")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Alprostadil Injection Attenuates Coronary Microembolization-Induced Myocardial Injury Through GSK-3β/Nrf2/HO-1 Signaling-Mediated Apoptosis Inhibition
Authors Qin Z, Kong B, Zheng J, Wang X, Li L
Received 20 July 2020
Accepted for publication 19 September 2020
Published 22 October 2020 Volume 2020:14 Pages 4407—4422
DOI https://doi.org/10.2147/DDDT.S272877
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Zhenbai Qin, Binghui Kong, Jing Zheng, Xiantao Wang, Lang Li
Department of Cardiology, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China
Correspondence: Lang Li
Department of Cardiology, The First Affiliated Hospital of Guangxi Medical University, No. 21 Shuangyong Road, Qingxiu District, Nanning, Guangxi 530021, China
Tel +86 0771-5331171
Email [email protected]
Objective: Coronary microembolization (CME) results in progressive contractile dysfunction associated with cardiomyocyte apoptosis. Alprostadil injection improves microcirculation, which is effective in treating various cardiovascular disorders. However, the therapeutic effects of alprostadil in CME-induced myocardia injury remain unknown. Therefore, we evaluated the effects of alprostadil injection on cardiac protection in a rat model of CME and explored the underlying mechanisms.
Methods: A rat model of CME was established by injecting polyethylene microspheres into the left ventricle. After injection of microspheres, rats in the alprostadil group received alprostadil via tail vein within 2 minutes. Cardiac function, histological alterations in myocardium, serum c-troponin I (cTnI) levels, myocardium adenosine triphosphate (ATP) concentrations, the activity of superoxide dismutase (SOD) and malondialdehyde (MDA) content in myocardium, and myocardial apoptosis-related proteins were detected 12 hours after CME modeling.
Results: Compared with the Sham group, ATP concentrations, SOD activity in the myocardium, and cardiac function were significantly decreased in a rat model of CME. In addition, serum cTnI levels, MDA content, expression levels of pro-apoptotic proteins, and the number of TUNEL-positive nuclei were remarkably higher in CME group than those in the Sham group. However, alprostadil treatment notably reduced serum cTnI levels and expression levels of pro-apoptotic proteins, while noticeably improved cardiac function, and accelerated SOD activity in the myocardium following CME. Additionally, it was unveiled that the protective effects of alprostadil injection inhibit CME-induced myocardial apoptosis in the myocardium potentially through regulation of the GSK-3β/Nrf2/HO-1 signaling pathway.
Conclusion: Alprostadil injection seems to significantly suppress oxidative stress, alleviate myocardial apoptosis in the myocardium, and improve cardiac systolic and diastolic functions following CME by regulating the GSK-3β/Nrf2/HO-1 signaling pathway.
Keywords: coronary microembolization, alprostadil injection, apoptosis, GSK-3β/Nrf2/HO-1 signaling pathway
Introduction
Coronary microembolization (CME) is closely associated with periprocedural myocardial injury and no-reflow phenomenon after percutaneous coronary interventions (PCI).1,2 CME may occur during thrombolytic therapy and coronary interventions, resulting in serious clinical issues including contractile dysfunction, malignant arrhythmias, and even sudden cardiac death.3–5 Clinically, it is a great challenge for interventionists to prevent and treat CME. Studies have reported that myocardial apoptosis was involved in the progression of CME.6,7 Thus, regulating myocardial apoptosis during CME can be a promising method to reduce myocardial injury. Glycogen synthase kinase 3β (GSK-3β) is a serine-threonine kinase of the glycogen synthase kinase subfamily, which is ubiquitously expressed in a variety of organs and regulates multiple signaling pathways.8,9 Nuclear factor erythroid 2-related factor 2 (Nrf2) is an important factor in maintaining cellular redox homeostasis regulating antioxidant response elements (ARE) expression, including heme oxygenase-1 (HO-1), NAD(P)H quinone oxidoreductase-1 (NQO1), and glutathione S-transferase (GST).10,11 Studies have revealed that GSK-3β can inhibit Nrf2 transcriptional activity through phosphorylation, which leads to Nrf2 degradation and weakens its antioxidant ability.12,13
Alprostadil, also known as liposome prostaglandin E1 (Lipo-PGE1), is a drug approved by FDA, which displays a variety of pharmacologic actions, such as vasodilation, inhibiting platelet aggregation, and improving deformation of erythrocytes.14 Accumulating evidence has demonstrated that alprostadil protects different organs from ischemia and reperfusion injury, including lungs, liver, kidneys, and heart.15–18 Moreover, a recent clinical study showed that intracoronary infusion of alprostadil can effectively improve coronary perfusion and ameliorate coronary slow-flow phenomenon following myocardial infarction.19 Therefore, we hypothesized that alprostadil treatment can reduce CME-induced myocardial injury. In the current study, we aimed to assess the protective effects of alprostadil on CME and the possible role of the GSK-3β/Nrf2/HO-1 pathway in the cardioprotection of alprostadil in rats. Our findings demonstrated that alprostadil injection significantly attenuated myocardial injury post-CME through inhibiting GSK-3β activity and repressing cardiomyocyte apoptosis in the myocardium.
Materials and Methods
Animal Grouping and Modeling
Herein, 24 healthy adult male Sprague-Dawley (SD) rats (weight, 250–300g) were obtained from the Experimental Animal Center of Guangxi Medical University (License number: SYXK-GUI- 2020–0001) and adapted to the facility 1 week before the experiment. According to the random number table method, rats were randomly divided into four groups (n=6 per group): Sham, CME, CME plus alprostadil (2 μg/kg, low-dose), and CME plus alprostadil (4 μg/kg, high-dose). The doses used in our study were based on Zhang et al’s20,21 study and our preliminary experiment result (data were not published), and the concentrations of alprostadil were comparable to those for human use, which were converted for rats by using the intraspecies dose conversion formula. The animals received normal rat chow and water under a 12-hour light/dark cycle at 23±2°C. The study protocol was approved by the Ethics Committee of Guangxi Medical University (Application number: 201901010) and in compliance with the ARRIVE Guidelines for the Use of Laboratory Animals. A CME model of rats was established as described previously.22 In brief, rats received an anesthetic dose of pentobarbitone sodium (30–40 mg/kg) intraperitoneally, and then an animal ventilator was connected to assist respiration. Afterwards, the skin on the left chest was sheared and sterilized with 75% alcohol, followed by cutting open along the left edge of the sternum between the second to fourth intercostal spaces until clearly exposing the heart. Subsequently, the ascending aorta was separated and the pericardium was removed, and, then, a total of 4,500 polyethylene microspheres per rat (diameter, 42 μm; Biosphere Medical Inc., Rockland, MA, USA) dissolved in 0.15 mL normal saline were injected into the left ventricle together with a proper animal hemostatic clip clamping the ascending aorta for 12 seconds. Rats in the Sham group also underwent similar procedures and received 0.15 mL normal saline. Furthermore, the CME plus alprostadil (Chongqing Yaoyou Pharmaceutical Co., Ltd., Chongqing, China) group was established by injection of alprostadil at doses of 2 and 4 μg/kg within 2 minutes following CME modeling, and rats in the Sham and CME groups both received the same volume of normal saline.
Echocardiography
Our previous experiments found that cardiac function of rats reached the lowest level after 12 hours of CME.23 Hence, at 12 hours after operation, rats received an anesthetic dose of pentobarbitone sodium (30–40 mg/kg) intraperitoneally. After that, cardiac function-based indices of rats, including left ventricular end-diastolic diameter (LVDd), left ventricular fractional shortening (LVFS), left ventricular end-systolic diameter (LVDs), cardiac output (CO), heart rate (HR), and left ventricular ejection fraction (LVEF) were assessed by using an animal specific ultrasound instrument (Phillips, Andover, MA, USA) according to the manufacturer’s instruction. The cardiac function examination was performed by an ultrasound specialist who was blinded to the study design, and the values were averaged from three cardiac cycles.
Tissue Sampling and Treatment
After 12 hours of CME modeling and cardiac function measurement, all rats received an anesthetic overdose of pentobarbitone sodium (60 mg/kg) intraperitoneally. Before being sacrificed, blood was gained from each rat for enzyme-linked immunosorbent assay (ELISA) through the abdominal aorta. Upon the cardiac arrest, it was quickly extracted and each heart tissue was segmented into three parts: the apex, middle, and bottom parallel to the atrioventricular sulcus. After that, the apex and middle parts were immediately stored at −80°C for quantitative reverse transcription polymerase chain reaction (RT-qPCR) and Western blot assays, respectively. Finally, the heart bottom was fixed in 4% paraformaldehyde, embedded into paraffin, and serially sliced into 4 μm sections for hematoxylin and eosin (H&E) staining, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, and hematoxylin-basic fuchsin-picric acid (HBFP) staining.
