")
Back to Journals » Journal of Inflammation Research » Volume 15
Alterations and Prediction of Functional Profiles of Gut Microbiota After Fecal Microbiota Transplantation for Iranian Recurrent Clostridioides difficile Infection with Underlying Inflammatory Bowel Disease: A Pilot Study
Authors Azimirad M, Jo Y, Kim MS, Jeong M, Shahrokh S, Aghdaei HA , Zali MR, Lee S , Yadegar A , Shin JH
Received 8 September 2021
Accepted for publication 28 December 2021
Published 6 January 2022 Volume 2022:15 Pages 105—116
DOI https://doi.org/10.2147/JIR.S338212
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Monika Sharma
Masoumeh Azimirad,1,* YoungJae Jo,2,* Min-Sueng Kim,2 Minsoo Jeong,2 Shabnam Shahrokh,3 Hamid Asadzadeh Aghdaei,4 Mohammad Reza Zali,3 Seungjun Lee,5 Abbas Yadegar,1 Jae-Ho Shin2
1Foodborne and Waterborne Diseases Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 2Department of Applied Biosciences, Kyungpook National University, Daegu, 41566, Republic of Korea; 3Gastroenterology and Liver Diseases Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 4Basic and Molecular Epidemiology of Gastrointestinal Disorders Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 5Department of Food Science and Nutrition, College of Fisheries Science, Pukyong National University, Busan, Republic of Korea
*These authors contributed equally to this work
Correspondence: Abbas Yadegar; Jae-Ho Shin Tel +98-21-22432518; +82 53-950-5716
Fax +98-21-22432527; +82 53-953-7233
Email [email protected]; [email protected]
Background and Purpose: Fecal microbiota transplantation (FMT) has emerged for the therapeutic treatment of recurrent Clostridioides difficile infection (rCDI) with concurrent inflammatory bowel disease (IBD). As the first Iranian population cohort, we examined how gut microbiota and their functional profiles change in Iranian rCDI patients with underlying IBD before and after FMT.
Patients and Methods: FMT was performed to eight IBD patients via colonoscopy. Profiles of gut microbiota from donors and recipients were investigated using 16S rRNA gene sequence analysis.
Results: Patients experienced no IBD flare-ups or other adverse effects, and all recovered to full health. Moreover, all rCDI patients lacked the Bacteroidetes present in donor samples. After FMT, the proportion of Bacteroidetes increased until a normal range was achieved. More specifically, the relative abundance of Prevotella was found to increase significantly following FMT. Prevotella was also found to correlate negatively with inflammation metrics, suggesting that Prevotella may be a key factor for resolving CDI and IBD. Gut microbiota diversity was found to increase following FMT, while dysbiosis decreased. However, the similarity of microbial communities of host and recipients did not increase, and wide variation in the extent of donor stool engraftment indicated that the gut bacterial communities of recipients do not shift towards those of donors.
Conclusion: FMT leads to significant alterations of the community structure of gut bacteria in rCDI patients with IBD. The change in relative abundance of Proteobacteria and bacterial diversity indicated that FMT promotes recovery from intestinal permeability and inflammation in rCDI patients. Moreover, strong negative correlation between Prevotella and inflammation index, and decreased dysbiosis index advocate that the improvement of CDI is possibly due to gut microbiome alteration. Collectively, our findings show that FMT would be a promising therapy to help reprogram the gut microbiome of Iranian rCDI patients with IBD.
Keywords: fecal microbiota transplantation, Clostridioides difficile infection, inflammatory bowel disease, gut microbiome, functional profiles, gut dysbiosis
Introduction
The gastrointestinal tract harbors a complex and dynamic microbial community that regulates host metabolic and immune functions.1 The gut microbes that inhabit the mammalian gut collectively provide myriad physiological and immunological functions that are distinct from the host’s own constitutive resources.2 However, disturbances to the composition and function of gut microbial inhabitants induced by antibiotics administration, comorbidities and other environmental factors can lead to life-threatening diseases.3 Imbalance and undesirable changes in the gut microbiome, sometimes referred to as intestinal dysbiosis, have been linked to infections such as Clostridioides difficile (C. difficile) infection (CDI), inflammatory bowel disease (IBD), metabolic syndrome, allergic diseases, and other systemic inflammatory disorders.4 A diversified gut microbiota can produce various beneficial products, aid in nutrient absorption, and maintain gut barrier integrity, which promotes colonization resistance through dynamic antagonistic interactions with pathogens like C. difficile.5
C. difficile is the leading cause of nosocomial diarrhea and remains a major healthcare concern contributing to high morbidity and mortality worldwide.6 Based on our previous 14-year-long cross-sectional study, there is an increasing prevalence of CDI (15.9%) in Tehran healthcare settings.7 Approximately 20–30% of patients experience recurrent CDI (rCDI) after cessation of antibiotic therapy for the initial infection, which can increase to 50–60% after a subsequent episode.8 Generally, CDI is associated with antibiotic use that disturbs the normal composition and structure of intestinal microbiota, thereby resulting in the colonization and overgrowth of C. difficile and consecutive toxin production.9 Patients with IBD are easily infected by C. difficile, and the incidence of CDI among the IBD population is reported to be higher than in patients without IBD.10 The imbalance of the gut microbiota in patients with IBD, characterized by a reduction in overall microbial biodiversity and perturbation of their regular functions, has been suggested to markedly impact the periodicity of disease progression and remission.11
Fecal microbiota transplantation (FMT), which restores the normal composition and functionality of the gut microbiota, has been proven as a potent therapeutic option for rCDI with success rates of >90%.12 In addition to CDI, some studies have demonstrated that FMT could emerge as a promising treatment approach to cure IBD by effectively correcting underlying dysbiosis, inducing clinical remission.13–15 Although the exact beneficial mechanisms of FMT remain poorly understood, FMT can reverse antibiotic-induced or disease-associated alterations in the disrupted gut microbiome by replacing it with a normal intestinal microbiota resembling that of healthy donors.16
Recently, our previous study demonstrated that FMT could be an effective and safe therapeutic alternative for treatment of rCDI in patients with concurrent IBD.14 The objective of this study is to examine how gut microbiota and their related functional profiles change in Iranian rCDI patients with underlying IBD before and after FMT.
