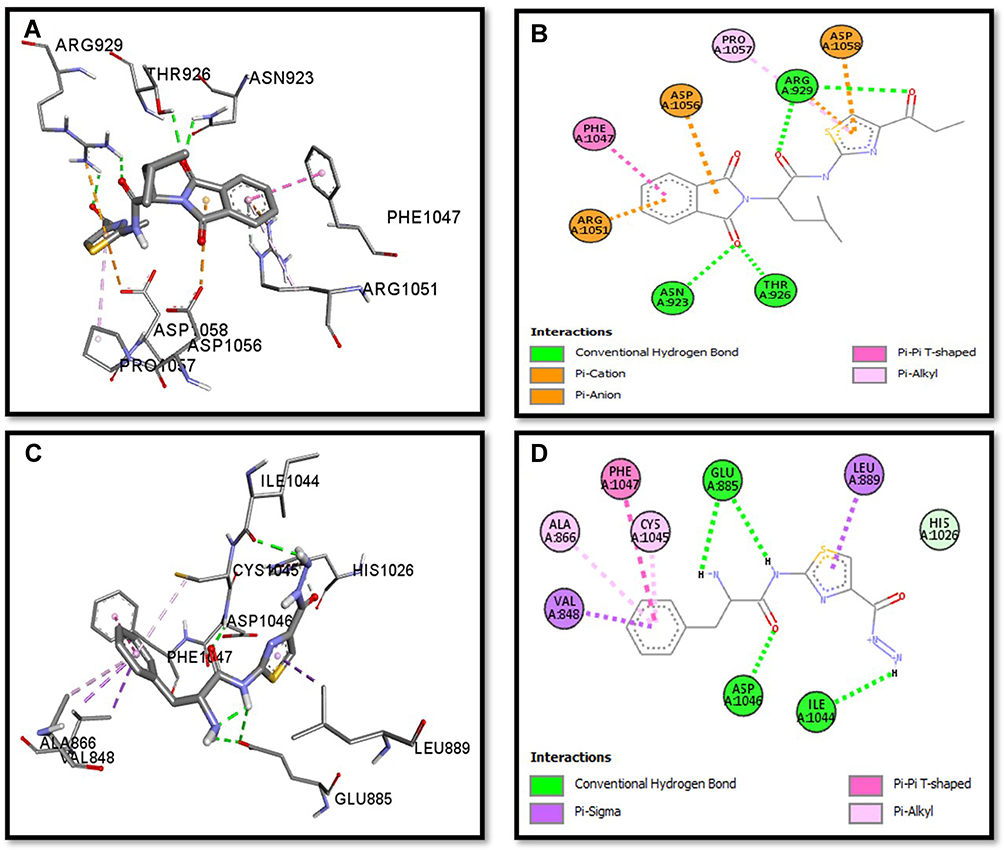 |
Figure 8 Post docking analysis visualized by Discovery Studio Visualizer in both 2D and 3D poses in the protein structures of VEGFR. Interaction between S3c and VEGFR (A and B), S5b and VEGFR (C and D). 3D poses (A and C) and 2D (B and D). |
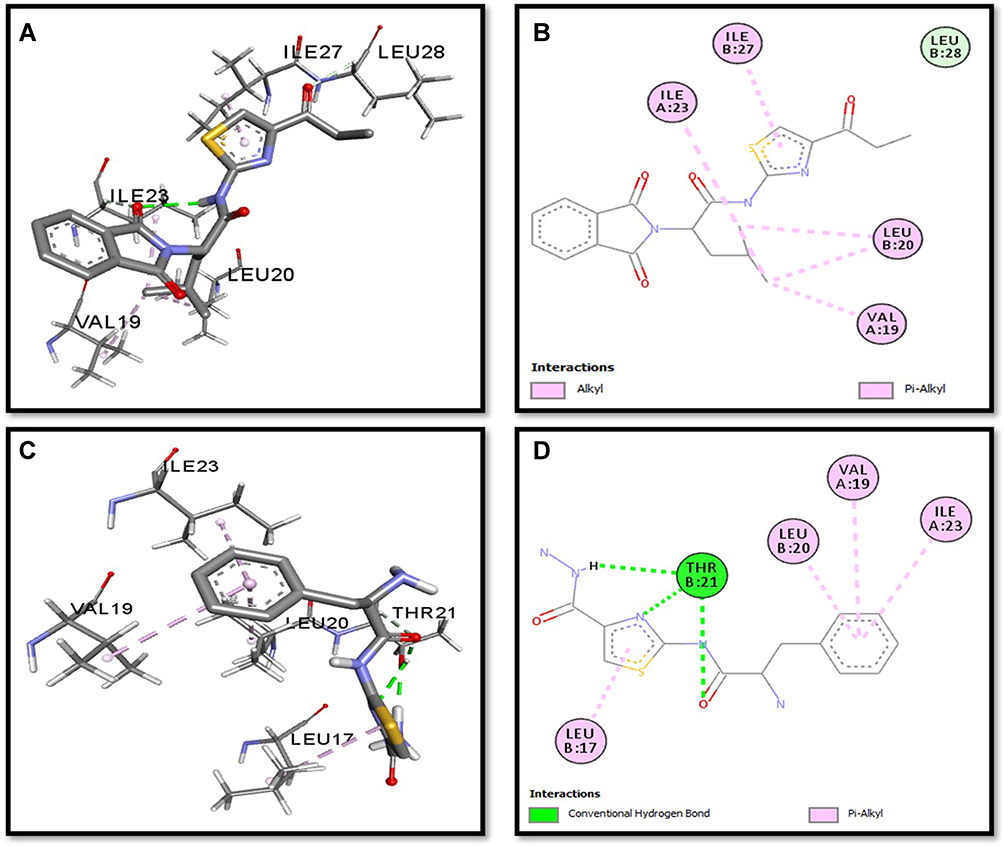 |
Figure 9 Post docking analysis visualized by Discovery Studio Visualizer in both 2D and 3D styles with PDGFR. Interaction between S3c and PDGFR (A and B), S5b and PDGFR (C and D). 3D poses (A and C) and 2D (B and D). |
Discussion
In this study, we demonstrated the synthesis of new 2-aminothiazole and 2-aminopyridine derivatives, which possess amino acid moieties to execute antioxidant and anticancer potential. Interest in the chemotherapeutic activity of thiazoles was potentiated by the discovery of natural antineoplastic agents such as tiazofurin, bleomycin, netropsin, and thiazole netropsin.44 Moreover, many clinically useful anticancer drugs possess thiazole and pyridine as active moieties including dactinomycin and Imatinib mesylate respectively.29,35
The network-medicine approach implies a promising strategy by targeting several proteins in a network by rewiring network structures particularly proteins, as denatured or misfolded protein will induce vulnerability for chemoresistance and subsequent failure of cancer therapies.45 We demonstrated that our synthesized compounds can target multiple protein targets such as EGFR, VEGFR, PDGFR, p110a/PI3K, RET, BCR-ABL, c-KIT, c-Raf, B-Raf, CTLA-4, and ALK and thereby validating the concept of network-based approach. Derivative S3c showed an equivalent capacity of inhibition against PDGFR, c-KIT, and ALK target with lowest binding energy values −5.2 Kcal/mol, −8.1 Kcal/mol, and −7.5 Kcal/mol respectively. Moreover, compound S5b exhibited strong bindings against cancer targets PDGFR, VEGFR, c-Raf, and CTLA-4 with energy values of −5.3 Kcal/mol, −8.6 Kcal/mol, −8.1 Kcal/mol, −5.7 Kcal/mol respectively. The p110a isoform of PI3K is the main target involved in the development of ovarian cancer,8 and all three ligands (S3c, S5b, and S6c) displayed a good binding affinity with this target with binding energy values of −9.0 Kcal/mol, −8.5 Kcal/mol and −7.9 Kcal/mol respectively.