ELISA
Blood samples (2 mL) obtained from the abdominal aorta at 12 hours after operation were centrifuged at 5,000 g for 5 minutes at room temperature, and then the serum was collected for ELISA detection. Serum cTnI concentrations were determined by using a cTnI-specific ELISA kit (Roche, Inc., Basel, Switzerland) according to the manufacturer’s instructions.
Measurement of SOD Activity and MDA Content in Myocardium
The activity of SOD and MDA content in the myocardium were determined using SOD assay kit (A001-3; Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and MDA assay kit (A003-1-2; Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's manual. The levels of SOD and MDA were measured by a micro-plate reader and the OD value was measured at a wavelength of 450 nm and 532 nm, separately. Finally, SOD activity and MDA content were calculated according to the formula given in the instructions.
ATP Assay
The level of ATP in heart tissues was examined with a commercial ATP detection kit (S0026; Beyotime Institute of Biotechnology, Shanghai, China) following the manufacturer’s instructions. Briefly, 30 mg tissues from the middle of the heart were minced and homogenized with 400 μL ATP lysis buffer. After that, samples were centrifuged at 4°C for 5 minutes at 12,000 g, and the supernatants were collected for further detection. Next, 100 μL ATP working solution and 20 μL supernatant from each rat were added to a 96-well plate, and then a luminometer was applied to determine the relative light unit value. Finally, a standard curve was plotted and the ATP concentration in the heart samples was calculated according to the standard curve.
Evaluation of Apoptosis in Tissue Sections by TUNEL Assay
Myocardial apoptosis was determined by using TUNEL assay with a commercial TUNEL assay kit (Roche, Inc., Basel, Switzerland) according to the manufacturer’s instruction. Under a light microscope, the numbers of TUNEL-positive nuclei were counted in five random non-overlapping microscopic fields. TUNEL index was calculated as the percentage of TUNEL-positive nuclei in the total number of 200 counted nuclei.24
Measurement of Myocardial Microinfarct Size
In this study, myocardial microinfarct regions were detected by HBFP staining. The cytoplasm of normal myocardiocytes was stained yellow and their nuclei were stained blue, while the ischemic myocardium and erythrocytes were stained red. Five non-overlapping fields were randomly selected from each section to calculate the infarction area by using a DMR-Q550 pathological image analyzer (Leica, Germany). The percentage of infarction size was calculated as the percentage of infarction area over the total observed area.
Double Immunofluorescence Staining
After 12 hours of CME modeling, rats were sacrificed. Sections with a thickness of 4 μm were prepared for immunofluorescence staining. Double immunofluorescence staining was carried out according to the manufacturer's manual. Briefly, the sections were washed three times with phosphate-buffered saline (PBS) (pH 7.4) and blocked in 3% bovine serum albumin (BSA) for 30 minutes at room temperature. After blocking, sections were incubated at 4°C overnight with rabbit monoclonal anti-HO-1 antibody (ab189491; 1:250; Abcam, Cambridge, UK) and rabbit monoclonal anti-Nrf2 antibody (ab137550; 1:500; Abcam). After incubation with primary antibody, sections were rinsed with PBS five times, and then, incubated with fluorescent secondary antibodies for 50 minutes at room temperature. Nuclei were then counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for 7 minutes. The images were acquired using a fluorescence microscope (Olympus, Tokyo, Japan).
Electron Microscopy of Heart Tissues
Rats were sacrificed at 12 hours after operation. Myocardial tissue samples were cut into about 1 mm3 pieces and fixed with 3% glutaraldehyde overnight at 4°C. After that, the pieces were washed three times with 0.1 mol/L PBS (pH=7.4) and post-fixed in 1% osmium tetroxide for 2 hours. Next, the samples were rinsed again with PBS several times and dehydrated with a series of graded ethanol (50~100%). Subsequently, specimens were embedded into resin, continually sliced into ultrathin sections (50 nm), stained with uranyl acetate and lead citrate, and finally examined and photographed with a Hitachi H-7650 electron microscope (Hitachi, Tokyo, Japan).
Gene Expression Analysis
Total RNA was extracted from the apex of the heart by using TRIzol reagent (TaKaRa, Shiga, Japan) according to the manufacturer's manual. The concentration and purity of RNA were identified by using a NanoDrop2000 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA), and reversely transcribed into cDNA using a PrimeScript RT Reagent kit (TaKaRa). Gene expression levels were detected by RT-qPCR using a TB Green Premix Ex Taq Ⅱ kit on StepOnePlus system (Applied Biosystems, Foster City, CA, USA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as an internal control. The primer sequences are summarized in Table 1. The relative expression of mRNA was calculated using 2-ΔΔCt method.
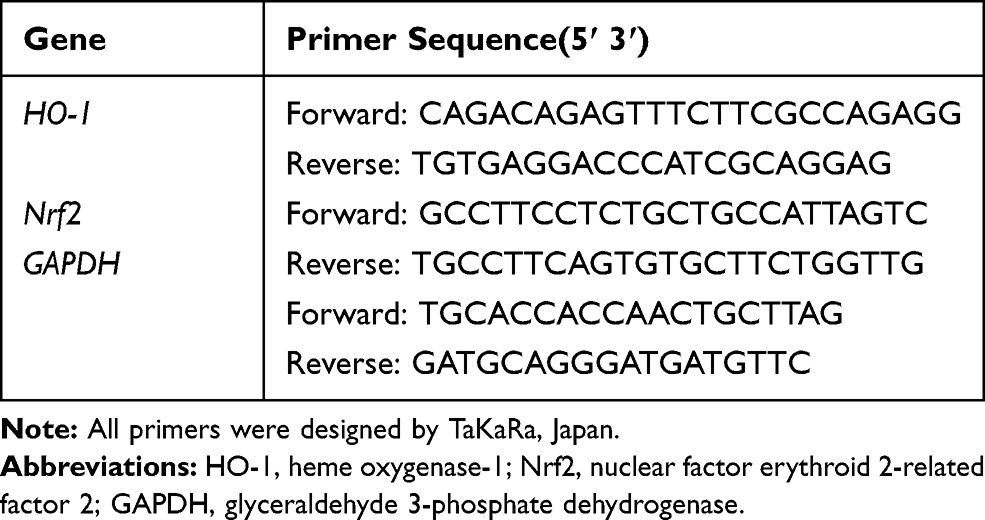 |
Table 1 Primer Used for qPCR |
Isolation of Mitochondria and Cytosol Fractions
To detect the cytochrome c (Cyt-c) expression level in the myocardium cytoplasm, mitochondria and cytoplasm of the heart were separated by using a commercial cytoplasmic and mitochondrial protein extraction kit (C500051; Sangon Biotechnology Inc., Shanghai, China) according to the manufacturer's manual.
Western Blotting
Total protein was extracted from heart tissues and the concentration of protein was determined by using a bicinchoninic acid (BCA) assay kit (P0012S; Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer's manual. Subsequently, 25 μg of protein was separated by 10~12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred onto polyvinylidene difluoride (PVDF) membranes. Afterwards, PVDF membranes were washed three times with 1×TBST buffer, then blocked with 5% skim milk at room temperature for 1 hour, and finally incubated at 4°C overnight with primary antibodies as follows: cleaved-caspase-9 (9504T; 1:1000; Cell Signaling Technology, Inc., Danvers, MA, USA); cleaved-caspase-3 (ab49822; 1:500; Abcam); Bax (ab32503; 1:1000; Abcam); Bcl-2 (ab59348; 1:500; Abcam); Nrf2 (ab137550; 1:1000; Abcam); HO-1 (ab189491; 1:2000; Abcam); GSK-3β (9315S; 1:1000; Cell Signaling Technology, Inc.); p-GSK-3β (Ser-9) (9322S; 1:1000; Cell Signaling Technology, Inc.); Cyt-c (ab13575; 1:2000; Abcam); and GAPDH (ab8245; 1:10000; Abcam). After an overnight incubation, membranes were washed and incubated with corresponding secondary antibodies conjugated with horseradish peroxidase (HRP) (ab6721; 1:10000; Abcam) for 2 hours at room temperature. Finally, immunoreactive bands were detected by enhanced chemiluminescence reagents (Pierce, Rockford, IL, USA). The Image J software was adopted to analyze the gray value of protein bands, and GAPDH served as an internal control.
Statistical Analysis
SPSS 26.0 software (IBM, Armonk, NY, USA) was used for data analysis. Data were expressed as mean±standard deviation. One-way analysis of variance (ANOVA) followed by post hoc test was employed to examine the differences among four groups. P<0.05 was considered statistically significant.