Patients and Methods
Patient Selection
Eight patients with IBD and rCDI aged over 18 years were participated in this one-year study from November 2018 to April 2019. They were offered options at two teaching hospitals in Tehran for an FMT procedure to cure their rCDI.14 The treatment protocol for FMT in patients with rCDI was approved by the Institutional Ethical Review Committee of Research Institute for Gastroenterology and Liver Diseases at Shahid Beheshti University of Medical Sciences (Project No. IR.SBMU.RIGLD.REC.1396.185), and the study was also conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all eligible subjects prior to participation in the FMT procedure. Fecal samples were obtained from donors and patients one week (wk) prior to FMT and again from patients 2 and 8 wk following FMT. Clinical follow-up of patients was performed 2 and 8 wk after FMT to monitor for cure, failure and adverse events in accordance with the approved treatment protocol for FMT in patients with rCDI in Iran. This study in Tehran was part of a clinical project of our FMT research group that was conducted at the College of Agriculture and Life Sciences, Kyungpook National University in South Korea.
Clinical Diagnosis
The criteria for receiving FMT were 1) at least three or more documented episodes of mild-to-moderate CDI, or at least two episodes of severe CDI requiring hospitalization and 2) failure of a 6- to 8-week course of therapy with tapered vancomycin.14,17,18 Clinical, endoscopic, and histopathologic findings and disease activity pertaining to ulcerative colitis and Crohn’s disease were measured using the Mayo score and the Crohn’s disease activity index, respectively. Primary and secondary endpoints were defined on clinical grounds as the resolution of diarrhea while off antibiotics for CDI at 8 weeks after last FMT and resolution of clinical symptoms subsequent to repeat FMT after failure of the initial FMT.14
Research Participants
Eligible participants were IBD patients with moderate to severe CDI despite treatment with anti-TNF alpha agents, corticosteroids, immunomodulators and high-dose vancomycin for at least 10 days. Patients were excluded if they were pregnant, unable to provide informed consent, had a history of disease severity requiring hospitalization and had received invasive mechanical ventilation, or because of recent chemotherapy, advanced human immunodeficiency virus (HIV) or other severe immunodeficiency.
Healthy family members or friends (adults ≥ 18 years) were screened based on stool and serology for possible risk factors for potentially transmittable diseases including bacterial, parasitic, and viral pathogens. A complete overview of the donor screening procedure is documented in our previous study.14
FMT Procedure
As we previously reported, the donors took a single dose of osmotic laxative (magnesium hydroxide) the night before the stool donation and were instructed to collect 50 to 100 g of fresh feces and immediately transport it to the laboratory on the day of the scheduled FMT. Approximately 50 g of donor stool was dissolved in 300 mL of sterile physiological saline (0.9% w/v of NaCl) and filtered through gauze to make a liquid slurry. A total of 300 mL of the fecal suspension was administered into the recipient’s terminal ileum or cecum via the colonoscope working channel within 6 hours after collection via colonoscopy following bowel purge preparation (polyethylene glycol) the day before the procedure.
16S rRNA Gene Sequencing of Bacterial Community in Fecal Samples
Total bacterial genomic DNA was extracted from 35 stool samples using the QIAamp® DNA Mini Kit (QIAGEN, Hilden, Germany) following the manufacturer’s protocol. Extracted DNA was kept in 70% alcohol and shipped to Kyungpook National University in South Korea for 16S amplicon sequencing. The V4-V5 hypervariable regions in the 16S rRNA gene were amplified using the forward primer 515F (5′-individual barcode-GTGCCAGCMGCCGCGG-3′) and reverse primer 907R (5′-individual barcode-CCGTCAATTCMTTTRAGTTT-3′). PCR was implemented according to the conditions described previously.19 Briefly, the PCR mixture consisted of 25 μL Emerald AMP GT PCR 1× Master Mix (Takara Bio, Shiga, Japan), 1 μL (10 μM) of each barcoded PCR primer pair, 1 μL of DNA template (1–50 ng DNA) and 22 μL of ultrapure water. The samples were incubated at 95°C for 5 min, followed by 35 cycles of 95°C for 30 s, 57°C for 30 s, and 72°C for 30 s, and finally 72°C for 5 min. PCR products were then held at 4°C until analysis. The concentration of PCR products was measured using a Qubit® 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). The amplicon products were purified using an AMPure XP bead purification kit (Beckman Coulter, Brea, CA, USA) and pooled in equal concentrations. A 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) was used to verify the ideal concentration. The prepared libraries were sequenced on an Ion S5 (Thermo Fisher Scientific Korea Inc., Seoul, Korea).
Bioinformatic and Statistical Analyses
Analysis of the generated raw sequences was conducted using the software package Quantitative Insights Into Microbial Ecology 2 (QIIME2, v. 2020.8).20 Sequence reads were designated to samples according to their individual barcodes. Samples with an average Phred quality score lower than Q30 (Phred ≥ Q30) were removed. To generate amplicon sequence variants (ASVs), a trimmed length of 250 bp was denoised and chimeric sequences were removed using DADA2.21 ASVs were selected using a naive Bayes QIIME2 classifier based on a distance of 0.01 (≥99% identity) with the Greengenes database (v.13_8).22 The depth of the feature table was rarefied at 1578 reads.
Overall fecal microbiomes were analyzed using RStudio 1.3.1093 (https://www.rstudio.com/), the Calypso web application (v. 8.84),23 and GraphPad Prism 8 (GraphPad Software, Inc., San Diego, CA). Taxonomic classifications were assessed at phylum, order, and genus level. Using the R packages phyloseq,24 vegan,25 microbiome,24 and picante,26 alpha diversity indices (Observed features, Chao1, and Simpson) with Faith’s phylogenetic diversity were evaluated. The Wilcoxon signed-rank test was conducted, and p values less than 0.05 were regarded as statistically significant.
To track whether the microbial compositions of CDI patients and donors were more similar following FMT, beta diversities were calculated based on two distance matrices, weighted and unweighted UniFrac.27 The Wilcoxon signed-rank test was then carried out to calculate the statistical significance of differences between the pre- and post-FMT distance values.
In addition, since CDI and IBD are associated with dysbiosis,28 a microbial dysbiosis (MD) index was used to investigate the dominance of individual taxa associated with the disease condition to identify dysbiosis severity based on the changes in microbial community structure in the patient’s fecal microbiota. As defined by Gevers et al29 a high value of the MD index indicates a positive correlation with clinical disease seriousness. The Wilcoxon signed-rank test was performed to compare the MD index values between the communities.