Redox imbalance and oxidative stress are hallmarks of cancers, so, antioxidants play a significant role in cancer treatment by ameliorating levels of free radicals and oxidative stress.37,38 Therefore, the antioxidant activity of synthesized compounds was performed to preliminary screen the free radical scavenging potential. Based on our results, five compounds S3b, S3c, S4c, S5b, and S6c with an EC50 value of 16.25 ug/mL, 14.16 ug/mL, 17.03 ug/mL, 12.97 ug/mL, and 15.19 ug/mL respectively were selected for evaluation of the in vitro anticancer potential in comparison to ascorbic acid.
Cisplatin is clinically used in a wide spectrum of cancers, however, its therapeutic implications are limited by its toxicities, and tumor resistance.9 To overcome, we incorporated amino acids conjugation into thiazole and pyridine scaffold to minimize toxicities by enhancing selective cellular permeability of the heterocyclic scaffold.41 Cellular mechanisms responsible for cisplatin resistance are multifactorial that restrict their clinical use.12,13 Our results revealed that compounds S3c, S5b, and S6c were found to be more active in cisplatin-resistant cell lines than the parent cells by displaying less IC50 values, 11.52 µM, 14.32 µM, and 15.41 µM respectively relative to the standard cisplatin which was 16.43 µM along with stable RF values of 0.74, 0.56, and 0.49 respectively. RF determines the ability of a compound to induce cell death in resistant cells and RF value less than 2 is an indication of significant activity of a compound against resistant cell lines.46 The RF for all the tested compounds S3b, S3c, S4c, S5b, and S6c were noteworthy ie 1.42, 0.74, 1.31, 0.56, and 0.49 respectively, while, for cisplatin is 26.93. This may be due to their variable effects on genes and proteins which are differentially expressed in sensitive and resistant cells such as oncogenes (c-fos, c-myc, H-ras, c-jun, and c-abl), tumor suppressor genes (p53), transcription factors, and regulatory proteins involved in signal transduction pathways.14 Moreover, compound S3c demonstrated equal effectiveness in both parent and cisplatin-resistant ovarian cancer cells with an IC50 value of 15.57 µM, 11.52 µM respectively.
Therefore, it can be inferred that compounds S3c, S5b, and S6c can be considered as potential leads or hits for the development of new anticancer agents for cisplatin-resistant ovarian cancer. More detailed studies are required to delineate the detailed underlying mechanism of these compounds in different cancer cell lines and targets.
Conclusion
In conclusion, our findings demonstrated that derivatives S3c, S5b and S6c were found to be more active in cisplatin-resistant ovarian cancer cell lines than cisplatin-sensitive cells. Therefore, these compounds may be used as potential anticancer lead candidates for further screening because they might reduce oxidative stress in cancerous cells and also possess multiple target inhibitory potential.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported by Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions No: 2019SHIBS0004, Shenzhen, 518055, China.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Ghoneum A, Said N. PI3K-AKT-mTOR and NFκB pathways in ovarian cancer: implications for targeted therapeutics. Cancers. 2019;11:949. doi:10.3390/cancers11070949
2. Wu YH, Huang YF, Chen CC, Huang CY, Chou CY. Comparing PI3K/Akt inhibitors used in ovarian cancer treatment. Front Pharmacol. 2020;11:206. doi:10.3389/fphar.2020.00206
3. Lee MJ, Ye AS, Gardino AK, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149(4):780–794. doi:10.1016/j.cell.2012.03.031
4. Zhang F, Ren C, Lau KT. A network medicine approach to build a comprehensive atlas for the prognosis of human cancer. Brief Bioinform. 2016;17(6):1044–1059. doi:10.1093/bib/bbw076
5. Liu X, Wu J, Zhang D, et al. Network pharmacology-based approach to investigate the mechanisms of Hedyotis diffusa willd. in the treatment of gastric cancer. Evid Based Complement Alternat Med. 2018. doi:10.1155/2018/7802639
6. Nasser AA, Eissa IH, Oun MR. Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFRWT and EGFRT790M. Org Biomol Chem. 2020;18(38):7608–7634. doi:10.1039/D0OB01557A
7. Pawson T, Linding R. Network medicine. FEBS Lett. 2008;582:1266–1270. doi:10.1016/j.febslet.2008.02.011
8. Liu R, Chen Y, Liu G, et al. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020;11:797. doi:10.1038/s41419-020-02998-6
9. Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016;106:27–36. doi:10.1016/j.phrs.2016.01.001
10. Navya PN, Kaphle A, Daima HK. Nanomedicine in sensing, delivery, imaging and tissue engineering: advances, opportunities and challenges. Nanoscience. 2019;5:30–56.
