")
Back to Journals » OncoTargets and Therapy » Volume 13
α-MSH-PE38KDEL Kills Melanoma Cells via Modulating Erk1/2/MITF/TYR Signaling in an MC1R-Dependent Manner
Authors Liu X , Li H, Cong X , Huo D, Cong L, Wu G
Received 19 June 2020
Accepted for publication 9 November 2020
Published 3 December 2020 Volume 2020:13 Pages 12457—12469
DOI https://doi.org/10.2147/OTT.S268554
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Xilin Liu,1,* Hong Li,2,* Xianling Cong,3 Da Huo,1 Lele Cong,4 Guangzhi Wu1
1Department of Hand Surgery, China Japan Union Hospital of Jilin University, Changchun City, Jilin Province 130033, People’s Republic of China; 2Emergency Medical Department, China Japan Union Hospital of Jilin University, Changchun City, Jilin Province 130033, People’s Republic of China; 3Tissue Bank, China Japan Union Hospital of Jilin University, Changchun City, Jilin Province 130033, People’s Republic of China; 4Department of Dermatology, China Japan Union Hospital of Jilin University, Changchun City, Jilin Province 130033, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guangzhi Wu
Department of Hand Surgery, China Japan Union Hospital of Jilin University, Changchun City, Jilin Province 130033, People’s Republic of China
Tel +86-13944181019
Email [email protected]
Background/Objective: The immunotoxin α-MSH-PE38KDEL consisting of α-MSH and PE38KDEL showed high cytotoxicity on MSH receptor-positive melanoma cells, suggesting that α-MSH-PE38KDEL might be a potent drug for the treatment of melanoma. Herein, we explored whether the Erk1/2/MITF/TYR signaling, a verified target of α-MSH/MC1R, was involved in α-MSH-PE38KDEL-mediated cytotoxicity.
Methods: Human melanoma cell line A375, mouse melanoma cell line B16-F10, human breast cancer cell line MDA-MB-231 and human primary epidermal melanocytes (HEMa) with different expression levels of MC1R were used in this study. Cell apoptosis and viability were determined by using flow cytometry and MTT assays. Protein expressions were tested by Western blotting.
Results: The expression levels of MC1R in A375 and B16-F10 cells were significantly higher than that of MDA-MB-231 and HEMa. α-MSH-PE38KDEL treatment induced a significant inhibition in cell viability in A375 and B16-F10 cells, while showed no obvious influence in the viability of MDA-MB-231 and HEMa cells. However, knockdown of MC1R abolished α-MSH-PE38KDEL role in promoting cell apoptosis in A375 and B16-F10 cells, and upregulation of MC1R endowed α-MSH-PE38KDEL function to promote cell apoptosis in MDA-MB-231 and HEMa cells. Additionally, α-MSH-PE38KDEL treatment increased the phosphorylation levels of Erk1/2 and MITF (S73), and decreased MITF and TYR expressions in an MC1R-dependent manner. All of the treatments, including inhibition of Erk1/2 with PD98059, MC1R downregulation and MITF overexpression weakened the anti-tumor role of α-MSH-PE38KDEL in melanoma.
Conclusion: Collectively, this study indicates that α-MSH-PE38KDEL promotes melanoma cell apoptosis via modulating Erk1/2/MITF/TYR signaling in an MC1R-dependent manner.
Keywords: MC1R, melanoma, α-MSH-PE38KDEL, apoptosis, viability, Erk1/2/MITF/TYR
Introduction
Melanoma, as a type of skin cancers, is known as “the king of cancers” due to its high malignancy. Although melanoma only accounts for 10% of skin cancers, about 80% of the cancer-related mortality is caused by melanoma.1 Moreover, melanoma is the most rapidly growing tumor among all malignant tumors, with a growing incidence rate of 3–5% every year and a propensity to affect young patients.2–4 As a result, it is needful to further clarify the molecular mechanisms underlying melanoma progression.
It is well identified that environmental ultraviolet ray exposure, fair skin, high number of melanocytic nevi and family history are the risk factors for melanoma.5 Molecular and genetic findings indicate that variations in the coding region of the melanocortin-1-receptor (MC1R), the receptor of α-MSH (α-melanocyte-stimulating hormone), exert crucial roles in the occurrence and development of melanoma.6 Noticeably, there are less than 100 α-MSH receptor sites on human normal cells or epidermal melanocytes, but about 900–5700 α-MSH receptor sites on the surface of human melanoma cells,7 indicating that MC1R might serve as an effective therapeutic target for melanoma.
Recombinant immunotoxins are chimeric proteins that are constructed via binding the antigen-binding domain of an antibody to a bacterial toxin, such as Pseudomonas exotoxin A (PE). Bacterial toxins can suppress the synthesis of cellular proteins, resulting in the apoptotic death of cancer cells.8,9 PE contains three domains, domain I, II and III. Domain III consists of a REDL sequence which is responsible for the combination with the mammalian KDEL receptor and the catalytic activity of the toxin. Its C-terminal amino acid sequence induces the movement of the toxin from the Golgi apparatus to the endoplasmic reticulum.8,10 Domain I is responsible for the transport of toxin to cells via binding to CD91.11,12 Usually, domain I is replaced with the antibody/ligand to make it higher specificity.13 PE38KDEL is a mutated and truncated form of PE, in which the domain Ia has been removed and replaced with an antiparallel β structure for cell recognition.14 In our previous study,15 we found that the immunotoxin α-MSH-PE38KDEL equipped α-MSH (α-melanophore-stimulating hormone) to PE38KDEL showed high cytotoxicity on MSH receptor-positive melanoma cells, suggesting a prospect for the treatment of melanoma.
Tyrosinase (TYR) is a key enzyme that regulates melanin synthesis,16 which is regulated by a microphthalmia-associated transcription factor (MITF).17 It is reported that Erk1/2 activation can phosphorylate MITF at Ser73 and 409 sites, leading to a temporary increase in the trans-activation activity and subsequent degradation of MITF.18 As α-MSH can activate the ERK1/2 signaling via binding to MC1R,19,20 we conjecture that ERK1/2/MITF/TYR signaling may be involved in α-MSH-PE38KDEL-mediated apoptosis of melanoma cells.
In the current study, we aimed to uncover the mechanism underlying α-MSH-PE38KDEL-mediated apoptosis in melanoma cells and to determine whether α-MSH-PE38KDEL binds to MC1R to induce cell apoptosis via modulating the ERK1/2/MITF/TYR signaling. In addition, the in vivo tumor models were established in male athymic BALB/C nude mice according to previous studies.21,22
Materials and Methods
α-MSH-PE38KDEL Obtainment
The immunotoxin α-MSH-PE38KDEL was constructed by connecting the α-MSH gene to PE38KDEL with the flexible Linker SGGGGS (TaKaRa, Dalian, China), as we previously described.15
Cell Culture and Treatment
Human melanoma cell line A375 (ATCC® CRL-1619™), mouse melanoma cell line B16-F10 (ATCC® CRL-6475), human breast cancer cell line MDA-MB-231 (ATCC® HTB-26), and human primary epidermal melanocytes (HEMa) (ATCC® PCS-200-013™) were all purchased from American Type Culture Collection (VA, USA). A375, B16-F10 and HEMa cells were cultured in Dulbecco’s Modified Eagle’s Medium, and MDA-MB-231 cells were in Leibovitz’s L-15 Medium, all supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. All used reagents were obtained from Thermo Fisher Scientific (MA, USA).
The cells were treated with 100 ng/mL α-MSH-PE38KDEL for 24 hours. To repress the activation of Erk1/2 signaling, A375 cells were treated with 100 μM PD98059 (Byotime, Jiangsu, China) for 4 hours.