The SourceTracker2 program was utilized to estimate the proportion of the donor’s original fecal bacterial community present in the patient’s intestinal environment after transplant.30 The gut microbiomes of all donors and pre-FMT patients were designated as sources and those of the post-FMT patients as sinks. For all samples at 2 weeks and 8 weeks post-FMT, SourceTracker2 calculated the proportion of the patients’ microbiome communities that were attributed to each source microbiome, first for the donor (described as Donor sample), second for the patient’s pre-FMT microbiome (described as Patient sample), and third for unknown community. To better understand the engraftment results, we conducted microbial source tracking based on the ASV and genus levels as well.
Metagenomic functional prediction was carried out using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 (PICRUSt2 v. 2.1.3).31 The prediction of abundances for each gene family group was generated from the ASV table file, and we followed the PICRUSt2 default scripts. The obtained prediction of metagenomic functional abundances was combined with descriptions from the KEGG ORTHOLOGY (KO) database, and the log2-transformed abundances of particular genes involved in oxidative phosphorylation were described in a Sankey diagram.
Pan-genome analysis was used to compare the taxonomic and genetic differences in Prevotella genera between donor and patient samples. Using “seq” tool in seqkit software, the donor-specific Prevotella ASVs detectable only in donor samples and patient specific ones were extracted.32 We then acquired the deeper taxonomy of each ASVs in NCBI blast with default parameters. Subsequently the PICRUSt2 analysis was performed to investigate the gene components of Prevotella genera differentially detectable in donor and patient guts respectively and generate the functional gene lists. Core genes (detected in all strains), accessory genes (shared at least by two strains) and unique genes (strain-specific genes) were identified by the rules reported in the previous study.33 The core genes in donor-specific Prevotella group were extracted and those functional genes were annotated from KO database.
Results
FMT Has Shown Successful Resolution of rCDI Symptoms
Six male and two female rCDI patients with IBD (one with CD and seven with UC) participated in this study. The average age of the patients was 35 years (22–60 years). Patients’ BMIs ranged from 18.3 to 29.6 kg/m2; thus, none recorded a BMI over 30 kg/m2, which Boulange et al define as obese. One patient was hospitalized during FMT treatment, while the remaining seven patients received FMT as outpatients. Four patients suffered watery-bloody stools and the other four experienced watery stools. Transplant was completed via colonoscopy with fresh donor stools. Six related and two unrelated donors contributed stool for FMT. Six patients were diagnosed with healthy status after the first FMT procedure and the other two were diagnosed as healthy after a second. No correlation was found between the number of FMT procedures needed and the type of IBD. C-reactive protein (CRP) values, a marker of inflammation, decreased by a median value of 7.25 (range 15.4–1). Table S1 provides specific information about the characteristics and clinical data of patients and donors as well as the antibody therapies applied before FMT.
FMT Induces Alteration of Gut Microbiome in Transplant Recipients: Transfer of Fecal Microbiota Achieves Healthier Intestinal Microbial Community in CDI Patients
Our previous study with clinical data demonstrated that all patients were diagnosed with healthy status after FMT procedures.14 In the present study, the effects of FMT on gut microbiota in patients were examined. To assess the changes in the gut microbiome following FMT, stool samples were collected prior to FMT and 2 and 8 weeks following FMT, described as “Pre”, “2 wk”, and “8 wk”, respectively. The taxonomic classifications of individual rCDI patients’ microbiomes showed varying proportions of Bacteroidetes, Firmicutes, and Proteobacteria at the phylum level (Figure S1A and Bacteroidales, Clostridiales and Enterobacteriales at the order level (Figure S1B). While rCDI patients overall exhibited a deficiency of Bacteroidetes, accounting for only approximately 18% of the total fecal bacterial community pre-FMT, this was increased to 32% at 2 wk and 38% at 8 wk following transplant (Figure 1A). Simultaneously, at the order level, the relative abundance of Bacteroidales surged from 18.62% to 37.59% while that of Enterobacteriales dropped from 18.97% to 0.04% (Figure 1B).
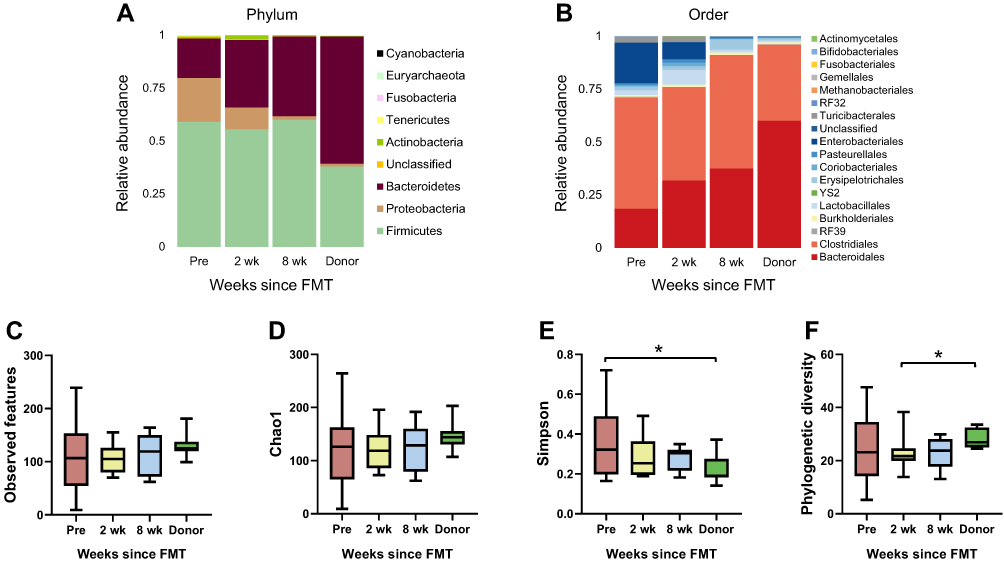 |
Figure 1 (A and B) Changes in recipient fecal bacterial composition following FMT. Relative abundances of taxonomic groups were described in 4 groups (recipients a week prior to FMT and 2 and 8 weeks after FMT and donors). Bacteroidetes increased and Proteobacteria decreased at the phylum level after FMT, and the bacterial composition at the order level showed increasing Bacteroidales and diminishing Enterobacteriales. (C–F) Changes in alpha diversity according to various indices. Significant differences among groups were not found for observed features or Chao1 but were observed for Simpson diversity (*Wilcoxon signed-rank test p < 0.05 for Pre and Donor) and phylogenetic diversity (*Wilcoxon signed-rank test p < 0.05 for 2 wk and Donor). |
To compare the alpha diversity of gut microbiomes among the groups, several indices were used, including observed features and Chao1 for richness, Simpson for evenness, and phylogenetic diversity (Figure 1C–F). Regarding Chao1 and observed features, although recipients’ diversity increased slightly, no significant change was detected. The rCDI patients prior to FMT exhibited the highest Simpson diversity index values, with a significant difference between the Pre and Donor groups. The phylogenetic diversity index also demonstrated a significant difference between the 2 wk and Donor groups, with the latter being higher. The various indices of fecal microbial diversity shifted toward the donors’ values following treatment, though the differences between pre- and post-FMT recipient values were not statistically significant.