11. Navya PN, Kaphle A, Srinivas SP, Bhargava SK, Rotello VM, Daima HK. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converge. 2019;6:23.
12. Chen S, Chang J. New insights into mechanisms of cisplatin resistance: from tumor cell to microenvironment. Int J Mol Sci. 2019;20(17):4136. doi:10.3390/ijms20174136
13. Ferreira JA, Peixoto A, Neves M, et al. Mechanisms of cisplatin resistance and targeting of cancer stem cells: adding glycosylation to the equation. Drug Resist. 2016;24:34–54.
14. Zhou J, Kang Y, Chen L, et al. The drug-resistance mechanisms of five platinum-based antitumor agents. Front Pharmacol. 2020;11:343.
15. Vitaku E, Smith DT, Njardarson JT. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem. 2014;57:10257–10274. doi:10.1021/jm501100b
16. Bosenbecker J, Bareno VDO, Difabio R, et al. Synthesis and antioxidant activity of 3-(pyridin-2-yl,ethyl)-1,3-thiazinan(thiazolidin)-4-ones. J Biochem Mol Toxic. 2014;28:425–432. doi:10.1002/jbt.21581
17. Sraa AM, Mastoura ME, Heba HS, et al. Synthesis and biological evaluation of some novel thiazole-based heterocycles as potential anticancer and antimicrobial agents. Molecules. 2019;24:539. doi:10.3390/molecules24030539
18. Hussein AM, Khames AA, El-Adasy AA, et al. Design, synthesis and biological evaluation of new 2-aminothiazole scaffolds as phosphodiesterase type 5 regulators and COX-1/COX-2 inhibitors. RSC Adv. 2020;10(50):29723–29736. doi:10.1039/D0RA05561A
19. Rodríguez-Rangel S, Bravin AD, Ramos-Torres KM, Brugarolas P, Sánchez-Rodríguez JE. Structure-activity relationship studies of four novel 4-aminopyridine K+ channel blockers. Sci Rep. 2020;10(1):52. doi:10.1038/s41598-019-56245-w
20. Hersi F, Omar HA, Al-Qawasmeh RA, et al. Design and synthesis of new energy restriction mimetic agents: potent anti-tumor activities of hybrid motifs of aminothiazoles and coumarins. Sci Rep. 2020;10(1):2893. doi:10.1038/s41598-020-59685-x
21. El-Naggar AM, Eissa IH, Belal A, El-Sayed AA. Design, eco-friendly synthesis, molecular modeling and anticancer evaluation of thiazol-5(4H)-ones as potential tubulin polymerization inhibitors targeting the colchicine binding site. RSC Adv. 2020;10(5):2791–2811. doi:10.1039/C9RA10094F
22. Eissa IH, Metwaly AM, Belal A, et al. Discovery and antiproliferative evaluation of new quinoxalines as potential DNA intercalators and topoisomerase II inhibitors. Arch Pharm. 2019;352(11):e1900123. doi:10.1002/ardp.201900123
23. El‐Helby AA, Sakr H, Eissa IH, Al‐Karmalawy AA, El‐Adl K. Benzoxazole/benzothiazole‐derived VEGFR‐2 inhibitors: design, synthesis, molecular docking, and anticancer evaluations. Arch Pharm. 2019;352(12):e1900178. doi:10.1002/ardp.201900178
24. Abu-Melha S, Edrees MM, Riyadh SM, Abdelaziz MR, Elfiky AA, Gomha SM. Clean grinding technique: a facile synthesis and in silico antiviral activity of hydrazones, pyrazoles, and pyrazines bearing thiazole moiety against SARS-CoV-2 main protease (Mpro). Molecules. 2020;25(19):4565. doi:10.3390/molecules25194565