Lentivirus Obtainment and Stable Cell Line Establishment
The lentivirus vector shRNAs which are applied to silence MC1R (sh-MC1R) in melanoma A375 and B16-F10 cells and the negative controls (sh-NC) were synthesized by the GenePharma Co., LTD. (Shanghai, China).
To establish the stablly sh-MC1R transfected cell lines, B16-F10 cells were transfected with the lentivirus vector and maintained in 5 μg/mL puromycin for 2 weeks, with medium being changed every other day. After that, the survival cells were inoculated into 6-well plates and used for the in vivo assay.
Cell Transfection
To upregulate MC1R, MITF and TYR expression, the cells were transfected with the overexpressing plasmid of MC1R, MITF and TYR (OE-MC1R; OE-MITF; OE-TYR; GenePharma) with the help of Lipofectamine 2000 reagent (Invitrogen, Waltham, MA, USA) based on the manufacturer’s instructions.
Real-Time Quantitative PCR (qPCR) Assay
Total RNA was extracted from cells with Trizol reagent (Thermo Fisher Scientific, MA, USA) in accordance with the manufacturer’s instructions. After quantification with Nanodrop (Thermo Fisher Scientific), a total of 1 μg RNA was reversely transcripted into cDNA with a First Strand cDNA Synthesis Kit purchased from CWBIO (Jiangsu, China). Subsequently, the mRNA level of MC1R was assessed by using the qPCR assay with an SYBR Green reagent (Takara, Dalian, China) in an ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). GAPDH level was applied to normalize the mRNA level of MC1R. The primer sequences of MC1R and GAPDH were synthesized by Sangon Biotech (Shanghai, China) and were as follows:
Human-MC1R: sense-5ʹ- ACTTCTCACCAGCAGTCGTG-3ʹ,
MC1R-antisense-5ʹ- CATTGGAGCAGACGGAGTGT-3ʹ;
Human-GAPDH: sense-5ʹ-CCACTAGGCGCTCACTGTTCT-3ʹ,
GAPDH-antisense-5ʹ- GCATCGCCCCACTTGATTTT-3ʹ;
Mouse-MC1R: sense-5ʹ- GAGTAGCACCTGGGGTGAAC-3ʹ,
MC1R-antisense-5ʹ-CTGCAGGAATCCCAGGTTGT-3ʹ;
Mouse-GAPDH: sense-5ʹ- CCCTTAAGAGGGATGCTGCC-3ʹ,
GAPDH-antisense-5ʹ-ACTGTGCCGTTGAATTTGCC-3ʹ.
Western Blotting Assay
To determine protein expressions, Western blotting assay was carried out. RIPA lysis buffer (Beyotime, Jiangsu, China) was applied to isolate total protein from tissues and cells. Then, the supernatant was harvested following centrifugation at 4°C for 20 min at a speed of 1500 g. Next, the protein concentrations were assessed with a BCA kit (Thermo Fisher Scientific). After that, proteins were degenerated at 100°C for 10 min and an equal amount of proteins from every sample were separated by 10% SDS-PAGE and then transferred into the PVDF membranes (Millipore, Billerica, MA, USA). The membranes were then incubated with the indicated primary antibodies, including MC1R (No. PA5-75,342, Thermo Fisher Scientific), Bax (No. sc-7480, Santa Cruz Biotechnology, Dallas, Texas, USA), Bcl-2 (No. sc-7382, Santa Cruz Biotechnology), caspase-3 (No. ab13847, Abcam, Cambridge, MA, USA), cleaved caspase3 (No. ab2302, Abcam), caspase 9 (No. ab52298, Abcam), cleaved caspase 9 (No. ab2324, Abcam), Erk1/2 (No. ab184699), p-Erk1/2 (No. ab223500, Abcam), MITF (No. ab122982), p-MITF (S73) (No. ab59201, Abcam), TYR (No. ab180753, Abcam), Na, K-ATPase (No. 3010, Cell Signaling Technology, MA, USA) and GAPDH (1:5000 dilution; No. TA-08, Zhongshanjinqiao, Beijing, China) overnight at 4°C. After that, the membranes were probed with the corresponding secondary antibodies (Proteintech, Wuhan, China) for 1 hour at room temperature. Protein expression signaling was detected by using the gel imaging instrument (Eberhardzell, Germany) following incubation with an enhanced chemiluminescence reagent (ECL; Millipore). ImageJ software was applied for protein quantitation.
MTT Assay
Cell proliferation was detected by using MTT reagent (Merck KGaA, Darmstadt, Germany) in the light of the manufacturer’s instructions. In detail, 3×103 cells were seeded into 96-well plates with a 200 μL culture medium. At 48 hours post transfection/infection, cell culture medium was removed and the cells were incubated with 20 μg MTT in 200 μL medium at 37°C for 4 hours. After incubation with 150 μL dimethyl sulfoxide (DMSO) for 10 min, the absorbance at 570 nm for each well was tested.
Flow Cytometry Assay
Flow cytometry assay was performed to assess cell apoptosis. Annexin V Apoptosis Detection kit (BD Biosciences, San Diego, CA, USA) was used to test cell apoptosis. After 48 hours of transfection/infection, cells were harvested and washed with PBS for one time. Then, the cells were incubated with 5 μL of Annexin V and 5 μL PI (propidium iodide) for 15 min in the dark. After that, the cells were washed in 1× binding buffer and resuspended in 500 μL of 1× binding buffer. Then, the cells were subjected to detection on a Beckman FC500 flow cytometer (Beckman Coulter, Inc., Brea, California, USA) and analyzed with the help of FlowJo 7.6 software.
Xenotransplantation Assay
Four-week-old male athymic BALB/C nude mice obtained from Weitonglihua. Co., LTD (Beijing, China) were applied to assess α-MSH-PE38KDEL role in cell tumorigenesis. In detail, 1×106 stably transfected B16-F10 cells with α-MSH-PE38KDEL treatment (40 mg/kg)/PD98059 (10 mg/kg) or without were resuspended in 200 μL PBS, and then injected into the armpit area of nude mice, with 6 mice in each group. Twenty-eight days post transplantation, mice were sacrificed and the tumors were collected for weighing. This animal experiment was performed in accordance with the principles and procedures of the NIH Guide for the Care and Use of Laboratory Animals and approved by the ethical committee of China Japan Union Hospital of Jilin University.
Statistical Analysis
Each experiment in the current study was performed three times. Data were indicated as mean ± standard deviation (SD). Statistical analyses were performed by SPSS23.0 with one-way analysis of variance (one-way ANOVA) followed by Tukey’s tests for multiple groups and student’s t tests for two groups. P<0.05 was considered as statistically significant.
Results
α-MSH-PE38KDEL Induces Cell Apoptosis via Identification of MC1R
To explore whether MC1R is essential for α-MSH-PE38KDEL-mediated cytotoxicity, we chose several cell lines with different expression patterns of MC1R. As shown in Figure 1A, compared with human melanocytes HEMa, the expression levels of MC1R in cell membrane and total cell lysis of human melanoma cell line A375 and mouse melanoma cell line B16-F10 were significantly increased, while was decreased in human breast cancer cell line MDA-MB-231. α-MSH-PE38KDEL treatment induced significant inhibition in cell viability in A375 and B16-F10 cells, while showed no obvious change in the viability of MDA-MB-231 and HEMa cells (Figure 1B). In addition, α-MSH-PE38KDEL treatment significantly increased the expression levels of cleaved caspase3/9 and Bax and decreased Bcl-2 expression in A375 and B16-F10 cells, but showed no obvious influence in the expression levels of cleaved caspase 3/9, Bax and Bcl-2 in MDA-MB-231 and HEMa cells (Figure 1C–F). Consistently, α-MSH-PE38KDEL treatment significantly increased the apoptosis of A375 and B16-F10 cells (Figure 1G). These results indicated that α-MSH-PE38KDEL promoted cell apoptosis in MC1R-positive cells.