After FMT, The Fecal Microbial Diversity of rCDI Patients Shifted to Be Nearly Equivalent to That of Donors, but the Bacterial Composition Changed Distinctively
Beta diversity analysis was performed to determine whether the patients’ gut microbial composition structure resembled that of the donors following FMT. Weighted and unweighted UniFrac matrices were used to monitor the distances between the microbial compositions of the recipient and Donor groups. After patients received FMT, the weighted UniFrac demonstrated that the microbial community distance was smaller at 2 wk but increased at 8 wk (Wilcoxon signed-rank test, p > 0.05, Figure 2A). Conversely, the distance according to the unweighted UniFrac matrix increased at 2 wk and decreased at 8 wk (Wilcoxon signed-rank test, p > 0.05, Figure 2B). Consequently, the beta diversity between patients and donors did not change significantly. Source tracking was used to evaluate the proportion of donors’ microbiota that was engrafted in patients by revealing the extent to which patients’ fecal bacterial community came to resemble that of donors. Among the recipients, the extent of bacterial engraftment following FMT varied from 4% to 86% (at 2 weeks) and 15 to 79% (at 8 weeks) based on ASV (Figure 2D), and 3% to 97% (at 2 weeks) and 1% to 98% (at 8 weeks) based on the genus level per SourceTracker2 (Figure 2C).
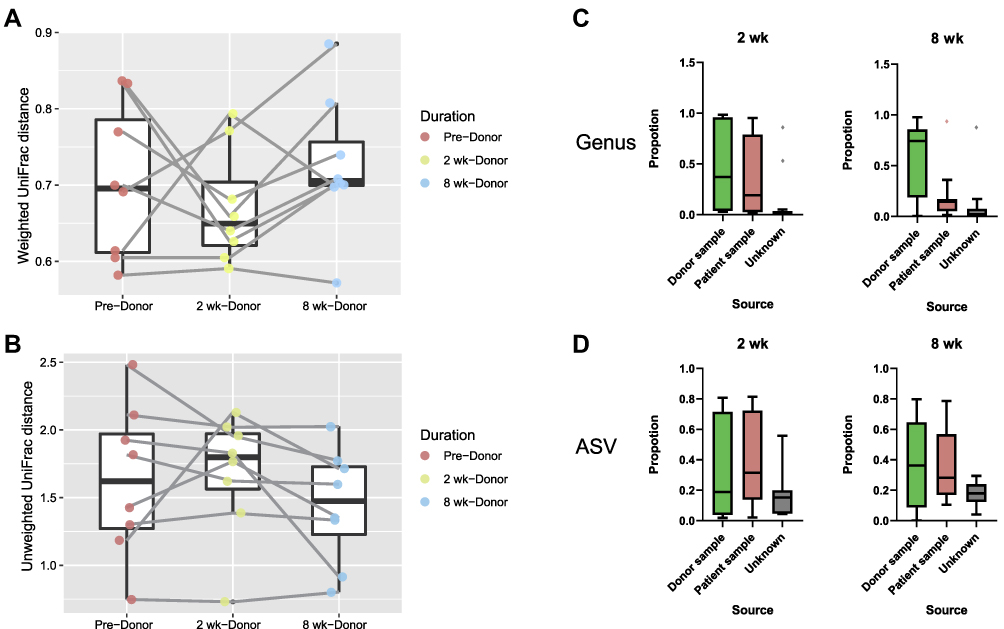 |
Figure 2 (A and B) Tracking the distance in beta diversity between FMT recipients and donors. According to the weighted UniFrac matrix, 2 weeks after FMT, the distance between the bacterial community structures of patients and donors decreased, but at 8 weeks the distance increased. Conversely, the distance between patients and donors first increased and then decreased according to the Unweighted UniFrac matrix. The beta diversities in the weighted and unweighted UniFrac matrices did not reveal a consistent pattern (Wilcoxon signed-rank test p > 0.05). (C and D) Proportions of origin of the fecal bacterial communities of recipients and bacterial engraftment following FMT as determined using Sourcetracker2. The proportions were calculated for the ASV and genus levels, respectively. |
Significant Changes in Taxa from FMT are Associated with Alleviation of Inflammation
A total of 79 genera were detected in the feces of rCDI patients and donors, and the relative abundances of the top 30 genera are shown as a heatmap in Figure 3A. Among the genera, only Prevotella exhibited a significant difference at both 2 weeks and 8 weeks after transplantation. To examine how the significant changes in taxon relative abundance affected patients’ inflammation, correlations between Prevotella and the ratio of Bacilli over Clostridia, CRP, and erythrocyte sedimentation rate (ESR) were examined.34–36 Prevotella was observed to negatively correlate with the ratio of Bacilli/Clostridia (Spearman’s r = −0.426, p = 0.038, Figure 3B), but distinct correlations were not found with CRP (Spearman’s r = −0.034, p = 0.873, Figure 3C) or ESR (Spearman’s r = −0.202, p = 0.344, Figure 3D). The value of the MD index was highest in the Pre group and lowest in the Donor group and was found to decrease gradually in the recipients following FMT. Significant differences were found between the Pre and Donor groups as well as the 2 wk and Donor groups (Wilcoxon signed-rank test, p < 0.05, Figure 3E), suggesting that the CDI patients at 8 wk did not differ significantly from donors.