Results
Alprostadil Injection Ameliorates CME-Induced Myocardial Morphological Aberrant
H&E staining showed that myocardial fibers in the Sham group were uniformly aligned, with clear staining and complete morphology. In the CME group, after embolizing microspheres, the cytoplasm was deeply stained by eosin; and there was loosening or edema, enlargement, degeneration, dissolving or necrosis of myocardial cells, and inflammatory cell infiltration. In the CME plus alprostadil (2 μg/kg) group, the conditions of myocardial degeneration and edema were ameliorated, with fewer histomorphological abnormalities and relatively uniform alignment. Additionally, there was no myocardial degeneration and necrosis in the CME plus alprostadil (4 μg/kg) group (Figure 1).
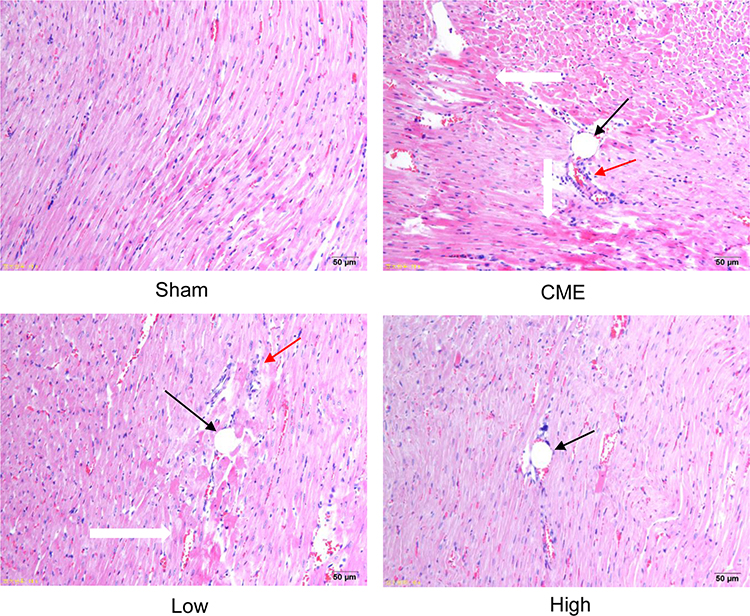 |
Figure 1 Alprostadil injection ameliorated myocardial morphological alterations of CME rats (magnification ×200, scale bar=50 μm) (n=6, per group). The black arrow indicates microspheres, red arrow represents inflammatory cell infiltration, and white arrow shows microinfarction focus. |
Alprostadil Injection Improved Cardiac Function Following CME
Twelve hours after CME modeling, the results of echocardiography were as shown in Table 2 and Figure 2. Compared with the Sham group, the cardiac systolic function of rats in the CME group was negatively influenced, as LVEF, FS, CO, and HR were remarkably attenuated, while LVDd and LVDs were elevated. Importantly, LVEF, FS, CO, and HR were remarkably increased after treatment with alprostadil at doses of 2 and 4 μg/kg, while LVDd and LVDs were significantly decreased. No significant difference was found between the Sham group and the CME plus alprostadil (4 μg/kg) group.
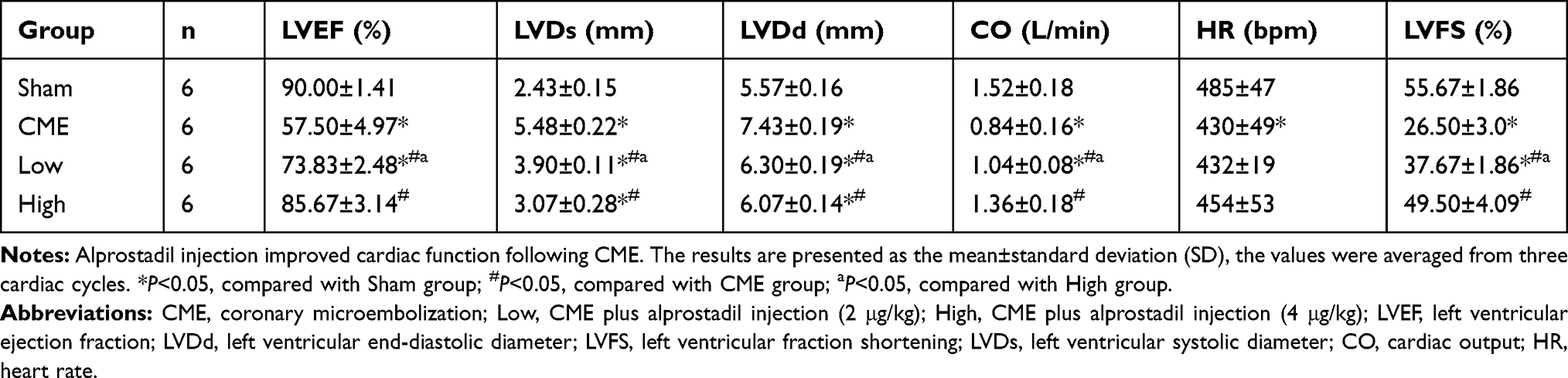 |
Table 2 Cardiac Function Measured by Echocardiography |
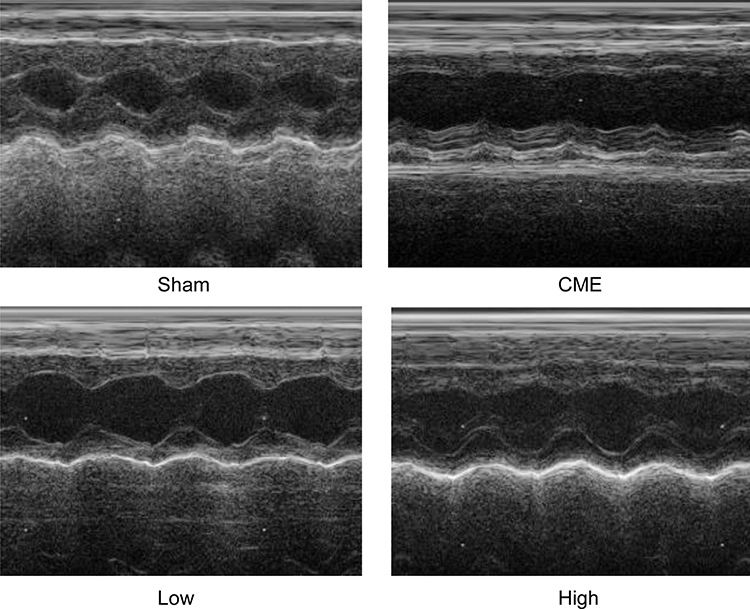 |
Figure 2 Cardiac function-based indices of rats were measured by echocardiography (n=6 per group). |
Alprostadil Injection Ameliorated Myocardial Injury Following CME
Compared with the Sham group, the serum cTnI level was markedly elevated in the CME group. However, treatment with alprostadil significantly decreased serum cTnI levels compared with the CME group (Table 3).
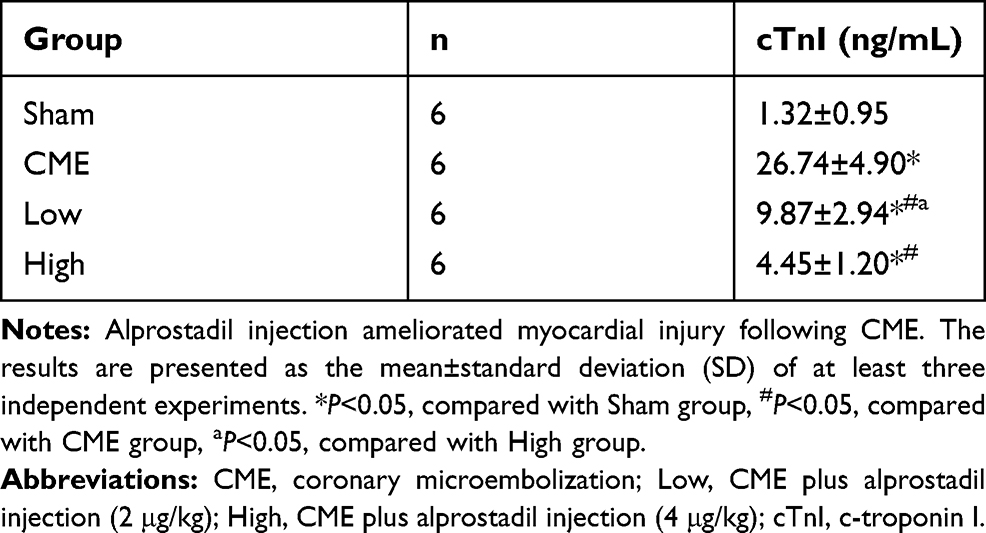 |
Table 3 Serum cTnI Concentrations of Each Group |
Alprostadil Injection Increased Myocardial Antioxidative Ability Following CME
SOD activity and MDA content were measured to assess the effect of alprostadil on myocardium oxidative stress. As shown in Table 4, compared with the Sham group, SOD activity was significantly decreased together with increased MDA content in the CME group. However, treatment with alprostadil (2 or 4 μg/kg) obviously reduced MDA content and improved SOD activity compared with the CME group. This result demonstrated that alprostadil injection treatment promoted myocardial antioxidant activity post-CME.