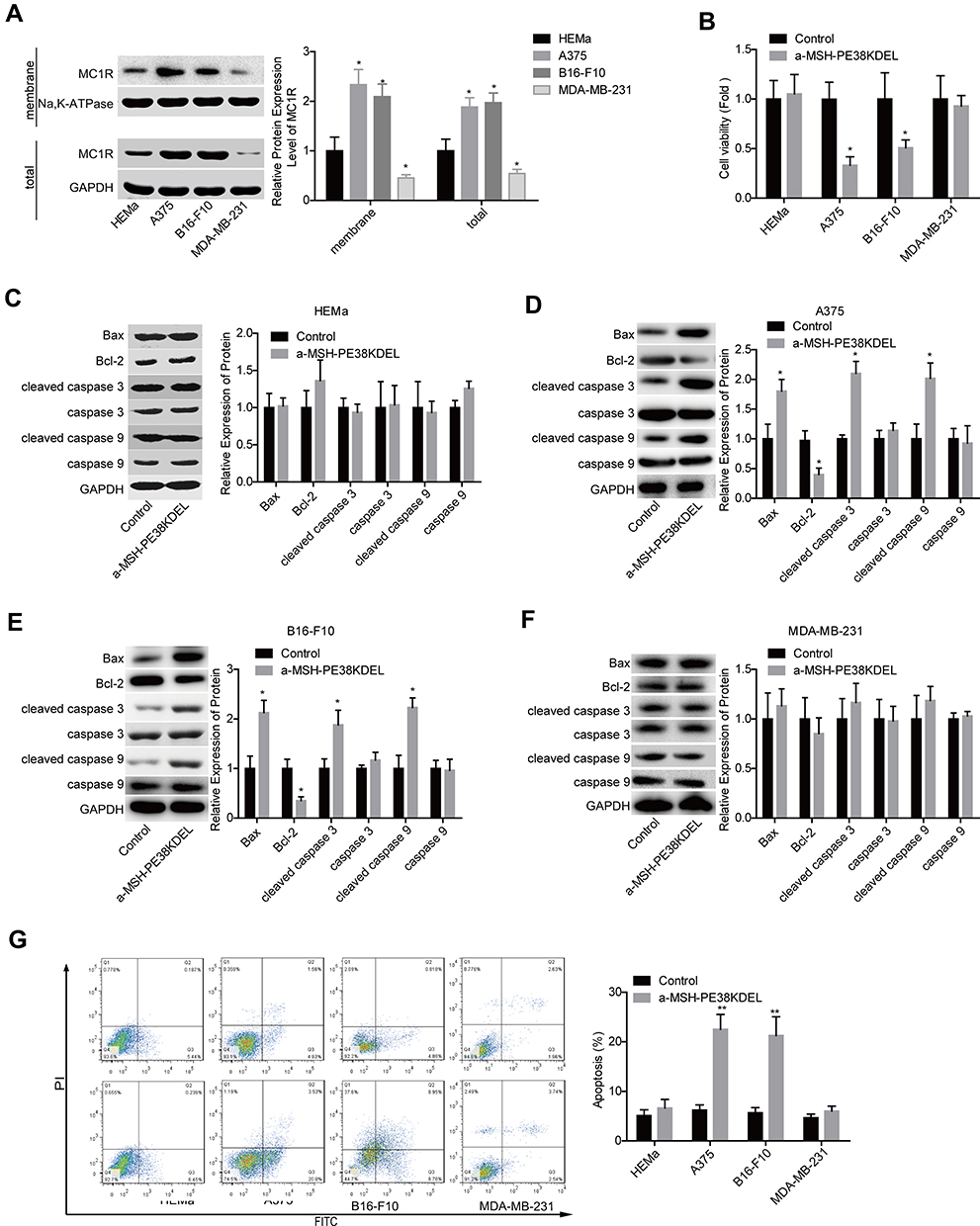 |
Figure 1 α-MSH-PE38KDEL induced cell apoptosis through identification of MC1R. (A) The expression levels of MC1R protein in cell membrane and total cell lysis of HEMa, A375, B16-F10, and MDA-MB-231 cells were detected by Western blotting assay. HEMa, A375, B16-F10 and MDA-MB-231 cells were treated with α-MSH-PE38KDEL (100 ng/mL) for 24 hours, the cells were then submitted to the following assays. (B) Cell viability was determined by MTT assay. (C–F) The expression levels of Bax, Bcl-2, caspase 3/9 and cleaved caspase 3/9 were determined by using Western blotting assay. (G) Cell apoptosis rates were tested by flow cytometry assay. (n=3, *P<0.05, **P<0.01). |
α-MSH-PE38KDEL Induces Cell Apoptosis in a MC1R-Dependent Manner
The loss-of-function assays were then performed to further elucidate whether MC1R is essential in α-MSH-PE38KDEL-mediated cell apoptosis. As shown in Figure 2A–B, MC1R expression was obviously reduced after cell infection with sh-MC1R in A375 and B16-F10 cells. Compared with the α-MSH-PE38KDEL group, the inhibitory role of α-MSH-PE38KDEL in cell growth was significantly weakened following cell infection with sh-MC1R (Figure 2C–D). In addition, knockdown of MC1R increased Bcl-2 expression and decreased the levels of Bax and cleaved caspase 3/9 (Figure 2E–F), as well as reduced cell apoptosis rate induced by α-MSH-PE38KDEL treatment (Figure 2G–H).