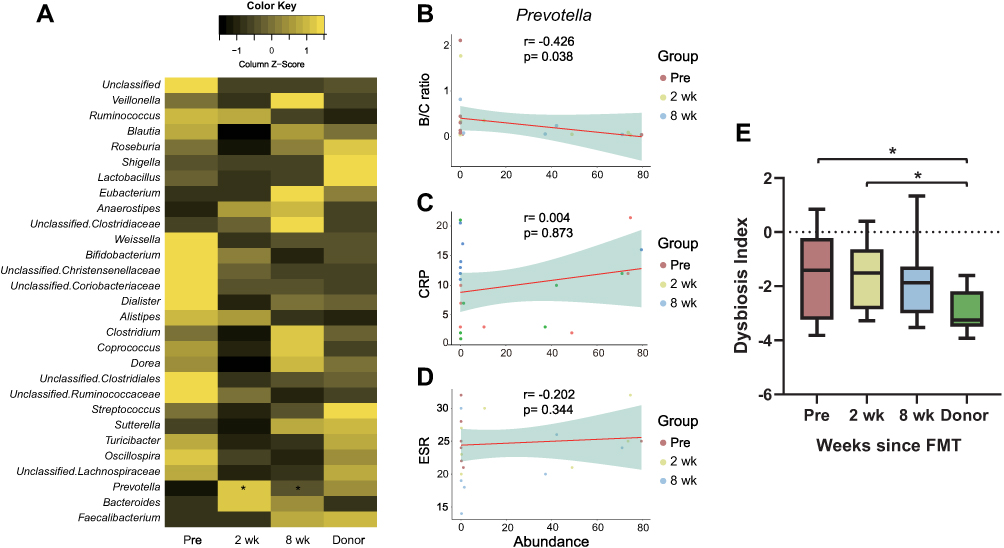 |
Figure 3 (A) Relative abundances of the top 30 genera in rCDI patients and donors. Prevotella exhibited significant increases in both the 2 wk and 8 wk groups compared to the Pre group (Wilcoxon signed-rank test p < 0.05). (B–D) Spearman correlations between Prevotella and the ratio of Bacilli over Clostridia (B/C ratio), CRP, and ESR (r = −0.426, p = 0.038 for B/C ratio; r = 0.034, p = 0.873 for CRP; r = −0.202, p = 0.344 for ESR, Spearman). (E) Shifts in the MD index following fecal microbiota transplant, revealing that the initially significantly higher value in rCDI patients decreased to be comparable to that to donors in the 8 weeks following FMT (*Wilcoxon signed-rank test p < 0.05). |
Moreover, via pan-genome analysis, total 33 ASVs assigned as Prevotella strains were monitored to understand the difference of functional genes between donors and patients (Figure S2). With the subsequent analysis comparing the origin of those strains, our results demonstrated that five of Prevotella strains specific at donor fecal samples (two Prevotella copri, Prevotella corporis and two uncultured Prevotella), six at patient samples prior to the FMT (two Prevotella bivia, Prevotella dentalis, Prevotella timonensis and two uncultured Prevotella) and 22 Prevotella strains which were detectable both at donor and patient samples (Figure S2A and B). Beyond the taxonomic difference between Prevotella strains among donor and patient samples, we focused on the predictable gene components of strains specific at either donors or patients to manifest the contribution of donors’ Prevotella in the remission of symptoms. By gene prediction analysis on strains, we could observe 717 core genes and 117 accessory genes in donor specific Prevotella strains while 661 core genes and 331 accessory genes in patient specific ones (Figure S2C). Subsequently, we deepened our sight into core gene components of donor specific Prevotella strains excluding the universally detectable Prevotella core gene components so that we could concentrate on the unique functional traits of donor specific Prevotella strains. Consequently, 66 donor-specific Prevotella core genes were detected while 10 in patient-specific Prevotella (Figure S2D). We also investigated the functions and contributions of 66 donor-specific Prevotella core genes acquired. Among the functions, ferritin coding gene (K02217) and pyridoxine kinase (K00868) were confirmed on sets of the donor-specific Prevotella core genes (Table S2).
Metagenomic Functional Prediction Shifts Following FMT
A total of 5566 predicted metagenomic functions were obtained using PICRUSt2 and annotated using KEGG Orthology (KO) groups. The described functions were classified according to the KEGG pathway. Of a total of 254 KEGG pathways, 40 pathways (16 for metabolism, 7 for genetic information processing, 3 for environmental information processing, and 8 for cellular processes) were selected for discrimination among the Pre, 2 wk, 8 wk, and Donor groups using the LEfSe (linear discriminant analysis effect size, LDA > 2.5) algorithm through the Huttenhower galaxy server.37 Although significant hits were not confirmed in the pathways, the abundances of individual predicted metagenomes in the recipients shifted toward the compositions of the donors (Wilcoxon signed-rank test, p > 0.05, Figure 4A). Additionally, while the proportion of oxidative phosphorylation was found to be low in the Pre group, the group was shown to contain a higher abundance of ntpG, ntpI, atpD, ntpB, ppaC and hydA genes (which belong to the oxidative phosphorylation pathway) than the other groups (Wilcoxon signed-rank test, p > 0.05, Figure 4B).