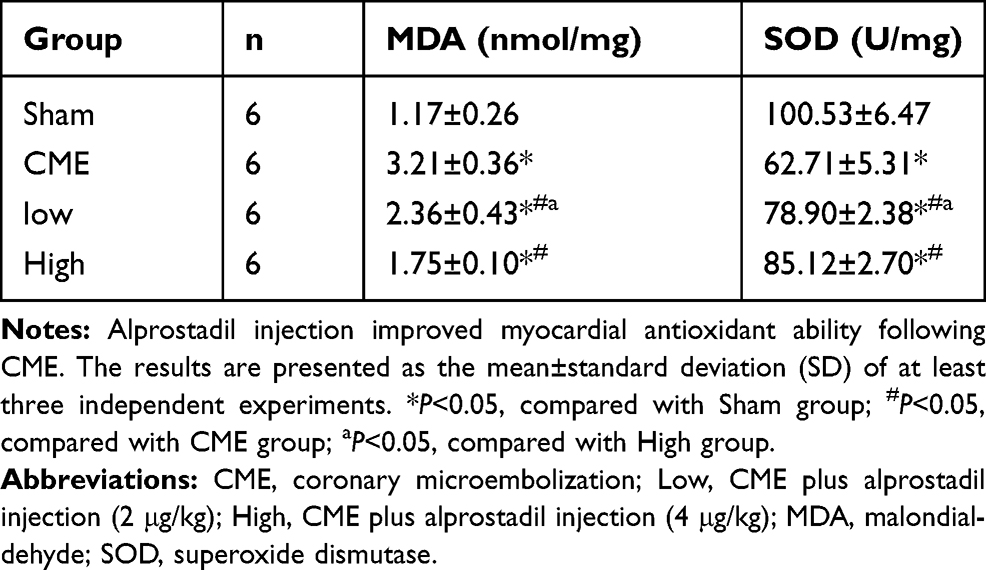 |
Table 4 SOD Activity and MDA Content of Each Group |
Alprostadil Injection Attenuated CME-Induced Mitochondrial Injury
Mitochondria possess >30% cellular volume densities in cardiomyocytes of rats and play a significant role in regulating cardiac ATP production.25,26 As mitochondrial metabolism is an important source for ATP generation, we tested ATP concentrations to reflect the integrity of mitochondrial function. It was found that ATP concentrations were remarkably reduced after CME, which could be reversed by alprostadil injection at 2 μg/kg and 4 μg/kg (Table 5). Besides, transmission electron microscopy examination of heart tissues showed that myocardial mitochondria had markedly vacuolated degeneration and swelling in the CME group. However, after alprostadil treatment, the ultrastructural changes observed in the CME group were remarkably ameliorated along with mitochondrial integrity preserved (Figure 3). This result indicated that alprostadil injection can restore mitochondrial function and ameliorate mitochondrial damages.
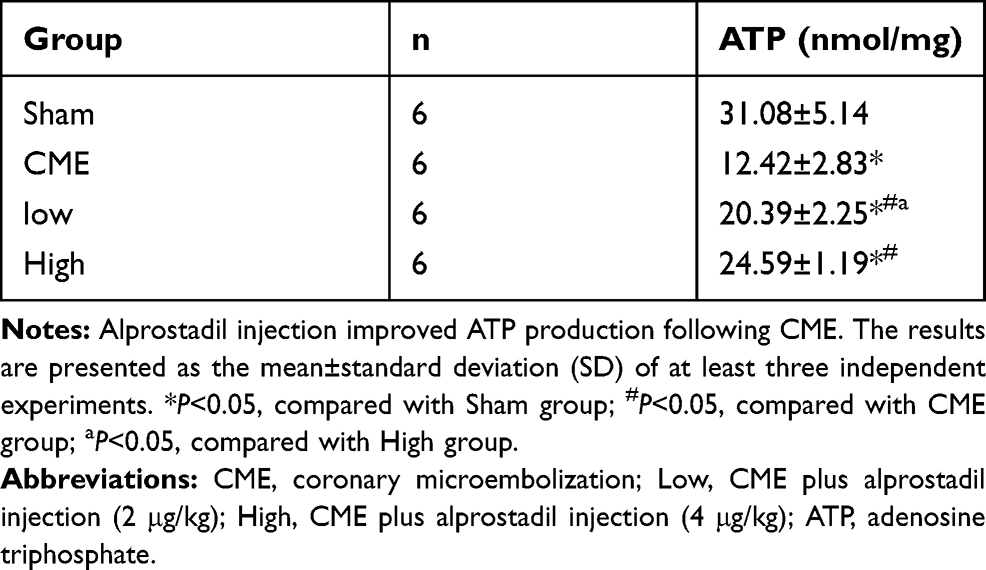 |
Table 5 ATP Concentrations of Each Group |
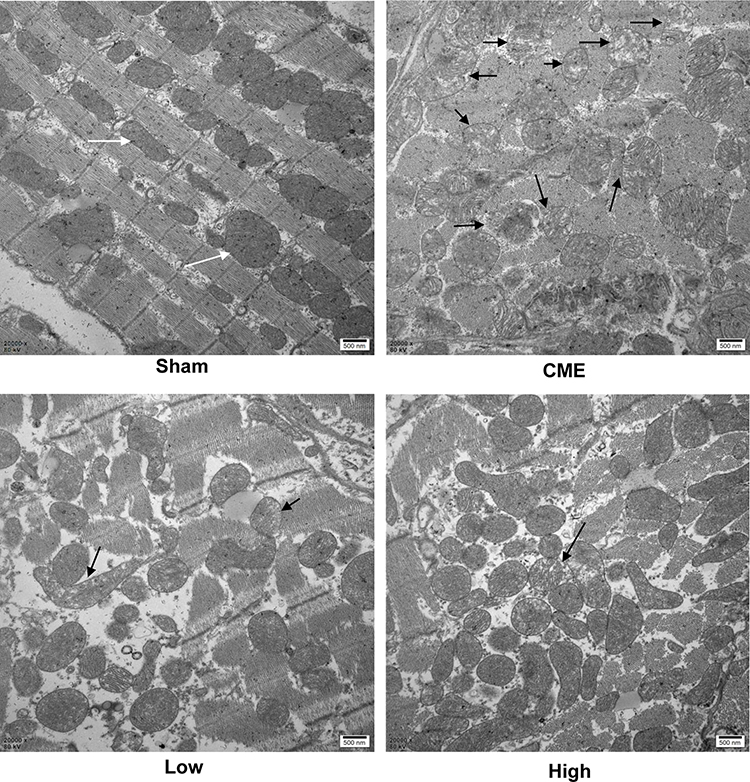 |
Figure 3 Myocardial mitochondrial morphology observed by transmission electron microscopy (magnification ×20,000, scale bar=500 nm). The black arrow represents the typical vacuolated degeneration and enlarged mitochondria, and white arrow indicates the normal mitochondrial morphology (n=3 per group). |
Alprostadil Injection Suppressed CME-Induced Myocardial Apoptosis
Cardiomyocyte apoptosis contributes to the progression of myocardial injury following CME. Thus, we evaluated whether administration of alprostadil might have anti-apoptotic effects following CME. As illustrated in Figure 4, a great number of TUNEL-positive nuclei were observed in the CME group. On the contrary, few apoptotic cells were seen in the alprostadil treatment groups. In addition, CME significantly increased the protein expressions of Bax, Cytosol-Cyt-c, cleaved-caspase-3, and cleaved-caspase-9, while decreased Bcl-2 protein expression compared with the Sham group. Furthermore, alprostadil treatment markedly suppressed CME-induced elevation in pro-apoptotic proteins, while increased Bcl-2 protein expression (Figure 5). The above-mentioned results indicated that alprostadil treatment repressed myocardial apoptosis.