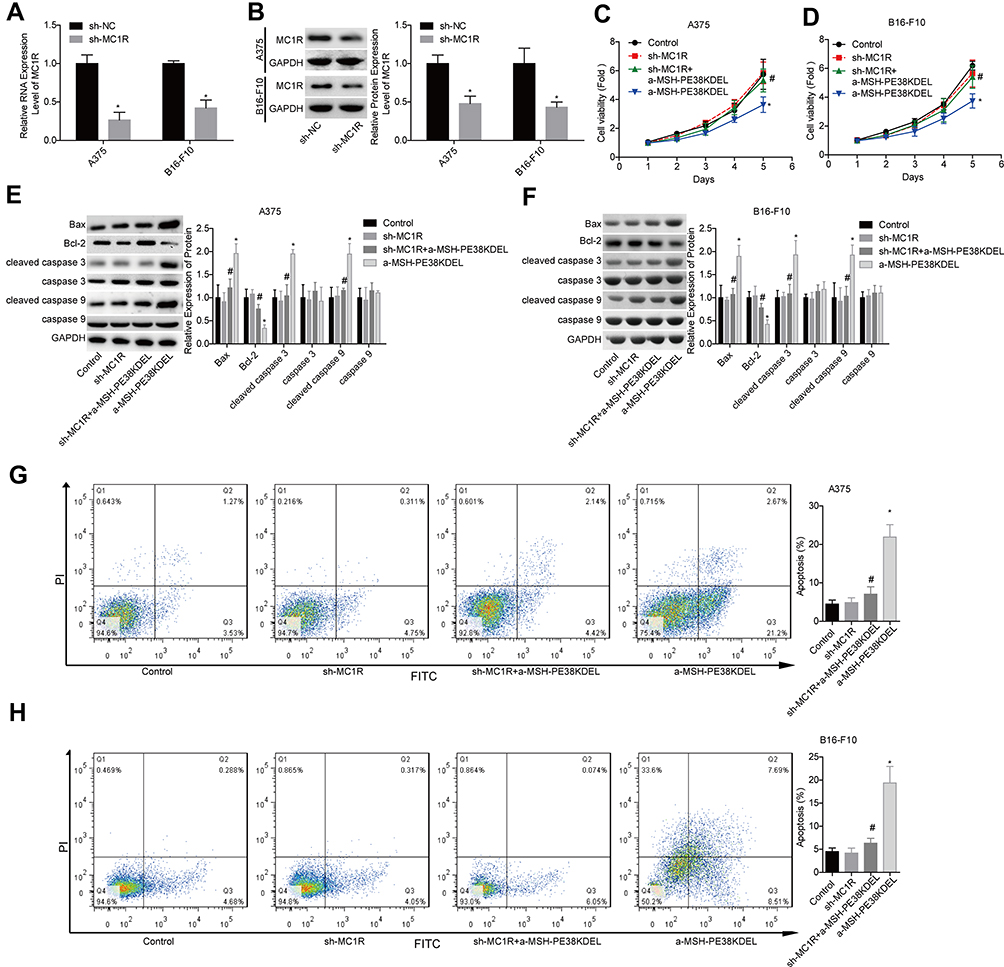 |
Figure 2 Knockdown of MC1R abolished α-MSH-PE38KDEL-induced cytotoxicity in A375 and B16-F10 cells. (A and B) MC1R expression was detected by qPCR and Western blotting assays following sh-MC1R/sh-NC infection. Then, the cells were divided into control, sh-MC1R, sh-MC1R+α-MSH-PE38KDEL and α-MSH-PE38KDEL groups, and the following assays were carried out. (C and D) Cell viability was determined by MTT assay. (E and F) The expression levels of Bax, Bcl-2, caspase 3/9 and cleaved caspase 3/9 were determined by using Western blotting assay. (G and H) Cell apoptosis rates were tested by flow cytometry assay. (n=3, *P<0.05, vs. control group; #P<0.05, vs. α-MSH-PE38KDEL group). |
Additionally, the gain-of-function assay was performed in HEMa and MDA-MB-231 cells. Compared with the OE-NC group, MC1R expression was apparently increased following cell transfection with OE-MC1R (Figure 3A–B). Overexpression of MC1R conferred α-MSH-PE38KDEL abilities to inhibit cell growth (Figure 3C–D) and induce cell apoptosis (Figure 3G–H). In addition, Bcl-2 expression was reduced while the expression levels of Bax and cleaved caspase 3/9 were increased when MC1R was upregulated inα-MSH-PE38KDEL-treated HEMa and MDA-MB-231 cells (Figure 3E–F). The above findings suggested that α-MSH-PE38KDEL triggered cell apoptosis in an MC1R-dependent manner.
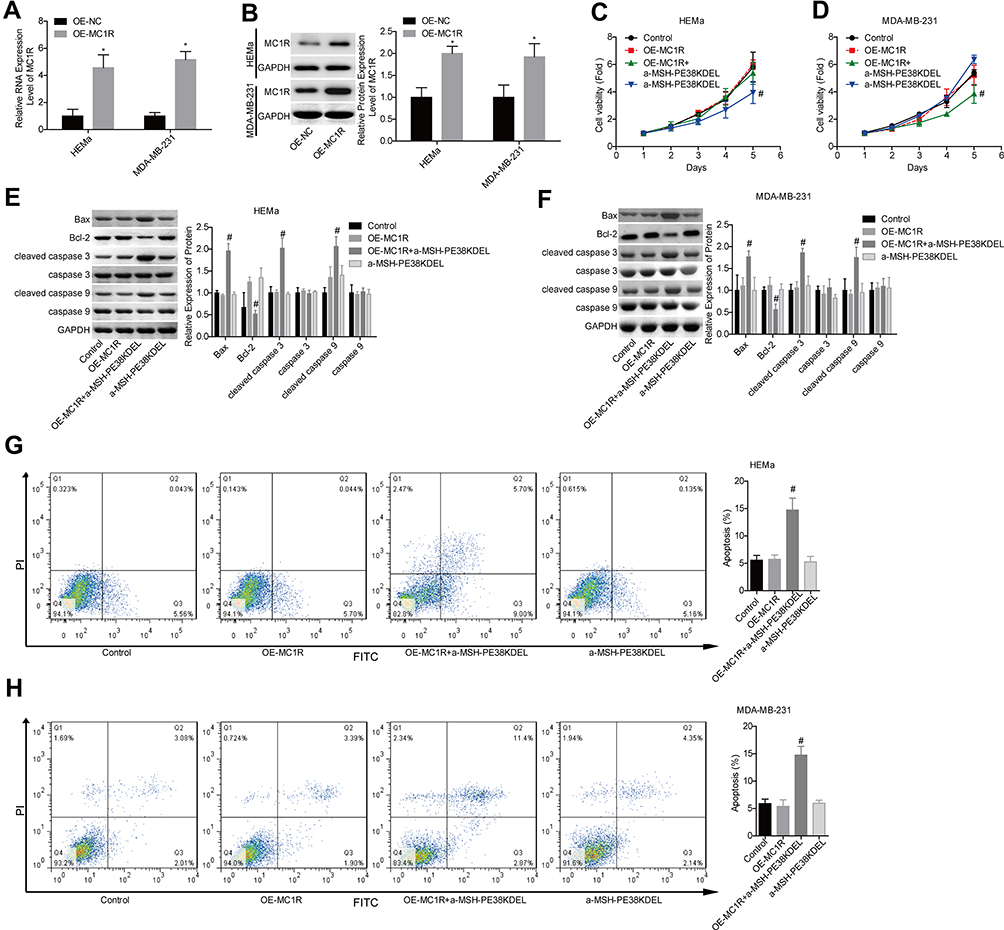 |
Figure 3 Upregulation of MC1R conferred α-MSH-PE38KDE cytotoxicity in HEMa and MDA-MB-231 cells. (A and B) MC1R expression was detected by qPCR and Western blotting assays following OE-MC1R/OE-NC transfection. Then, the cells were divided into control, OE-MC1R, OE-MC1R+α-MSH-PE38KDEL and α-MSH-PE38KDEL groups, and the following assays were carried out. (C and D) Cell viability was determined by MTT assay. (E and F) The expression levels of Bax, Bcl-2, caspase 3/9 and cleaved caspase 3/9 were determined by using Western blotting assay. (G and H) Cell apoptosis rates were tested by flow cytometry assay. (n=3, *P<0.05, vs. control group; #P<0.05, vs. α-MSH-PE38KDEL group). |
α-MSH-PE38KDEL Modulates the Activation of Erk1/2/MITF/TYR Signaling in a MC1R-Dependent Manner
Then, we explored the effect of α-MSH-PE38KDEL on the activation of Erk1/2/MITF/TYR signaling. Compared with the control group, the levels of p-Erk1/2 and p-MITF (S73) were obviously increased, while MITF and TYR expressions were decreased following α-MSH-PE38KDEL treatment in A375 (Figure 4A) and B16-F10 cells (Figure 4B). However, these above tendencies were significantly counteracted following MC1R downregulation in A375 and B16-F10 cells as compared with the α-MSH-PE38KDEL group (Figure 4A–B). These results illustrated that α-MSH-PE38KDEL modulated the Erk1/2/MITF/TYR signaling in an MC1R-dependent manner.