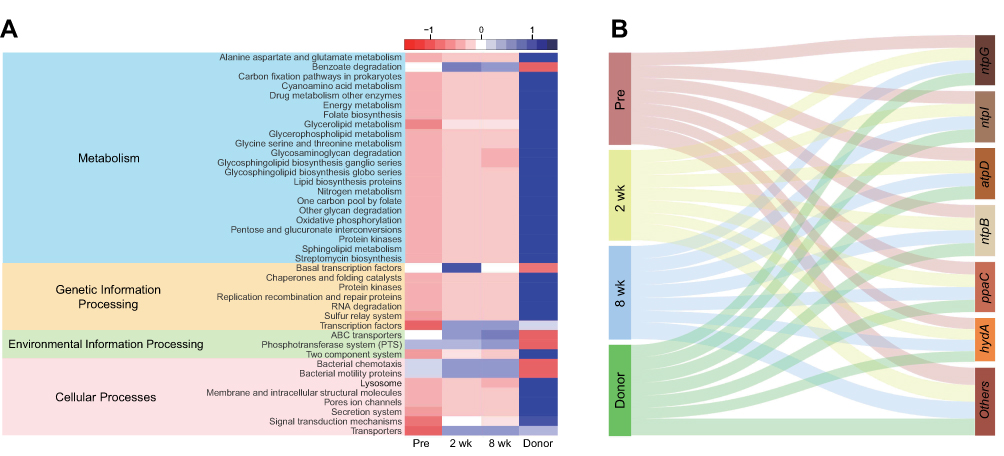 |
Figure 4 (A) Forty predicted metabolic pathways were selected from the results of PICRUSt2 using LEfSe (LDA > 2.5, Wilcoxon signed-rank test p > 0.05) and are described using a heatmap. (B) Sankey diagram showing genetic quantitative distribution for each group, suggesting that donors exhibit the largest amount of genes involved in oxidative phosphorylation, but it leans toward genes other than ntpG, ntpI, atpD, ntpB, ppaC and hydA, which were more abundant in the Pre group (no significant differences in the amount of each gene among groups, Wilcoxon signed-rank test p > 0.05). |
Discussion
Numerous successful responses to FMT treatment of CDI and IBD have been reported.38–41 A previous study postulated that FMT’s mechanism of action against CDI is that the directly transplanted bacteria change the intestinal environment, rendering it inhospitable to C. difficile. In this study, we administered FMT to Iranian rCDI patients with underlying IBD and for the first time examined their gut microbiota alterations and functional profiles before and after the FMT procedure. All eight rCDI patients with underlying IBD who participated in the study exhibited resolution of CDI symptoms, thereby reaffirming the efficacy of FMT. Additionally, we scrutinized the alterations of gut microbiota in the rCDI patients following FMT. Unlike previous studies in which FMT altered the gut microbiota of recipients to be similar to their donors,42–47 our results showed that rCDI patients recovered their own healthy gut environment independently. All eight rCDI patients showed successful response of CDI status to transplantation regardless of patients’ age, sex, BMI, or relation to donors. The alpha diversity of the original gut microbial composition was increased after FMT; however, in terms of beta diversity, the distance between patients and donors did not change significantly. Furthermore, the extent of engraftment of the donor fecal microbiota after FMT varied widely, and more evidence is required to determine that FMT causes the recipients’ gut microbiota to resemble that of their donors. A previous study asserted that an association between the host’s genetic factors and their gut microbiome composition possibly advocates this phenomenon.48
Following treatment, the proportion of Bacteroidetes in patients’ gut microbiota increased and the proportion of Proteobacteria decreased, aligning it more closely with the composition of donors. In addition, while gut microbial diversity did not change significantly, fecal microbial diversity tended to align closely with that of donors; these results are consistent with previous studies.49–51 Prevotella, the sole genus that increased significantly after FMT, showed a strong negative correlation with the Bacilli/Clostridia ratio, which is one of the metrics for inflammation. This association may indicate that Prevotella plays a key role in alleviating IBD severity. The substantial increment of Prevotella and the decreased dysbiosis index indicate that the changes in gut microbial composition following FMT influence the complete resolution of CDI symptoms. Interestingly, there was no statistically notable change in Proteobacteria in the Pre group compared to the 2 wk and 8 wk groups. Since Proteobacteria are gram-negative and include various pathogenic genera, the cell walls of which contain lipopolysaccharides (LPS) that are a crucial source of inflammation,52 taxonomic groups belonging to Proteobacteria were expected to decrease after FMT as Prevotella increased. This occurrence is consistent with the lower MD index score in the Pre group (mean value <0) and the fact that the rCDI severities of patients were recorded as mild in the metadata. Therefore, both the clinical diagnosis and fecal microbiome analysis suggest equivalent results regarding the changes in patients’ health status before vs after FMT. Using the pan-genome analysis to compare the difference in functionality of core genes between donors and rCDI patients, the results demonstrated that donors deriving Prevotella strains with ferritin gene may contribute to the clearance of pathogens in patients’ gut by overwhelming them in the competence of iron intakes.53 Moreover, regarding to pyridoxine kinase, it is reported that pyridoxal-5-phosphate catabolized by donor-specific Prevotella strains’ enzyme might contribute to the remission of inflammatory symptoms of recipients by Selhub et al.54 These findings with the interpretation of the ASV level of Prevotella strains suggest the potentially beneficial roles of donor-specific Prevotella strains compared to the strains originally residing in patients’ gut. Furthermore, advocating our hypothesis that Prevotella is the key factor for successful FMT, it suggests the predictable mode of actions which Prevotella can do in patients’ gut by enlisting the potentially beneficial genes.
In addition to effecting changes in bacterial abundances, FMT can also influence abundant genes and alter the features of the host metabolism, genetic information processing, environmental information processing, and cellular processes. Aside from the changes in genus relative abundance following FMT, there was no significant increase or decrease in the 40 KEGG pathways. However, we focused on oxidative phosphorylation with related genes and found that the abundances of ntpG, ntpI, atpD, ntpB, ppaC and hydA genes were highest in the Pre group, although donors exhibited the largest proportion of entire genes involving oxidative phosphorylation. Similar to Kulecka et al55 the tendency of those 5 genes to decrease after FMT suggests a possible association with the resolution of rCDI.
The major limitation of this study is that sample size was not sufficient to represent various aspects, such as IBD, age, BMI, and relationship with donors, it is cautious about representativeness. The small sample size might influence the absence of statistically significant results regarding microbial diversities, donor’s bacterial engraftment, and MD index. Nevertheless, our previous study revealed that all rCDI patients received successful transplant treatment.14 This study wants to focused on effects of FMT on rCDI patients from Iran. The highlight of this study is that the wide range of deviations in rCDI patients in various microbiome analyses was narrowed and the steady changes in fecal bacterial composition to become more similar to those of donors emphasize the efficacy of FMT biotherapy.
Conclusions
In this study, all scientific achievement conclusively shows that this research has a high potential as a pilot study. Regardless of age, relationship (related or unrelated), or gender, all Iranian rCDI patients with underlying inflammation overcame CDI symptoms after receiving FMT. Although the FMT donors differed in terms of health status, all patients recovered a healthy range of gut microbial community structure. Therefore, the key factor for successful response would be the health of the donor’s microbiota rather than the person’s overall physical condition. While FMT generates alterations in rCDI patients’ fecal bacterial diversity and the value of the MD index becomes similar to that of donors, the bacterial compositions do not come to resemble those of donors. Although additional study is needed to demonstrate that Prevotella is a key factor for resolving rCDI in Iranians, this study is expected to be a foundational reference for subsequent gut microbiome study aimed at the Iranian population.
Data Sharing Statement
All of the raw 16S rRNA gene sequence data for this current study were deposited with the National Center for Biotechnology Information’s BioProject under accession number PRJNA737199.