25. Sayed AR, Gomha SM, Taher EA, et al. One-pot synthesis of novel thiazoles as potential anti-cancer agents. Drug Des Devel Ther. 2020;14:1363–1375. doi:10.2147/DDDT.S221263
26. Abdelrazek FM, Gomha SM, Shaaban MEB, et al. One-pot three-component synthesis and molecular docking of some novel 2-thiazolyl pyridines as potent antimicrobial agents. Mini Rev Med Chem. 2019;19(6):527–538. doi:10.2174/1389557518666181019124104
27. Abdelrazek FM, Gomha SM, Abdelrahman AH, Metz P, Sayed MA. A facile synthesis and drug design of some new heterocyclic compounds incorporating pyridine moiety and their antimicrobial evaluation. Lett Drug Des Discov. 2017;14(7):752–762. doi:10.2174/1570180814666161128120240
28. Ali AR, El-Bendary ER, Ghaly MA, et al. Novel acetamidothiazole derivatives: synthesis and in vitro anticancer evaluation. Eur J Med Chem. 2013;69:908–919. doi:10.1016/j.ejmech.2013.08.021
29. Popsavin M, Spaic S, Svircev M, et al. Synthesis and antitumour activity of new tiazofurin analogs bearing a 2,3-anhydro functionality in the furanose ring. Bioorg Med Chem Lett. 2007;17:4123–4127. doi:10.1016/j.bmcl.2007.05.050
30. Schiavi B, Ahond A, Al-Mourabit A, et al. Synthesis of 5-deazathiogirollines: analogs of a natural anticancer agent. Tetrahedron. 2002;58:4201–4215. doi:10.1016/S0040-4020(02)00383-6
31. Yeh CB, Su CJ, Hwang JM, et al. Therapeutic effects of cantharidin analogues without bridging ether oxygen on human hepatocellular carcinoma cells. Eur J Med Chem. 2010;45:3981–3985. doi:10.1016/j.ejmech.2010.05.053
32. Abbas I, Gomha S, Elaasser M, Bauomi M. Synthesis and biological evaluation of new pyridines containing imidazole moiety as antimicrobial and anticancer agents. Turk J Chem. 2015;39(2):334–346. doi:10.3906/kim-1410-25
33. Gomha SM, Muhammad ZA, Abdel-aziz HM, Matar IK, El-Sayed AA. Green synthesis, molecular docking and anticancer activity of novel 1,4-dihydropyridine-3,5-Dicarbohydrazones under grind-stone chemistry. Green Chem Lett Rev. 2020;13(1):6–17. doi:10.1080/17518253.2019.1710268
34. Gomha SM, Abdelrazek FM, Abdelrahman AH, Metz P. Synthesis of some new pyridine‐based heterocyclic compounds with anticipated antitumor activity. J Heterocycl Chem. 2018;55(7):1729–1737. doi:10.1002/jhet.3210
35. El-Naggar M, Almahli H, Ibrahim HS, Eldehna WM, Abdel-Aziz HA. Pyridine-ureas as potential anticancer agents: synthesis and in vitro biological evaluation. Molecules. 2018;23(6):1459. doi:10.3390/molecules23061459
36. Klaunig JE. Oxidative stress and cancer. Curr Pharm Des. 2018;24(40):4771–4778. doi:10.2174/1381612825666190215121712
37. Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative stress in cancer. Cancer Cell. 2020;38(2):167–197.