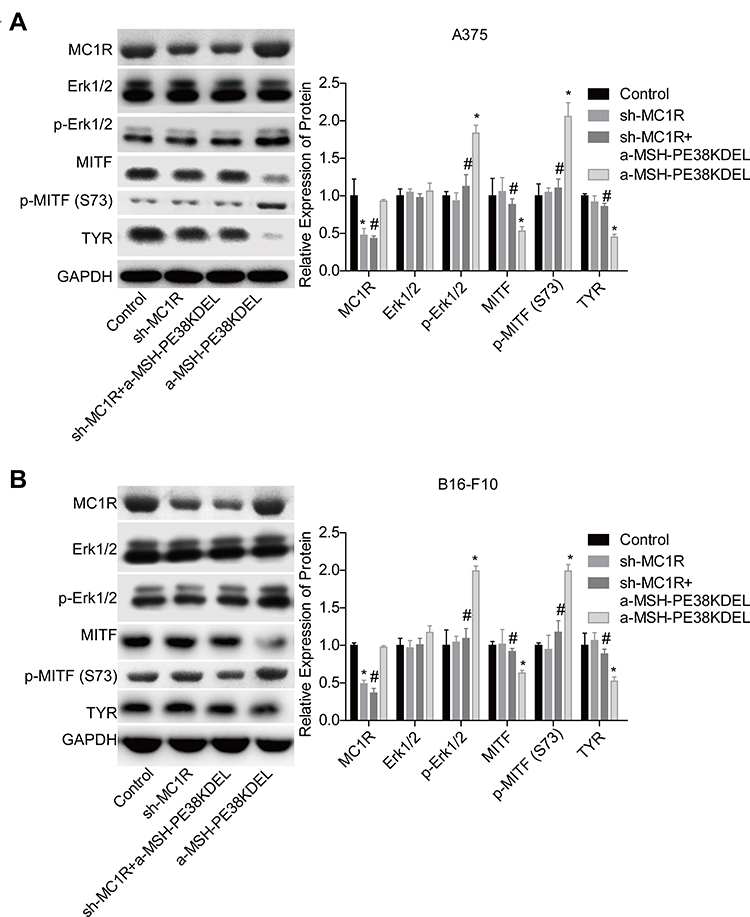 |
Figure 4 α-MSH-PE38KDEL modulated Erk1/2/MITF/TYR signaling in a MC1R-dependent manner. A375 and B16-F10 cells in control, sh-MC1R, sh-MC1R+α-MSH-PE38KDEL and α-MSH-PE38KDEL groups were submitted to the following assays. (A and B) The levels of MC1R, p-Erk1/2, Erk1/2, MITF, p-MITF (S73) and TYR were measured by Western blotting assay. (n=3, *P<0.05, vs. control group; #P<0.05, vs. α-MSH-PE38KDEL group). |
Inhibition of the Erk1/2 Signaling Abolishes α-MSH-PE38KDEL Role in Promoting Cell Apoptosis in A375 Cells
Then, we uncovered Erk1/2 role in α-MSH-PE38KDEL-mediated cytotoxicity in A375 cells. The expression of p-Erk1/2 was moderately decreased following PD98059 treatment for 4 hours (Figure 5A). In addition, PD98059 treatment enhanced A375 cell viability which was suppressed by α-MSH-PE38KDEL (Figure 5B), as well as increased Bcl-2 expression and decreased the expression levels of Bax and cleaved caspase 3/9 in A375 cells (Figure 5C). In addition, PD98059 treatment significantly weakened α-MSH-PE38KDEL role in promoting cell apoptosis in A375 cells (Figure 5D). This result demonstrated that α-MSH-PE38KDEL promoted melanoma A375 cell apoptosis via activating the Erk1/2 signaling.
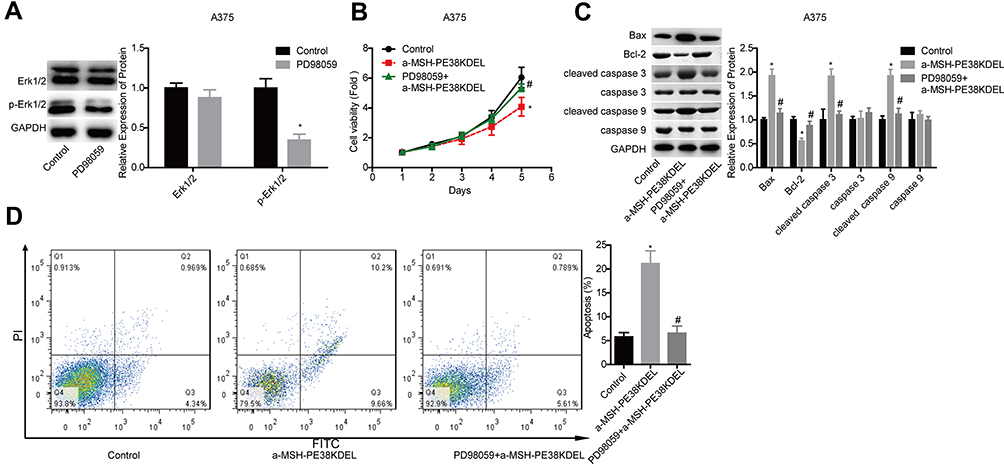 |
Figure 5 α-MSH-PE38KDEL induced cell apoptosis via activating Erk1/2 signaling. (A) Western blotting analysis of the inhibitory efficiency of PD98059 in A375 cells. A375 cells in control, α-MSH-PE38KDEL and PD98059+α-MSH-PE38KDEL groups were collected for the following detections. (B) MTT assay for cell viability evaluation. (C) Western blotting analysis of the expression of Bax, Bcl-2, caspase 3/9 and cleaved caspase 3/9. (D) Flow cytometry assay analysis of cell apoptosis rates. (n=3, *P<0.05, vs. control group; #P<0.05, vs. α-MSH-PE38KDEL group). |
Overexpression of MITF or TYR Abolishes α-MSH-PE38KDEL Roles in Promoting Cell Apoptosis in B16-F10 Cells
Additionally, we explored MITF and TYR role in α-MSH-PE38KDEL-mediated cytotoxicity in B16-F10 cells. The expression levels of MITF and TYR were significantly increased when the cells were transfected with OE-MITF or OE-TYR (Figure 6A–D). The inhibition in cell viability induced by α-MSH-PE38KDEL was neutralized when MITF or TYR was overexpressed (Figure 6E). Moreover, cell apoptosis induced by α-MSH-PE38KDEL treatment was decreased when MITF or TYR was overexpressed (Figure 6F–G). This result demonstrated that α-MSH-PE38KDEL promoted melanoma cell apoptosis via inhibiting MITF and TYR expression.