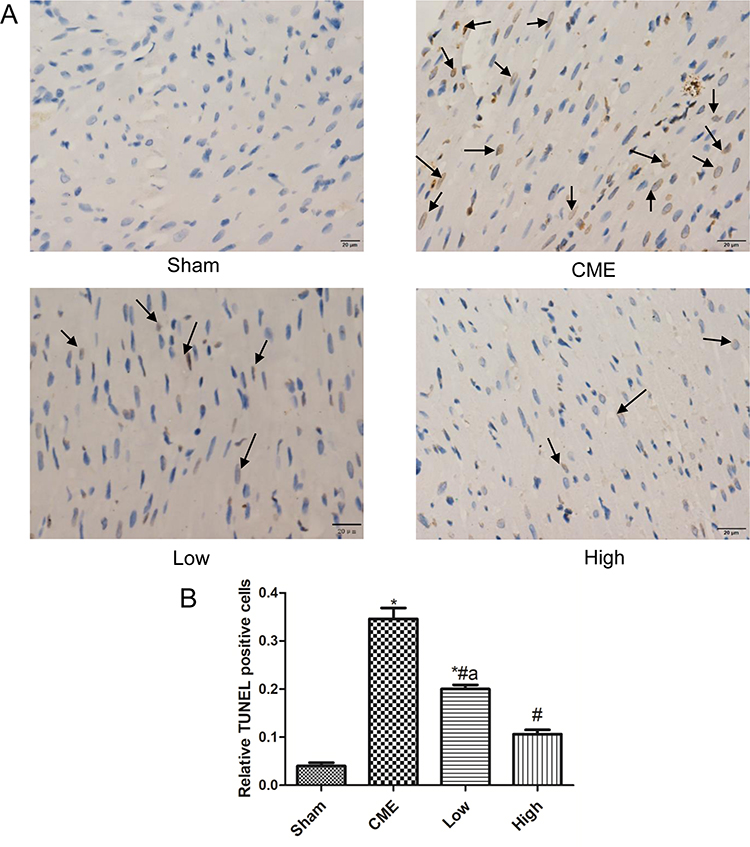 |
Figure 4 Alprostadil injection suppressed CME-induced myocardial apoptosis. (A) Representative images of TUNEL staining, in which apoptotic nuclei were yellow-brown stained, while normal nuclei were blue stained (magnification ×400, scale bar=20 μm). The black arrow indicates apoptotic cells. (B) Relative TUNEL-positive nuclei in each group (n=6 per group). *P<0.05, compared with Sham group, #P<0.05, compared with CME group, aP<0.05, compared with High group. |
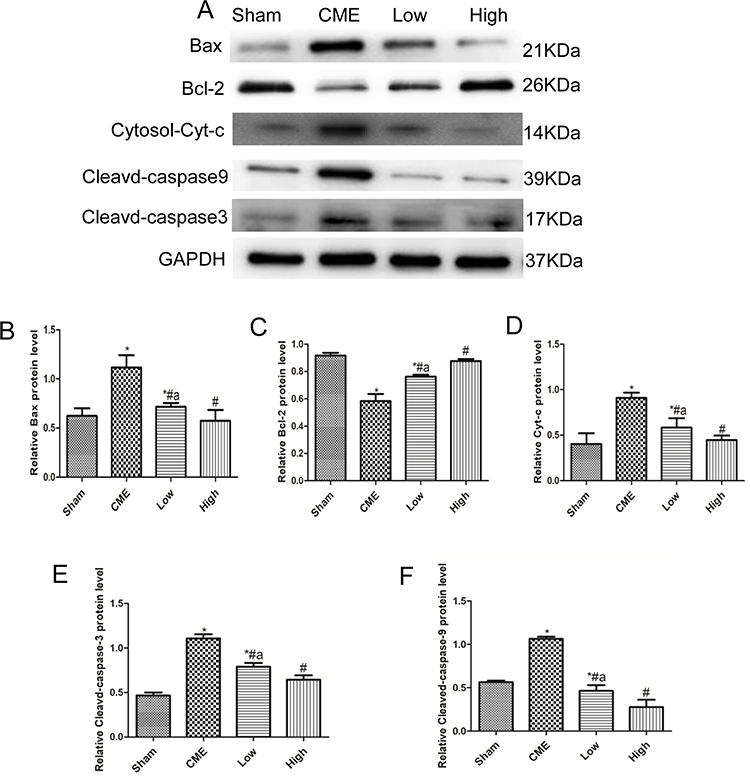 |
Figure 5 Alprostadil injection reduced expressions of pro-apoptotic proteins and improved anti-apoptotic protein expression levels. (A) The bands of Bax, Bcl-2, cytosol-Cyt-c, cleaved-caspase-9, and cleaved-caspase-3. (B–F) Quantification of expression levels of Bax, Bcl-2, cytosol-Cyt-c, cleaved-caspase-3, and cleaved-caspase-9. GAPDH served as an internal control (n=4 or 6, per group). *P<0.05, compared with Sham group; #P<0.05, compared with CME group; aP<0.05, compared with High group. |
Alprostadil Injection Reduced Microinfarction Area Following CME
HBFP staining revealed that there was no microinfarct observed in the Sham group, while it was significantly evident in the CME group. However, alprostadil injection significantly reduced the microinfarct area compared with the CME group (Figure 6). The above-mentioned results indicated that alprostadil injection reduced microinfarction area following CME.
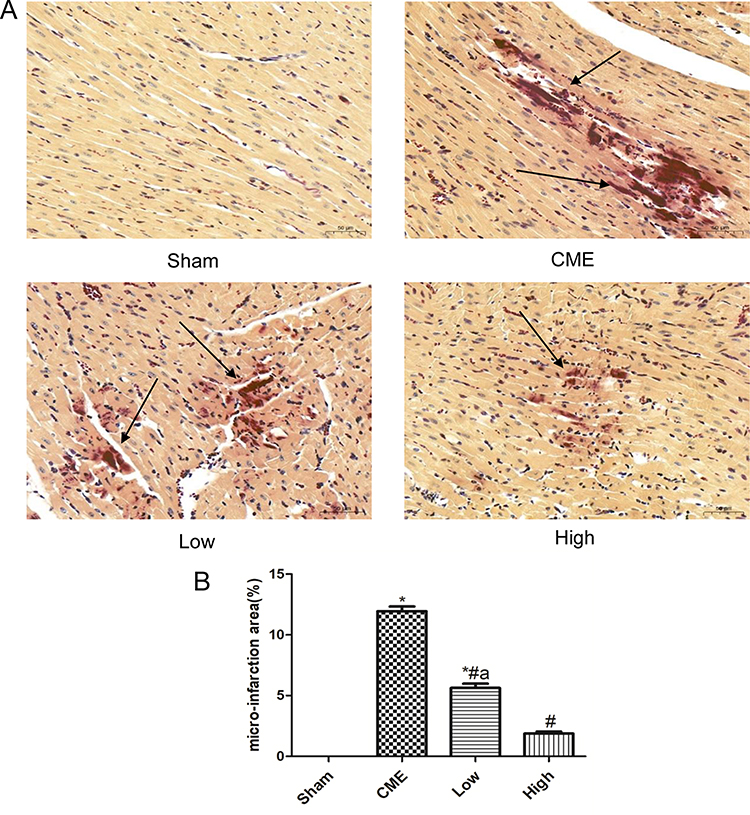 |
Figure 6 Alprostadil injection reduced myocardium micro-infarct size following CME (magnification ×200, scale bar=50 μm). (A) Representative images of HBFP staining; (B) The percentage of micro-infarct area in each group. The black arrow indicates micro-infarct focus. (n=6 per group). *P<0.05, compared with Sham group, #P<0.05, compared with CME group, aP<0.05, compared with High group. |
Alprostadil Injection Improved Nrf2 and HO-1 mRNA Levels Following CME
As shown in Figure 7A, compared with the Sham group, the Nrf2 mRNA level significantly declined in the CME group. Moreover, treatment with alprostadil (2 µg/kg) remarkably increased the Nrf2 mRNA level, which was further upregulated in the alprostadil injection (4 µg/kg) group. In comparison with the Sham group, the HO-1 mRNA level was increased moderately in the CME group, while it was markedly increased in the alprostadil injection treatment groups (Figure 7B).