38. Kruk J, Aboul-Enein HY. Reactive oxygen and nitrogen species in carcinogenesis: implications of oxidative stress on the progression and development of several cancer types. Mini Rev Med Chem. 2017;17(11):904–919. doi:10.2174/1389557517666170228115324
39. Alizadeh SR, Hashemi SM. Development and therapeutic potential of 2-aminothiazole derivatives in anticancer drug discovery. Med Chem Res. 2021;1–36. doi:10.1007/s00044-020-02686-2
40. Gomha SM, Muhammad ZA, Abdel-aziz MR, Abdel-aziz HM, Gaber HM, Elaassere MM. One-pot synthesis of new thiadiazolyl-pyridines as anticancer and antioxidant agents. J Heterocycl Chem. 2018;55(2):530–536. doi:10.1002/jhet.3088
41. Arun R, Ashok KCK. Synthesis, hydrolysis and pharmacodynamics profiles of novel prodrugs of mefenamic acid. Int J Curr Pharm Res. 2009;1:47–55.
42. Prakasha KC, Raghavendra GM, Harisha R, et al. Design, synthesis and antimicrobial screening of amino acids conjugated 2-amino-4-arylthiazole derivatives. Int J Pharm Pharm Sci. 2011;3:120–125.
43. Locatelli M, Gindro R, Travaglia F, et al. Study of DPPH-scavenging activity: development of a free software for the correct interpretation of data. Food Chem. 2009;114:889–897. doi:10.1016/j.foodchem.2008.10.035
44. Al-Saadi SM, Hassan MF, Sherif AFR. Synthesis and biological evaluation of some 2,4,5- trisubstituted thiazole derivatives as potential antimicrobial and anticancer agents. Arch Pharm Chem Life Sci. 2008;341:424–434. doi:10.1002/ardp.200800026
45. Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68. doi:10.1038/nrg2918
46. Laila A, Philip B, Charles C, et al. Synergism from combinations of tris(benzimidazole) monochloroplatinum(II) chloride with capsaicin, quercetin, curcumin and cisplatin in human ovarian cancer cell lines. Anticancer Res. 2014;34:5453–5464.
47. Madhu G, Nanjaian M, Darpan K. Facile synthesis and characterization of 2-aminothiazole-4-carbohydrazide. Int J Rec Adv Pharm Res. 2011;2:27–29.
48. Hassan M, Farzana S, Uzma Y, et al. Preparation of optically active amino acid derivatives of some methylated 5-amino azaheterocycles. Turk J Chem. 2000;24:165–175.
49. Cyril O, Didier ML, Johan W, et al. Synthesis and anticonvulsant activity of N,N-phthaloyl derivatives of central nervous system inhibitory amino acids. Arch Pharm Pharm Med Chem. 2001;334:323–331. doi:10.1002/1521-4184(200110)334:10<323::AID-ARDP323>3.0.CO;2-O
50. Abhilasha V, Nirupam D, Meenakshi D, et al. Conjugation of some NSAIDs with 5-phenyl-2-aminothiazole for reduced ulcerogenicity. Thai J Pharm Sci. 2010;34:49–57.
51. Sinead OMC, Joan EM, Stanley RM, et al. Intermediates in the Ing-Manske reaction. ARKIVOC. 2003;VII:180–189.
52. Molyneux P. The use of the stable free radical diphenyl picryl hydrazyl (DPPH) for estimating antioxidant activity. Songklanakarin J Sci Technol. 2004;26:211–219.
53. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi:10.1016/0022-1759(83)90303-4
54. O’Boyle NM, Banck M, James CA, et al. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:1–14. doi:10.1186/1758-2946-3-1
55. Rao SN, Head MS, Kulkarni A, et al. Validation studies of the site-directed docking program LibDock. J Chem Inf Model. 2007;47:2159–2171. doi:10.1021/ci6004299
56. Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a lamarckian genetic algorithm and and empirical binding free energy function. J Comput Chem. 1998;19:1539–1662. doi:10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.