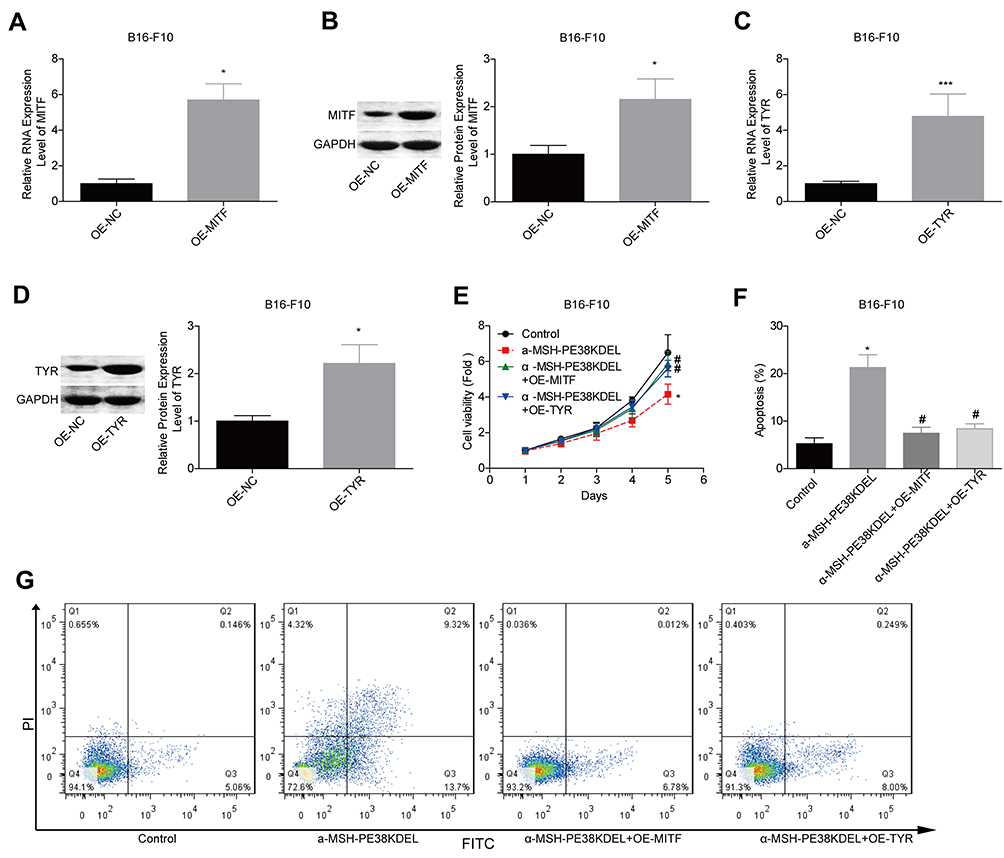 |
Figure 6 α-MSH-PE38KDEL induced cell apoptosis via downregulating MITF and TYR expression. (A–D) QPCR and Western blotting assays were carried out to analyze the transfected efficiencies of OE-MITF and OE-TYR in B16-F10 cells. (E) MTT assay for cell viability evaluation. (F–G) Flow cytometry assay analysis of cell apoptosis rates. (n=3, *P<0.05, ***P<0.001, vs. control group; #P<0.05, vs. α-MSH-PE38KDEL group). |
α-MSH-PE38KDEL Inhibits in vivo Tumor Formation via Modulating the MC1R/Erk1/2/MITF/TYR Signaling in B16-F10 Cells
Moreover, the in vivo assay was performed to further confirm the anti-tumor role of α-MSH-PE38KDEL in B16-F10 cells. Compared with the control group, tumor size and weight were significantly reduced in α-MSH-PE38KDEL group, while this effect was inhibited when MC1R was downregulated or Erk1/2 was inhibited with PD98059 (Figure 7A). In addition, both MC1R downregulation and PD98059 administration abolished α-MSH-PE38KDEL roles in promoting p-Erk1/2 and p-MITF (S73) expression and inhibiting MITF and TYR expression (Figure 7B). Furthermore, Bcl-2 expression was increased, while Bax and cleaved caspase3/9 expressions were decreased following sh-MC1R or PD98059 treatment as compared with the α-MSH-PE38KDEL group (Figure 7C). These results indicated that α-MSH-PE38KDEL treatment significantly inhibited the in vivo tumor formation ability of B16-F10 cells via modulating the MC1R/Erk1/2/MITF/TYR signaling.
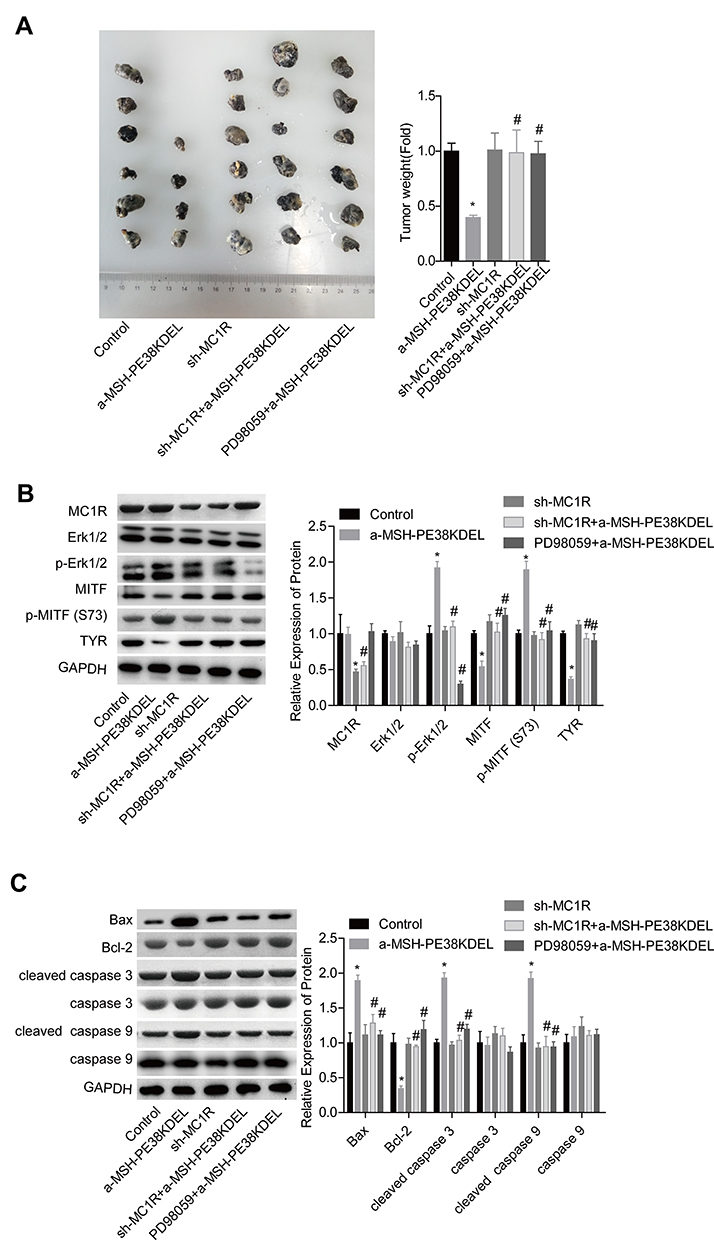 |
Figure 7 α-MSH-PE38KDEL inhibited in vivo tumor formation via modulating Erk1/2/MITF/TYR signaling in a MC1R-dependent manner. (A) In vivo tumor formation assay was used to detect the tumorigenesis of B16-F10 cells in different groups. (B) The levels of MC1R, p-Erk1/2, Erk1/2, MITF, p-MITF (S73) and TYR in tumor tissues were measured by Western blotting assay. (C) Western blotting analysis of the expression of Bax, Bcl-2, caspase 3/9 and cleaved caspase3/9 in tumor tissues. (*P<0.05, vs. control group; #P<0.05, vs. α-MSH-PE38KDEL group). |
Discussion
Although advanced progress has been achieved in therapies, including surgery, radiotherapy with scleral surface applicator, adjuvant therapy and systemic therapy, the incidence rate of melanoma is growing, with an annual growth rate of 3−5%.23 In China, it was estimated that the total number of new cases of melanoma was 6505 in 2011, with an incidence of 0.48/100,000.23 Recently, immunotoxins, as an anti-tumor drug, have drawn people’s attention. Hui et al15 reported that the immunotoxin α-MSH-PE38KDEL showed high cytotoxicity in MSH receptor-positive melanoma cells. Herein, we explored the mechanism underlying α-MSH-PE38KDEL-mediated cytotoxicity. The results demonstrated that α-MSH-PE38KDEL induced MC1R-positive cell apoptosis via modulating the Erk1/2/MITF/TYR signaling.