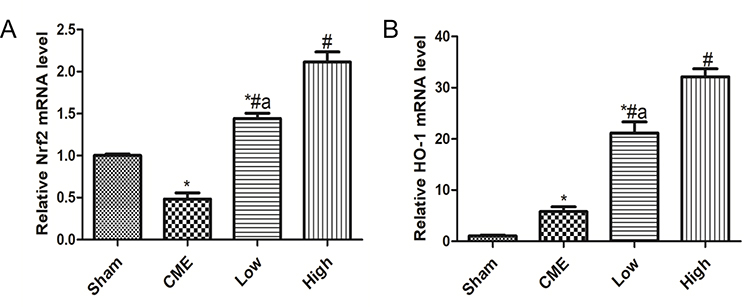 |
Figure 7 Alprostadil injection increased mRNA levels of Nrf2 and HO-1. (A) Nrf2; (B) HO-1 (n=6, per group). The results are presented as the mean±standard deviation (SD) of three independent experiments. *P<0.05, compared with the Sham group; #P<0.05, compared with CME group; aP<0.05, compared with High group. |
Alprostadil Injection Attenuated CME-Induced Myocardial Apoptosis in the Myocardium by Regulating GSK-3β/Nrf2/HO-1 Signaling Pathway
Activation of the GSK-3β/Nrf2/ARE signaling pathway has been demonstrated to attenuate cardiomyocyte apoptosis and improve cell viability.27 Recently, one study revealed that PGE1 inhibited coronary microcirculation dysfunction through activation of the GSK-3β-mitochondrial permeability transition pore (GSK-3β-mPTP) pathway.28 In view of the above findings, alprostadil protected the myocardia from CME-induced myocardial apoptosis. However, the specific pathway in alprostadil-mediated cardioprotection remains elusive. We hypothesized that the GSK-3β/Nrf2/HO-1 signaling pathway is involved in these protective effects. As expected, it was unveiled that the protein expression levels of p-GSK-3β (Ser-9) and Nrf2 were noticeably decreased in the CME group compared with the Sham group. In contrast, alprostadil injection significantly increased protein expressions levels of p-GSK-3β (Ser-9)/GSK-3β, Nrf2, and HO-1 (Figure 8A–D). Moreover, results of double immunofluorescence staining demonstrated that Nrf2 and HO-1 were highly expressed in the alprostadil injection groups compared with the CME group (Figure 9). Taken together, it can be concluded that the protective effects of alprostadil injection inhibit CME-induced myocardial apoptosis in the myocardium, potentially through regulation of the GSK-3β/Nrf2/HO-1 signaling pathway.
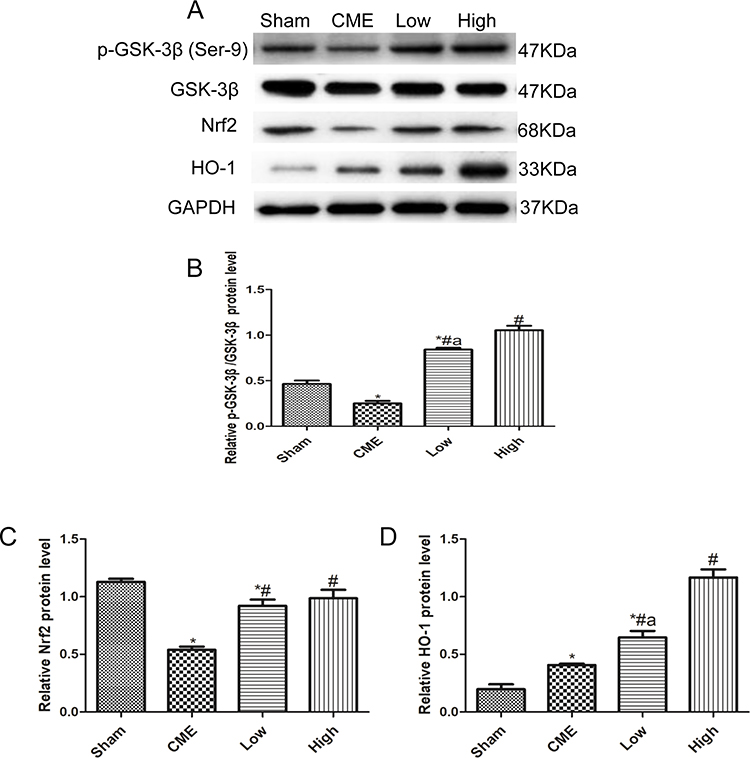 |
Figure 8 Effects of alprostadil injection on GSK-3β/Nrf2/HO-1 signaling pathway in CME-induced myocardial injury. (A) The expressions of GSK-3β, p-GSK-3β (Ser9), Nrf2, and HO-1 determined by Western blot. (B) Alprostadil injection increased the ratio of p-GSK-3β (Ser-9)/GSK-3β. (C) Quantification of Nrf2 expression levels. (D) Quantification of HO-1 expression levels (n=4 or 6, per group). *P<0.05, compared with the Sham group; #P<0.05, compared with the CME group; aP<0.05, compared with the High group. |
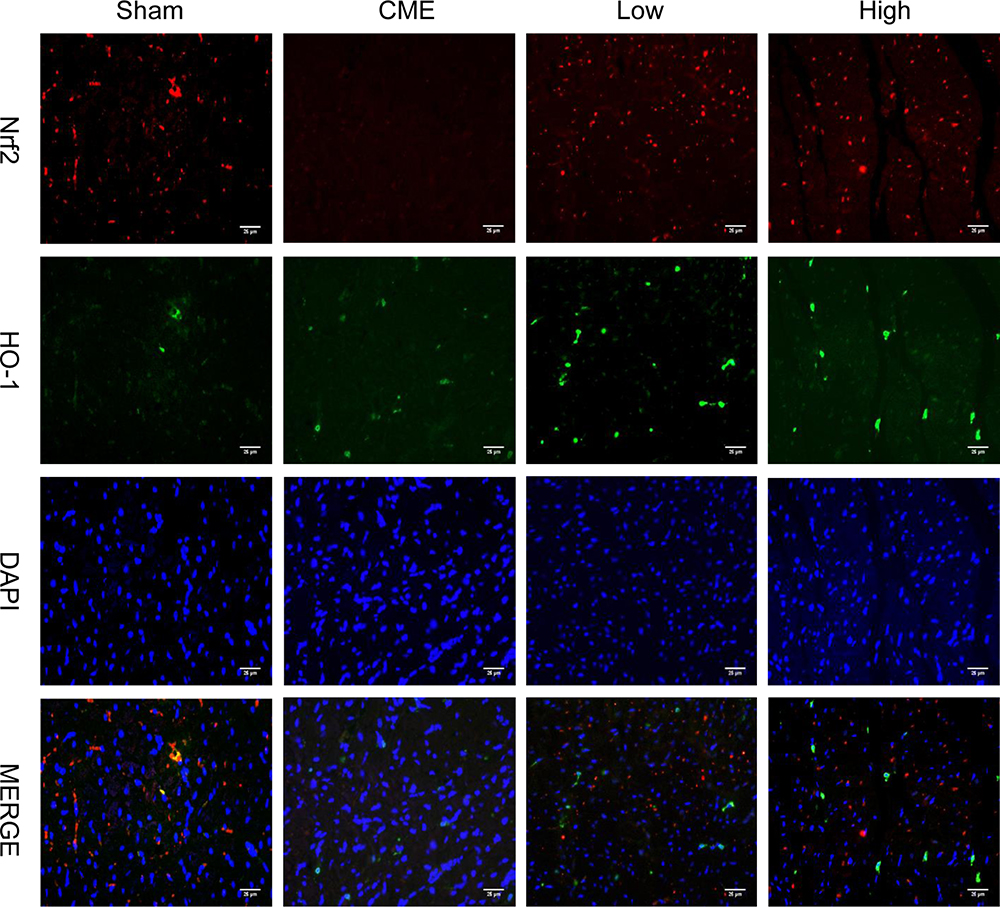 |
Figure 9 Double immunofluorescence staining of Nrf2 and HO-1 in the heart sections (magnification ×400, scale bar=25 μm) (n=6, per group). The red color indicates Nrf2, green color represents HO-1, and blue color shows nuclear. |
Discussion
The present study uncovered that alprostadil injection improved cardiac function, inhibited myocardial apoptosis, and enhanced myocardial antioxidant ability in a rat model of CME. In addition, alprostadil injection significantly increased ATP production and suppressed serum cTnI levels following CME. Furthermore, alprostadil significantly suppressed GSK-3β activity and activated the GSK-3β/Nrf2/HO-1 signaling pathway. Our findings indicated a potential mechanism for the therapeutic effects of alprostadil injection on CME.
CME is a common complication of percutaneous coronary interventions (PCI) and its incidence ranges from 0–70%, depending on detection methods.29 In recent years, numerous devices were designed to prevent CME during elective PCI and primary PCI. However, neither routine protective devices nor thrombolysis indicated satisfactory outcomes.30,31 Therefore, the development of effective therapeutic strategies is urgently needed. Alprostadil has multiple pharmacologic effects, including inhibiting platelet aggregation, improving microcirculation perfusion, and vasodilation. Injection of alprostadil is used clinically to improve cardiovascular microcirculation disorders caused by myocardial ischemia. Previous studies have found that intravenous administration of alprostadil in patients with ST elevation myocardial infarction undergoing primary PCI could improve myocardial microcirculation, reduce myocardium non-flow area, and decrease major adverse cardiac events (MACE).32 Interestingly, alprostadil injection also provides cardioprotection effects on elective PCI patients with unstable angina, which reduces the incidence rates of periprocedural myocardial injury, and decreases pro-inflammatory factor secretion.33 In human chronic ischemia heart disease, alprostadil effectively stimulates neoangiogenesis in infarct areas adjacent to the viable myocardium.34 In a canine thrombolysis model, alprostadil treatment accelerates the thrombolysis time, lowers the incidence rats of reocclusion, noticeably improves coronary blood flow, and reduces the myocardial infarct size.35 However, the role of alprostadil in CME-induced myocardial injury remains unclear. In the current study, it was found that administration of alprostadil injection through the tail vein after CME noticeably attenuated myonecrosis, preserved myocardial morphology, reduced serum markers of myocardial injury, and improved cardiac function in rats.