Acknowledgments
The authors wish to thank all laboratory staff of the Foodborne and Waterborne Diseases Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran. We are also very grateful to the NGS core facility in Kyungpook National University, Republic of Korea, for their support and assistance of next-generation sequencing in this project.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by a grant (no. RIGLD 992) from the Foodborne and Waterborne Diseases Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran. And the authors acknowledge financial supports from the Commercialization Promotion Agency for R&D Outcomes (COMPA) grant funded by the Ministry of Science and ICT (R&D project No. 1711139487), and the project to train professional personnel in biological materials by the Ministry of Environment, Republic of Korea.
Disclosure
No potential conflicts of interest are reported by the authors.
References
1. Khanna S, Vazquez-Baeza Y, González A, et al. Changes in microbial ecology after fecal microbiota transplantation for recurrent C. difficile infection affected by underlying inflammatory bowel disease. Microbiome. 2017;5(1):1–8. doi:10.1186/s40168-017-0269-3
2. Carlucci C, Petrof EO, Allen-Vercoe EJE. Fecal microbiota-based therapeutics for recurrent Clostridium difficile infection, ulcerative colitis and obesity. Ebiomedicine. 2016;13:37–45. doi:10.1016/j.ebiom.2016.09.029
3. Francino MP. Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances. Front Microbiol. 2016;6(1543). doi:10.3389/fmicb.2015.01543
4. Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. J Clin Gastroenterol. 2018;11(1):1–10. doi:10.1007/s12328-017-0813-5
5. Khanna S, Pardi DS. hepatology: clinical implications of antibiotic impact on gastrointestinal microbiota and Clostridium difficile infection. Expert Rev Gastroenterol Hepatol. 2016;10(10):1145–1152. doi:10.1586/17474124.2016.1158097
6. Martin JS, Monaghan TM, Wilcox MH. Clostridium difficile infection: epidemiology, diagnosis and understanding transmission. Nat Rev Gastroenterol Hepatol. 2016;13(4):206–216. doi:10.1038/nrgastro.2016.25
7. Azimirad M, Krutova M, Yadegar A, et al. Clostridioides difficile ribotypes 001 and 126 were predominant in Tehran healthcare settings from 2004 to 2018: a 14-year-long cross-sectional study. EMI. 2020;9(1):1432–1443. doi:10.1080/22221751.2020.1780949
8. Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825–834. doi:10.1056/NEJMoa1408913
9. Schäffler H, Breitrück A. Clostridium difficile – from colonization to infection. Front Microbiol. 2018;9:646. doi:10.3389/fmicb.2018.00646
10. Tan P, Li X, Shen J, Feng Q. Fecal microbiota transplantation for the treatment of inflammatory bowel disease: an update. FrontPharmacol. 2020;11:1409.
11. Pittayanon R, Lau JT, Leontiadis GI, et al. Differences in gut microbiota in patients with vs without inflammatory bowel diseases: a systematic review. Gastroenterol. 2020;158(4):930–946.e931. doi:10.1053/j.gastro.2019.11.294
12. Khanna S, Pardi DS, Kelly CR, et al. A novel microbiome therapeutic increases gut microbial diversity and prevents recurrent clostridium difficile infection. J Infect Dis. 2016;214(2):173–181. doi:10.1093/infdis/jiv766
13. Costello SP, Soo W, Bryant RV, Jairath V, Hart AL, Andrews JM. Systematic review with meta-analysis: faecal microbiota transplantation for the induction of remission for active ulcerative colitis. Aliment Pharmacol Ther. 2017;46(3):213–224. doi:10.1111/apt.14173
14. Azimirad M, Yadegar A, Gholami F, et al. Treatment of recurrent Clostridioides difficile Infection using fecal microbiota transplantation in Iranian patients with underlying inflammatory bowel disease. J Inflamm Res. 2020;13:563–570. doi:10.2147/JIR.S265520
15. Fischer M, Kao D, Kelly C, et al. Fecal microbiota transplantation is safe and efficacious for recurrent or refractory Clostridium difficile infection in patients with inflammatory bowel disease. IBD. 2016;22(10):2402–2409.
16. Martinez-Gili L, McDonald JA, Liu Z, et al. Understanding the mechanisms of efficacy of fecal microbiota transplant in treating recurrent Clostridioides difficile infection and beyond: the contribution of gut microbial-derived metabolites. Gut Microbe. 2020;12(1):1810531. doi:10.1080/19490976.2020.1810531
17. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. AJG. 2013;108(4):
18. Moore T, Rodriguez A, Bakken JS. Fecal microbiota transplantation: a practical update for the infectious disease specialist. Clin Infect Dis. 2014;58(4):541–545. doi:10.1093/cid/cit950
19. Jo YJ, Tagele SB, Pham HQ, et al. In situ profiling of the three dominant phyla within the human gut using TaqMan PCR for pre-hospital diagnosis of gut dysbiosis. Int J Mol Sci. 2020;21(6):1916. doi:10.3390/ijms21061916
20. Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–857. doi:10.1038/s41587-019-0209-9
21. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from illumina amplicon data. Nat Method. 2016;13(7):581–583. doi:10.1038/nmeth.3869
22. McDonald D, Price MN, Goodrich J, et al. An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME. 2012;6(3):610–618. doi:10.1038/ismej.2011.139
23. Zakrzewski M, Proietti C, Ellis JJ, et al. Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics. 2017;33(5):782–783. doi:10.1093/bioinformatics/btw725
24. McMurdie PJ, Holmes S, Watson M. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. doi:10.1371/journal.pone.0061217