Toxin α-MSH-PE38KDEL is a fusion protein that was constructed by assembling α-MSH gene to PE38KDEL through the flexible Linker SGGGGS.15 α-MSH belongs to the melanocortin family, with an acetyltridecapeptide structure,24 and combines with five G-protein-coupled melanocortin receptors (MC1R, MC2R, MC3R, MC4R and MC5R) to play different roles. In detail, α-MSH binds to MC1R to promote melanogenesis and thermoregulation, as well as the proliferation and differentiation of melanocytes,25,26 while binds to MC4R and MC5R to mediate food intake, energy expenditure, and fatty acid oxidation within skeletal muscle.27,28 MC1R is prevailingly expressed in melanocytes and melanoma cells.29 According to the ENSEMBL database (http://asia.ensembl.org/Homo_sapiens/Search/Results?q=MC1R;site=ensembl;facet_species=Human;page=1;facet_feature_type=Transcript), three protein-coding transcripts of MC1R were found, two of them yield the canonical 317 aa MC1R and one encodes a 382 aa form with an additional 65 aa C-terminal extension. Herein, we detected MC1R expression using the antibody from Thermo Fisher Scientific (cat. no. PA5-75,342). The results demonstrated that MC1R showed a higher expression pattern in A375 and B16-F10 cells as compared with MDA-MB-231 and HEMa with a molecular weight of ~36KDa and showed no dimeric and oligomeric form. This result suggests that MC1R expresses in the above cell lines may be the canonical 317 aa type.
Additionally, Robinson et al30 found that α-MSH could repress the growth and impair the adherence of melanoma B16G4F cells which were functionally null at MC1R following cell transfection with the wide type of MC1R. However, variant type of MC1R (Arg151Cys, Arg160Trp, and Asp294His) transfection abolished the role of α-MSH in cell growth and adherence inhibition.30 Cao et al31 reported that the wild-type (WT) of MC1R, but not red hair color (RHC)-associated MC1R variants, protected PTEN from degradation, and mediated the inactivation of PI3K/AKT signaling which plays an important role in the progression of melanoma.32 These results indicate that α-MSH plays an anti-tumor role in melanoma via binding to the wide type of MC1R. Therefore, we conjectured that MC1R was indispensable in α-MSH-PE38KDEL-mediated cytotoxicity. Consistently, the results showed that α-MSH-PE38KDEL administration led to a significant inhibition in cell viability and induced cell apoptosis in B16-F10 and A375 cells with high expression of MC1R, while showed no obvious influence in the activity and apoptosis of HEMa and MDA-MB-231 cells with lower expression of MC1R. Moreover, knockdown of MC1R abrogated α-MSH-PE38KDEL roles in inhibiting cell viability and promoting cell apoptosis. Interestingly, we found that the modest knockdown of MC1R caused an obvious effect on counteracting the anti-cancer role of α-MSH-PE38KDEL in melanoma, indicating that α-MSH-PE38KDEL may be effective in a heterozygotic background.
In general, keratinocytes produce α-MSH when exposed to UV radiation, leading to the activation of MC1R and the subsequent increase in the level of cyclic adenosine monophosphate (cAMP) and the transcription of MITF and TYR.33 Noticeably, α-MSH-induced MC1R activation activates the Erk1/2 pathway via activating cAMP in mouse melanocytes, but MC1R-mediated Erk1/2 activation is independent of cAMP activation in human melanocytes.19,20 Additionally, Liu et al34 demonstrated that the activation of Erk1/2 phosphorylated MITF at serine 73 after UV radiation, leading to the proteasome-mediated degradation of MITF in human melanocytes and melanoma cells. Taken together, we conjectured that α-MSH-PE38KDEL might serve as a regulator of the Erk1/2/MITF signaling via binding to MC1R. Consistently, our results showed that α-MSH-PE38KDEL treatment increased the phosphorylated levels of p-Erk1/2 and p-MITF (ser73), and reduced the levels of MITF and TYR in B16-F10 cells and even in A375 cells in which ERKs are already hyperphosphorylated as they harbor a V600E mutation in the BRAF protein kinase gene. Accumulated evidence shows that MITF plays a key role in the occurrence and development of melanoma via different pathways, such as stimulating angiogenesis via oxygen-induced factor 1,35 promoting cell growth via increasing the transcription activity of BCL-2 and CDK2,36,37 and improving the oxidative damage by activating the Ape/Ref-1.38 These results further confirmed that MITF repression may be an effective therapeutic method for melanoma.
In addition, we explored whether the Erk1/2/MITF/TYR signaling was involved in α-MSH-PE38KDEL-mediated anti-tumor role. Compared with the α-MSH-PE38KDEL group, inhibition of the Erk1/2 signaling or overexpression of MITF/TYR, abrogated α-MSH-PE38KDEL roles in promoting cell apoptosis and inhibiting cell growth, indicating the anti-oncogenic role of α-MSH-PE38KDEL in melanoma is dependent on the activation of Erk1/2 and inhibition of MITF/TYR.
In conclusion, this study demonstrates that α-MSH-PE38KDEL induces melanoma cell apoptosis via modulating the Erk1/2/MITF/TYR signaling in an MC1R-dependent manner. This study enriches the theoretical foundation of α-MSH-PE38KDEL in the treatment of MC1R-positive melanoma.
Acknowledgment
Co-first authors: Xilin Liu and Hong Li contributed equally to this study.
Funding
This study was supported by Industrial Independent Innovation Capability Project of Jilin Development and Reform Commission (No. 2019C005); Jilin Scientific and Technological Development Program (No. 20190905003SF) and Jilin Provincial Key Laboratory of precision medicine for cutaneous malignant melanoma (No. 20200601010JC).
Disclosure
The authors declare no conflicts of interest.
References
1. Melanoma OB. Editorial introductory research support, Non-U.S. Gov’t. Nature. 2014;515(7527):S109. doi:10.1038/515S109a
2. Ko JS. The immunology of melanoma. Clin Lab Med. 2017;37(3):449–471. doi:10.1016/j.cll.2017.06.001
3. Ribero S, Glass D, Bataille V. Genetic epidemiology of melanoma. Eur J Dermatol. 2016;26(4):335–339. doi:10.1684/ejd.2016.2787
4. Guy GP, Jr, Thomas CC, Thompson T, Watson M, Massetti GM, Richardson LC. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR. Morbidity and mortality weekly report. 2015;64(21):591–596.
5. Miller AJ, Mihm MC
6. Raimondi S, Sera F, Gandini S, et al. MC1R variants, melanoma and red hair color phenotype: a meta-analysis. Meta-analysis. Int J Cancer. 2008;122(12):2753–2760. doi:10.1002/ijc.23396
7. Froidevaux S, Calame-Christe M, Tanner H, Sumanovski L, Eberle AN. A novel DOTA-alpha-melanocyte-stimulating hormone analog for metastatic melanoma diagnosis. Research support, Non-U.S. Gov’t. J Nucl Med. 2002;43(12):1699–1706.
8. Weldon JE, Pastan I. A guide to taming a toxin–recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. Research support, N.I.H., intramural research support, Non-U.S. Gov’t review. FEBS J. 2011;278(23):4683–4700. doi:10.1111/j.1742-4658.2011.08182.x
9. Michalska M, Wolf P. Pseudomonas exotoxin a: optimized by evolution for effective killing. Front Microbiol. 2015;6:963. doi:10.3389/fmicb.2015.00963
10. Jackson ME, Simpson JC, Girod A, Pepperkok R, Roberts LM, Lord JM. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. Research support, Non-U.S. Gov’t. J Cell Sci. 1999;112(Pt 4):467–475.