It is reported that mitochondrial pro-apoptosis protein Cytochrome c (Cyt-c) is released into the cytoplasm in response to apoptotic stimuli, which is associated with formation of apoptosomes induced by pro-caspase-9, triggering the activation of caspase-3 and apoptosis.36 However, the release of Cyt-c can be reversed by anti-apoptosis protein Bcl-2, and its sequester Bax or other pro-apoptosis factors are integrated into the mitochondrial outer membrane.37,38 There are also trustable indicators in oxidative stress, such as MDA contents and SOD activity, which are usually used to evaluate lipid peroxidation and inner antioxidant ability, respectively. During CME, the myocardium generates oxidants, which inhibit antioxidant defense enzymes, cause oxidative stress, and lead to damage in lipids.39 Besides, oxidants can increase mitochondrial depolarization and induce mitochondrial release of Cyt-c into the cytosol, leading to apoptosis.40 Therefore, in the current study, we assessed the effects of alprostadil injection on myocardial apoptosis and oxidative stress through detecting the expression levels of these molecules. It was found that myocardial apoptosis and MDA contents were increased post-CME but reversed by alprostadil treatment. Additionally, transmission electron microscopy showed that the myocardium mitochondria of CME rats were swollen and fractured, and mitochondria cristae were dissolved and disappeared. However, after alprostadil treatment, mitochondrial damage was alleviated and the integrity of the mitochondria was largely preserved. The potential mechanisms may be that CME-induced myocardium oxidative stress causes damage to the mitochondria and release of pro-apoptosis factor Cyt-c into the cytoplasm, which sequentially activates caspase-3 and caspase-9, resulting in myocardial apoptosis. Collectively, the above-mentioned findings suggested that alprostadil injection exerted antioxidant and anti-apoptosis effects on a rat model of CME.
The activity of GSK-3β is negatively regulated by N-terminal phosphorylation of serine-9 (Ser-9) residues of the enzyme.41 GSK-3β mediated phosphorylation of substrates usually leads to inhibition of those substrates, and the end-result of stimulus-induced inhibition of GSK-3β is typically activation of its downstream targets.42 Nrf2 is one of the key protein substrates of GSK-3β, and GSK-3β can accelerate Nrf2 degradation, while inhibition of GSK-3β activity through Ser-9 residue phosphorylation leads to a substantial reduction in Nrf2 phosphorylation.43 Jin et al44 reported that inactivating GSK-3β alleviated hypoxia-induced cardiomyocyte apoptosis via an enhancing Nrf2/ARE signaling pathway. Besides, a recent study showed that alprostadil protects coronary microvascular function via the GSK-3β/m-PTP pathway in rat hearts subjected to sodium laurate-induced CME.28 In this study, they focused on the relation between GSK-3β and mPTP. However, in addition to the mPTP opening and inflammation, mitochondrial apoptosis and oxidative stress are also important in the progression of CME. In the current research, we proposed another role of alprostadil, the inhibition of mitochondrial apoptosis, which was confirmed by the decreased Cyt-c release into the cytoplasm, down-regulation of cleaved-caspase-3, cleaved-caspase-9, and Bax, as well as up-regulation of Bcl-2. Additionally, we further evaluated the possible role of GSK-3β/Nrf2/HO-1 in the protective effect of alprostadil. In our research, GSK-3β activity was determined by detecting the ratio of p-GSK-3β (Ser-9)/GSK-3β, and the increased value indicated inhibition of GSK-3β activity. Consistently, we found that the expression levels of p-GSK-3β (Ser-9) and Nrf2 were notably reduced in the CME group, while treatment with alprostadil injection greatly reversed those changes. In addition, Nrf2 and HO-1 mRNA and protein expression levels were markedly elevated in alprostadil treatment groups. Moreover, double immunofluorescence staining also indicated that Nrf2 and HO-1 were highly expressed after alprostadil treatment. Accumulating literature demonstrated that Nrf2 plays an important role in maintaining mitochondria integrity and improving the synthesis of ATP, especially under conditions of oxidative stress.45–48 Our results showed that myocardium ATP concentrations were remarkably decreased following CME, while alprostadil treatment significantly elevated ATP production. Taken together, these findings indicated that alprostadil injection results in significant cardio-protective effects on a rat model of CME, which may be achieved by activating the GSK-3β/Nrf2/HO-1 signaling pathway.
The present study has several limitations. Firstly, we established a rat model of CME by injecting plastic microspheres into the left ventricle together with clamping the ascending aorta. Therefore, potential differences between clinical and animal models should be acknowledged. Secondly, GSK-3β regulated the expression levels of multiple genes, and apoptosis may be regulated by complex mechanisms. It is necessary to conduct GSK-3β or Nrf2 knock-out rat experiments for further verification of our findings in the future. Finally, we only evaluated cardiac function at 12 hours after CME and the long-term effects of alprostadil on CME-mediated myocardial injury were not assessed. Hence, further studies are required to follow-up rats for at least 1 month and assess the cardiac function.
Conclusions
In conclusion, it was demonstrated that alprostadil injection attenuated cardiomyocyte apoptosis and improved myocardial antioxidant stress ability in CME-induced myocardial injury in vivo via modulation of the GSK-3β/Nrf2/HO-1 signaling pathway. Our findings revealed that alprostadil injection may be a promising medication for cardioprotection towards CME in clinical practice, while further study is required to further elucidate the effects of alprostadil injection on cardioprotection. Our findings may also provide a theoretical basis to investigate the effects of alprostadil injection on patients with CME.
Abbreviations
CME, coronary microembolization; cTnI, serum c-troponin I; ATP, adenosine triphosphate; SOD, superoxide dismutase; MDA, malondialdehyde; PCI, percutaneous coronary interventions; GSK-3β, glycogen synthase kinase 3β; Nrf2, nuclear factor erythroid 2-related factor 2; HO-1, heme oxygenase-1; Cyt-c, cytochrome c; p-GSK-3β (Ser-9), phosphorylation of serine-9 residues of GSK-3β.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No.81770346), The Project for Innovative Research Team in Guangxi Natural Science Foundation (Grant No.2018GXNSFGA281006) and CMVD Fund of XinXin Heart (SIP) Foundation - China Cardiovascular Association (Grant No. 2018-CCA-CMVD-09).
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Heusch G, Skyschally A, Kleinbongard P. Coronary microembolization and microvascular dysfunction. Int J Cardiol. 2018;258:17–23. doi:10.1016/j.ijcard.2018.02.010
2. Heusch G. The coronary circulation as a target of cardioprotection. Circ Res. 2016;118(10):1643–1658. doi:10.1161/CIRCRESAHA.116.308640
3. Jaffe R, Charron T, Puley G, et al. Microvascular obstruction and the no-reflow phenomenon after percutaneous coronary intervention. Circulation. 2008;117(24):3152–3156. doi:10.1161/CIRCULATIONAHA.107.742312
4. Ferrari R, Balla C, Malagù M, et al. Reperfusion damage-a story of success, failure, and hope. Circ J. 2017;81(2):131–141. doi:10.1253/circj.CJ-16-1124
5. Bolognese L, Carrabba N, Parodi G, et al. Impact of microvascular dysfunction on left ventricular remodeling and long-term clinical outcome after primary coronary angioplasty for acute myocardial infarction. Circulation. 2004;109(9):1121–1126. doi:10.1161/01.CIR.0000118496.44135.A7
6. Liu YC, Li L, Su Q, et al. Trimetazidine pretreatment inhibits myocardial apoptosis and improves cardiac function in a Swine model of coronary microembolization. Cardiology. 2015;130(2):130–136. doi:10.1159/000369246
7. Zhu HH, Wang XT, Sun YH, et al. MicroRNA-486-5p targeting PTEN protects against coronary microembolization-induced cardiomyocyte apoptosis in rats by activating the PI3K/AKT pathway. Eur J Pharmacol. 2019;855:244–251. doi:10.1016/j.ejphar.2019.03.045
8. Juhaszova M, Zorov DB, Yaniv Y, et al. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104(11):1240–1252. doi:10.1161/CIRCRESAHA.109.197996
9. Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7(6):1321–1327. doi:10.1016/S1097-2765(01)00253-2
10. Alam J, Stewart D, Touchard C, et al. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274(37):26071–26078. doi:10.1074/jbc.274.37.26071
11. Li Y, Paonessa JD, Zhang Y. Mechanism of chemical activation of Nrf2. PLoS One. 2012;7(4):e35122. doi:10.1371/journal.pone.0035122
12. Hayes JD, Chowdhry S, Dinkova-Kostova AT, Sutherland C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of β-TrCP and GSK-3. Biochem Soc Trans. 2015;43(4):611–620. doi:10.1042/BST20150011
13. Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2-an update. Free Radic Biol Med. 2014;66:
14. Kara H, Ozer A, Arpaci H, et al. Effect of alprostadil on erythrocyte deformability in ischemia reperfusion injury. Bratisl Lek Listy. 2015;116(8):
15. Soares BL, Freitas MA, Montero EF, et al. Alprostadil attenuates inflammatory aspects and leucocytes adhesion on renal ischemia and reperfusion injury in rats. Acta Cir Bras. 2014;29(Suppl 2):55–60. doi:10.1590/S0102-8650201400140011
16. Hsieh CC, Hsieh SC, Chiu JH, Wu YL. Protective effects of N-acetylcysteine and a prostaglandin E1 analog, alprostadil, against hepatic ischemia: reperfusion injury in rats. J Tradit Complement Med. 2014;4(1):64–71. doi:10.4103/2225-4110.124351
17. Erer D, Özer A, Demirtaş H, et al. Effects of alprostadil and iloprost on renal, lung, and skeletal muscle injury following hindlimb ischemia-reperfusion injury in rats. Drug Des Devel Ther. 2016;10:2651–2658.