25. Oksanen J, Blanchet FG, Kindt R, et al. Community ecology package. 2013;2.
26. Kembel SW, Cowan PD, Helmus MR, et al. Picante: r tools for integrating phylogenies and ecology. Bioinformatics. 2010;26(11):1463–1464. doi:10.1093/bioinformatics/btq166
27. Lozupone CA, Hamady M, Kelley ST, Knight RJA. microbiology e: quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73(5):1576–1585. doi:10.1128/AEM.01996-06
28. Brusaferro A, Cavalli E, Farinelli E, Cozzali R, Principi N, Esposito S. Gut dysbiosis and paediatric crohn’s disease. J Infect. 2019;78(1):1–7. doi:10.1016/j.jinf.2018.10.005
29. Gevers D, Kugathasan S, Denson Lee A, et al. The treatment-naive microbiome in new-onset crohn’s disease. Cell Host Microbe. 2014;15(3):382–392. doi:10.1016/j.chom.2014.02.005
30. Mann AE, Sabin S, Ziesemer K, et al. Differential preservation of endogenous human and microbial DNA in dental calculus and dentin. Sci Rep. 2018;8(1):1–15. doi:10.1038/s41598-018-28091-9
31. Douglas GM, Maffei VJ, Zaneveld JR, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38(6):685–688. doi:10.1038/s41587-020-0548-6
32. Shen W, Le S, Li Y, Hu F, Zou Q. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11(10):e0163962. doi:10.1371/journal.pone.0163962
33. Zhong C, Han M, Yang P, et al. Comprehensive analysis reveals the evolution and pathogenicity of Aeromonas, viewed from both single isolated species and microbial communities. mSystems. 2019;4(5):e00252–e00319. doi:10.1128/mSystems.00252-19
34. Pearson-Leary J, Zhao C, Bittinger K, et al. The gut microbiome regulates the increases in depressive-type behaviors and in inflammatory processes in the ventral hippocampus of stress vulnerable rats. Mol Psychiatry. 2020;25(5):1068–1079. doi:10.1038/s41380-019-0380-x
35. Langhorst J, Elsenbruch S, Koelzer J, Rueffer A, Michalsen A, Dobos GJ. Noninvasive markers in the assessment of intestinal inflammation in inflammatory bowel diseases: performance of fecal lactoferrin, calprotectin, and PMN-elastase, CRP, and clinical indices. Off J Am Coll Gastroenterol. 2008;103(1):162–169.
36. Litao MK, Kamat D. Erythrocyte sedimentation rate and C-reactive protein: how best to use them in clinical practice. Pediatr Ann. 2014;43(10):417–420. doi:10.3928/00904481-20140924-10
37. Thang MW, Chua X-Y, Price G, Gorse D, Field MAJF. MetaDEGalaxy: galaxy workflow for differential abundance analysis of 16s metagenomic data. F1000 Res. 2019;8:726. doi:10.12688/f1000research.18866.2
38. Fischer M, Sipe BW, Rogers NA, et al. Faecal microbiota transplantation plus selected use of vancomycin for severe-complicated Clostridium difficile infection: description of a protocol with high success rate. Aliment Pharmacol Ther. 2015;42(4):470–476. doi:10.1111/apt.13290
39. Schneider KM, Wirtz TH, Kroy D, et al. Successful fecal microbiota transplantation in a patient with severe complicated Clostridium difficile infection after liver transplantation. Case Rep Gastroenterol. 2018;12(1):76–84. doi:10.1159/000481937
40. Fang S, Kraft CS, Dhere T, et al. Successful treatment of chronic pouchitis utilizing fecal microbiota transplantation (FMT): a case report. Int J Colorectal Dis. 2016;31(5):1093–1094. doi:10.1007/s00384-015-2428-y
41. Park H, Laffin MR, Jovel J, et al. The success of fecal microbial transplantation in Clostridium difficile infection correlates with bacteriophage relative abundance in the donor: a retrospective cohort study. Gut Microbe. 2019;10(6):676–687. doi:10.1080/19490976.2019.1586037
42. Weingarden A, González A, Vázquez-Baeza Y, et al. Dynamic changes in short- and long-term bacterial composition following fecal microbiota transplantation for recurrent Clostridium difficile infection. Microbiome. 2015;3(1):10. doi:10.1186/s40168-015-0070-0
43. Seekatz AM, Aas J, Gessert CE, et al. Recovery of the gut microbiome following fecal microbiota transplantation. mBio. 2014;5(3):e00893–e00814. doi:10.1128/mBio.00893-14
44. Weingarden AR, Chen C, Bobr A, et al. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am J Physiol Gastrointest Liver Physiol. 2013;306(4):G310–G319. doi:10.1152/ajpgi.00282.2013
45. Hamilton MJ, Weingarden AR, Unno T, Khoruts A, Sadowsky MJ. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbe. 2013;4(2):125–135. doi:10.4161/gmic.23571
46. Shahinas D, Silverman M, Sittler T, et al. Toward an understanding of changes in diversity associated with fecal microbiome transplantation based on 16S rRNA gene deep sequencing. mBio. 2012;3(5):e00338–e00312. doi:10.1128/mBio.00338-12
47. Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent clostridium difficile-associated diarrhea. J Clin Gastroenterol. 2010;44(5):354–360. doi:10.1097/MCG.0b013e3181c87e02
48. Kurilshikov A, Medina-Gomez C, Bacigalupe R, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156–165. doi:10.1038/s41588-020-00763-1
49. Hourigan SK, Chen LA, Grigoryan Z, et al. Microbiome changes associated with sustained eradication of Clostridium difficile after single faecal microbiota transplantation in children with and without inflammatory bowel disease. Aliment Pharmacol Ther. 2015;42(6):741–752. doi:10.1111/apt.13326
50. Paramsothy S, Nielsen S, Kamm MA, et al. Specific bacteria and metabolites associated with response to fecal microbiota transplantation in patients with ulcerative colitis. Gastroenterol. 2019;156(5):1440–1454.e1442. doi:10.1053/j.gastro.2018.12.001
51. Li X, Gao X, Hu H, et al. Clinical efficacy and microbiome changes following fecal microbiota transplantation in children with recurrent Clostridium Difficile infection. Front Microbiol. 2018;9(2622). doi:10.3389/fmicb.2018.02622
52. Ngkelo A, Meja K, Yeadon M, Adcock I, Kirkham PA. LPS induced inflammatory responses in human peripheral blood mononuclear cells is mediated through NOX4 and Giα dependent PI-3kinase signalling. J Inflamm. 2012;9(1):1. doi:10.1186/1476-9255-9-1
53. Moreira AC, Mesquita G, Gomes MS. Ferritin: an inflammatory player keeping iron at the core of pathogen-host interactions. Microorganisms. 2020;8(4):589. doi:10.3390/microorganisms8040589
54. Selhub J, Byun A, Liu Z, Mason JB, Bronson RT, Crott JW. Dietary vitamin B6 intake modulates colonic inflammation in the IL10-/- model of inflammatory bowel disease. J Nutr Biochem. 2013;24(12):2138–2143. doi:10.1016/j.jnutbio.2013.08.005
55. Kulecka M, Waker E, Ambrozkiewicz F, et al. Higher genome variability within metabolism genes associates with recurrent Clostridium difficile infection. BMC Microbiol. 2021;21(1):36. doi:10.1186/s12866-021-02090-9
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.