11. Zdanovsky AG, Chiron M, Pastan I, FitzGerald DJ. Mechanism of action of Pseudomonas exotoxin. Identification of a rate-limiting step. J Biol Chem. 1993;268(29):21791–21799.
12. Wedekind JE, Trame CB, Dorywalska M, et al. Refined crystallographic structure of Pseudomonas aeruginosa exotoxin A and its implications for the molecular mechanism of toxicity. Research support, non-U.S. Gov’t research support, U.S. Gov’t, Non-P.H.S. Research support, U.S. Gov’t, P.H.S. Journal of Molecular Biology. 2001;314(4):823–837. doi:10.1006/jmbi.2001.5195
13. Fleming BD, Ho M. Glypican-3 targeting immunotoxins for the treatment of liver cancer. Review research support, N.I.H., intramural. Toxins. 2016;8(10):274. doi:10.3390/toxins8100274
14. Wolf P, Elsasser-Beile U. Pseudomonas exotoxin A: from virulence factor to anti-cancer agent. Int J Med Microbiol. 2009;299(3):161–176. doi:10.1016/j.ijmm.2008.08.003
15. Hui Q, Ma J, Song J, et al. In vitro and in vivo studies of antitumor effects of the recombinant immunotoxin MSH-PE38KDEL on melanoma. Neoplasma. 2014;61(4):392–400. doi:10.4149/neo_2014_048
16. Shirasugi I, Kamada M, Matsui T, Sakakibara Y, Liu MC, Suiko M. Sulforaphane inhibited melanin synthesis by regulating tyrosinase gene expression in B16 mouse melanoma cells. Research support, Non-U.S. Gov’t. Biosci Biotechnol Biochem. 2010;74(3):579–582. doi:10.1271/bbb.90778
17. Steingrimsson E, Copeland NG, Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Research support, Non-U.S. Gov’t research support, U.S. Gov’t, P.H.S. Annu Rev Genet. 2004;38:365–411. doi:10.1146/annurev.genet.38.072902.092717
18. Lee HJ, Lee WJ, Chang SE, Lee GY. Hesperidin, a popular antioxidant inhibits melanogenesis via Erk1/2 mediated MITF degradation. Int J Mol Sci. 2015;16(8):18384–18395. doi:10.3390/ijms160818384
19. Herraiz C, Journe F, Abdel-Malek Z, Ghanem G, Jimenez-Cervantes C, Garcia-Borron JC. Signaling from the human melanocortin 1 receptor to ERK1 and ERK2 mitogen-activated protein kinases involves transactivation of cKIT. Research support, N.I.H., extramural research support, non-U.S. Gov’t. Mol Endocrinol. 2011;25(1):138–156. doi:10.1210/me.2010-0217
20. Herraiz C, Jimenez-Cervantes C, Zanna P, Garcia-Borron JC. Melanocortin 1 receptor mutations impact differentially on signalling to the cAMP and the ERK mitogen-activated protein kinase pathways. Research support, non-U.S. Gov’t. FEBS Lett. 2009;583(19):3269–3274. doi:10.1016/j.febslet.2009.09.023
21. Mohammad N, Malvi P, Meena AS, et al. Cholesterol depletion by methyl-beta-cyclodextrin augments tamoxifen induced cell death by enhancing its uptake in melanoma. Research support, non-U.S. Gov’t. Mol Cancer. 2014;13:204. doi:10.1186/1476-4598-13-204
22. Nandy A, Dey SK, Das S, Munda RN, Dinda J, Saha KD. Gold (I) N-heterocyclic carbene complex inhibits mouse melanoma growth by p53 upregulation. Research support, non-U.S. Gov’t. Mol Cancer. 2014;13:57. doi:10.1186/1476-4598-13-57
23. Guo J, Qin S, Liang J, et al. Chinese guidelines on the diagnosis and treatment of melanoma (2015 edition). Chin Clin Oncol. 2016;5(4):57. doi:10.21037/cco.2015.12.02
24. Imokawa G, Kobayashi T, Miyagishi M, Higashi K, Yada Y. The role of endothelin-1 in epidermal hyperpigmentation and signaling mechanisms of mitogenesis and melanogenesis. Pigment Cell Res. 1997;10(4):218–228. doi:10.1111/j.1600-0749.1997.tb00488.x
25. D’Agostino G, Diano S. Alpha-melanocyte stimulating hormone: production and degradation. Research support, N.I.H., extramural review. J Mol Med (Berl). 2010;88(12):1195–1201. doi:10.1007/s00109-010-0651-0
26. Swope VB, Medrano EE, Smalara D, Abdel-Malek ZA. Long-term proliferation of human melanocytes is supported by the physiologic mitogens alpha-melanotropin, endothelin-1, and basic fibroblast growth factor. Research support, Non-U.S. Gov’t research support, U.S. Gov’t, P.H.S. Exp Cell Res. 1995;217(2):453–459. doi:10.1006/excr.1995.1109
27. An JJ, Rhee Y, Kim SH, et al. Peripheral effect of alpha-melanocyte-stimulating hormone on fatty acid oxidation in skeletal muscle. Research support, Non-U.S. Gov’t. J Biol Chem. 2007;282(5):2862–2870. doi:10.1074/jbc.M603454200
28. Galusca B, Prevost G, Germain N, et al. Neuropeptide Y and alpha-MSH circadian levels in two populations with low body weight: anorexia nervosa and constitutional thinness. PLoS One. 2015;10(3):e0122040. doi:10.1371/journal.pone.0122040
29. Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Research support, non-U.S. Gov’t. Nature. 2006;444(7119):638–642. doi:10.1038/nature05327
30. Robinson SJ, Healy E. Human melanocortin 1 receptor (MC1R) gene variants alter melanoma cell growth and adhesion to extracellular matrix. Research support, non-U.S. Gov’t. Oncogene. 2002;21(52):8037–8046. doi:10.1038/sj.onc.1205913
31. Cao J, Wan L, Hacker E, et al. MC1R is a potent regulator of PTEN after UV exposure in melanocytes. Research support, N.I.H., extramural research support, non-U.S. Gov’t. Mol Cell. 2013;51(4):409–422. doi:10.1016/j.molcel.2013.08.010
32. Gong C, Xia H. Resveratrol suppresses melanoma growth by promoting autophagy through inhibiting the PI3K/AKT/mTOR signaling pathway. Exp Ther Med. 2020;19(3):1878–1886. doi:10.3892/etm.2019.8359
33. Busca R, Ballotti R. Cyclic AMP a key messenger in the regulation of skin pigmentation. Pigment Cell Res. 2000;13(2):60–69. doi:10.1034/j.1600-0749.2000.130203.x
34. Liu F, Singh A, Yang Z, Garcia A, Kong Y, Meyskens FL
35. Busca R, Berra E, Pouyssegur J, Ballotti R. Hypoxia inducible factor 1a is a new target of microphthalmia-associated transcription factor (MITF) in melanoma cells. Med Sci. 2006;22(1):10–13. doi:10.1051/medsci/200622110
36. Du J, Widlund HR, Horstmann MA, et al. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Research support, non-U.S. gov’t research support, U.S. Gov’t, P.H.S. Cancer Cell. 2004;6(6):565–576. doi:10.1016/j.ccr.2004.10.014
37. McGill GG, Horstmann M, Widlund HR, et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Research support, non-U.S. Gov’t research support, U.S. Gov’t, P.H.S. Cell. 2002;109(6):707–718. doi:10.1016/S0092-8674(02)00762-6
38. Liu F, Fu Y, Meyskens FL
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.