18. Li JH, Yang P, Li AL, et al. Cardioprotective effect of liposomal prostaglandin E1 on a porcine model of myocardial infarction reperfusion no-reflow. J Zhejiang Univ Sci B. 2011;12(8):638–643. doi:10.1631/jzus.B1101007
19. Sheng X, Ding S, Ge H, et al. Intracoronary infusion of alprostadil and nitroglycerin with targeted perfusion microcatheter in STEMI patients with coronary slow flow phenomenon. Int J Cardiol. 2018;265:6–11. doi:10.1016/j.ijcard.2018.04.119
20. Zhang L, Zhang Y, Yu X, et al. Alprostadil attenuates myocardial ischemia/reperfusion injury by promoting antioxidant activity and eNOS activation in rats. Acta Cir Bras. 2018;33(12):1067–1077. doi:10.1590/s0102-865020180120000004
21. Zhou ZC. A course in toxicology. Peking University Medical Press; 2006.
22. Su Q, Li L, Sun Y, Yang H, Ye Z, Zhao J. Effects of the TLR4/Myd88/NF-κB signaling pathway on NLRP3 inflammasome in coronary microembolization-induced myocardial injury. Cell Physiol Biochem. 2018;47(4):1497–1508. doi:10.1159/000490866
23. Su Q, Lv X, Sun Y, et al. Role of TLR4/MyD88/NF-κB signaling pathway in coronary microembolization-induced myocardial injury prevented and treated with nicorandil. Biomed Pharmacother. 2018;106:776–784. doi:10.1016/j.biopha.2018.07.014
24. Su Q, Lv X, Ye Z, et al. The mechanism of miR-142-3p in coronary microembolization-induced myocardiac injury via regulating target gene IRAK-1. Cell Death Dis. 2019;10(2):61. doi:10.1038/s41419-019-1341-7
25. Barth E, Stämmler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. 1992;24(7):669–681. doi:10.1016/0022-2828(92)93381-S
26. Boyman L, Karbowski M, Lederer WJ. Regulation of mitochondrial ATP production: Ca2+ signaling and quality control. Trends Mol Med. 2020;26(1):21–39. doi:10.1016/j.molmed.2019.10.007
27. Fang Y, Zhao Y, He S, et al. Overexpression of FGF19 alleviates hypoxia/reoxygenation-induced injury of cardiomyocytes by regulating GSK-3β/Nrf2/ARE signaling. Biochem Biophys Res Commun. 2018;503(4):2355–2362. doi:10.1016/j.bbrc.2018.06.161
28. Zhu H, Ding Y, Xu X, et al. Prostaglandin E1 protects coronary microvascular function via the glycogen synthase kinase 3β-mitochondrial permeability transition pore pathway in rat hearts subjected to sodium laurate-induced coronary microembolization. Am J Transl Res. 2017;9(5):2520–2534.
29. Herrmann J. Peri-procedural myocardial injury: 2005 update. Eur Heart J. 2005;26(23):2493–2519. doi:10.1093/eurheartj/ehi455
30. Hildebrandt HA, Kahlert P, Baars T, et al. Is there a need for distal protection during native vessel percutaneous coronary intervention in patients with stable coronary artery disease? J Cardiovasc Med (Hagerstown). 2014;15(2):170–172. doi:10.2459/JCM.0b013e3283619351
31. Ge J, Schäfer A, Ertl G, Nordbeck P. Thrombus aspiration for ST-segment-elevation myocardial infarction in modern era: still an issue of debate? Circ Cardiovasc Interv. 2017;10(10):e005739. doi:10.1161/CIRCINTERVENTIONS.117.005739
32. Wei LY, Fu XH, Li W, et al. Effect of intravenous administration of liposomal prostaglandin E1 on microcirculation in patients with ST elevation myocardial infarction undergoing primary percutaneous intervention. Chin Med J (Engl). 2015;128(9):1147–1150. doi:10.4103/0366-6999.156078
33. Fan Y, Jiang Y, Fu X, et al. Effects of liposomal prostaglandin E1 on periprocedural myocardial injury in patients with unstable angina undergoing an elective percutaneous coronary intervention. Coron Artery Dis. 2015;26(8):671–677. doi:10.1097/MCA.0000000000000294
34. Mehrabi MR, Serbecic N, Tamaddon F, et al. Clinical benefit of prostaglandin E1-treatment of patients with ischemic heart disease: stimulation of therapeutic angiogenesis in vital and infarcted myocardium. Biomed Pharmacother. 2003;57(3–4):173–178.
35. Feld S, Li G, Amirian J, et al. Enhanced thrombolysis, reduced coronary reocclusion and limitation of infarct size with liposomal prostaglandin E1 in a canine thrombolysis model. J Am Coll Cardiol. 1994;24(5):1382–1390. doi:10.1016/0735-1097(94)90124-4
36. Gustafsson AB, Gottlieb RA. Mechanisms of apoptosis in the heart. J Clin Immunol. 2003;23(6):
37. Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281(5381):
38. Cheng EH, Wei MC, Weiler S, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8(3):
39. Su Q, Lv X, Ye Z. Ligustrazine attenuates myocardial injury induced by coronary microembolization in rats by activating the PI3K/Akt pathway. Oxid Med Cell Longev. 2019;2019:6791457. doi:10.1155/2019/6791457
40. Övey İS, Naziroğlu M. Homocysteine and cytosolic GSH depletion induce apoptosis and oxidative toxicity through cytosolic calcium overload in the hippocampus of aged mice: involvement of TRPM2 and TRPV1 channels. Neuroscience. 2015;284:225–233. doi:10.1016/j.neuroscience.2014.09.078
41. Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signaling. Biochem J. 1993;296(Pt 1):15–19. doi:10.1042/bj2960015
42. Guo Y, Gupte M, Umbarkar P, et al. Entanglement of GSK-3β, β-catenin and TGF-β1 signaling network to regulate myocardial fibrosis. J Mol Cell Cardiol. 2017;110:109–120. doi:10.1016/j.yjmcc.2017.07.011
43. Rojo AI, Medina-Campos ON, Rada P, et al. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: role of glycogen synthase kinase-3. Free Radic Biol Med. 2012;52(2):473–487. doi:10.1016/j.freeradbiomed.2011.11.003
44. Jin A, Li B, Li W, Xiao D. PHLPP2 downregulation protects cardiomyocytes against hypoxia-induced injury through reinforcing Nrf2/ARE antioxidant signaling. Chem Biol Interact. 2019;314:108848. doi:10.1016/j.cbi.2019.108848
45. Holmström KM, Baird L, Zhang Y, et al. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open. 2013;2(8):761–770. doi:10.1242/bio.20134853
46. Greco T, Shafer J, Fiskum G. Sulforaphane inhibits mitochondrial permeability transition and oxidative stress. Free Radic Biol Med. 2011;51(12):2164–2171. doi:10.1016/j.freeradbiomed.2011.09.017
47. Zhu H, Jia Z, Strobl JS, et al. Potent induction of total cellular and mitochondrial antioxidants and phase 2 enzymes by cruciferous sulforaphane in rat aortic smooth muscle cells: cytoprotection against oxidative and electrophilic stress. Cardiovasc Toxicol. 2008;8(3):115–125. doi:10.1007/s12012-008-9020-4
48. Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88(PtB):179–188. . doi:10.1016/j.freeradbiomed.2015.04.036
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.