")
Back to Journals » Journal of Pain Research » Volume 10
α-lipoic acid suppresses neuronal excitability and attenuates colonic hypersensitivity to colorectal distention in diabetic rats
Authors Sun Y, Yang PP, Song ZY, Feng Y, Hu DM, Hu J, Xu GY , Zhang HH
Received 17 February 2017
Accepted for publication 16 May 2017
Published 14 July 2017 Volume 2017:10 Pages 1645—1655
DOI https://doi.org/10.2147/JPR.S135017
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Yan Sun,1,* Pan-Pan Yang,1,* Zhen-Yuan Song,2 Yu Feng,1 Duan-Min Hu,1 Ji Hu,1 Guang-Yin Xu,3 Hong-Hong Zhang1,3
1Department of Endocrinology, The Second Affiliated Hospital of Soochow University, Suzhou, People’s Republic of China; 2Department of Endocrinology, The East District of Suzhou Municipal Hospital, Suzhou, People’s Republic of China; 3Jiangsu Key Laboratory of Translational Research and Therapy for Neuro-Psycho-Diseases, Institute of Neuroscience, Soochow University, Suzhou, People’s Republic of China
*These authors contributed equally to this work
Aim: Patients with long-standing diabetes often demonstrate intestinal dysfunction, characterized as constipation or colonic hypersensitivity. Our previous studies have demonstrated the roles of voltage-gated sodium channels NaV1.7 and NaV1.8 in dorsal root ganglion (DRG) in colonic hypersensitivity of rats with diabetes. This study was designed to determine roles of antioxidant α-lipoic acid (ALA) on sodium channel activities and colonic hypersensitivity of rats with diabetes.
Methods: Streptozotocin was used to induce diabetes in adult female rats. Colonic sensitivity was measured by behavioral responses to colorectal distention in rats. The excitability and sodium channel currents of colon projection DRG neurons labeled with DiI were measured by whole-cell patch-clamp recordings. The expressions of NaV1.7 and NaV1.8 of colon DRGs were measured by western blot analysis.
Results: ALA treatment significantly increased distention threshold in responding to colorectal distension in diabetic rats compared with normal saline treatment. ALA treatment also hyperpolarized the resting membrane potentials, depolarized action potential threshold, increased rheobase, and decreased frequency of action potentials evoked by ramp current stimulation. Furthermore, ALA treatment also reduced neuronal sodium current densities of DRG neurons innervating the colon from rats with diabetes. In addition, ALA treatment significantly downregulated NaV1.7 and NaV1.8 expression in colon DRGs from rats with diabetes.
Conclusion: Our results suggest that ALA plays an analgesic role, which was likely mediated by downregulation of NaV1.7 and NaV1.8 expressions and functions, thus providing experimental evidence for using ALA to treat colonic hypersensitivity in patients with diabetic visceral pain.
Keywords: diabetes, colonic hypersensitivity, dorsal root ganglion, voltage-gated sodium channels, α-lipoic acid
Introduction
Gastrointestinal (GI) disorders are common chronic complications in patients with diabetes mellitus (DM). One of the most frequently complained about GI symptom is abdominal pain or discomfort.1,2 It increases the morbidity of DM and worsens the quality of life of diabetic individuals since the current treatment options are very limited. The pathogenesis of GI disorders in diabetes is complex and multifactorial, including autonomic neuropathy, enteric neuronal loss, and oxidative stress.3–6 There is growing evidence indicating that oxidative stress may play an important role in the etiology and pathogenesis of diabetes and its complications such as lens cataracts, nephropathy, and neuropathy.7 Hyperglycemia is one of the most important factors responsible for the development of oxidative stress, which underlies the major complications in patients with type 1 or type 2 DM.8–11 Oxidative stress is characterized by the depletion of antioxidant defense and excessive production of oxygen free radicals, potentially leading to cell damage and destruction, and ultimately contributing to, and even accelerating the development of DM complications.12
Emerging evidence suggests that GI disorders in diabetes may be the consequence of enhanced activities of primary afferent fibers, leading to an increased excitatory tone in spinal cord.13,14 Diabetic colonic hypersensitivity has been reported to be associated with the depressed Kv4.2 expression and function in primary sensory neurons innervating the colon.14 Our previous study has shown that diabetic rats, induced by streptozotocin (STZ), showed a significant colonic hypersensitivity, which is likely mediated by enhanced function and expression of voltage-gated sodium channel (VGSC) NaV1.7 and NaV1.8 subunits in dorsal root ganglion (DRG) neurons.15 These data strongly indicate that ion channels in the primary sensory neurons play a pivotal role in the development of colonic visceral hypersensitivity. Further studies have pointed out that reactive oxygen species (ROS) and reactive nitrogen species may change activities of voltage-gated Na+, Ca2+, and K+ channels in the aging process.16 ROS and reactive nitrogen species also activated and potentiated the TRPV1 and TRPM2 cation channels.17,18 Whether oxidative stress is involved in the modulation of NaV1.7 and NaV1.8 activities under diabetic conditions is not known.
α-lipoic acid (ALA), a powerful antioxidant, has been increasingly used as a dietary supplement recently. Various studies have shown the safety and efficacy of ALA in clinic application.19–22 Its antioxidant properties have made it a popular dietary supplement in several pathological conditions such as diabetic polyneuropathies and inflammation.23 However, the detailed mechanisms underlying the ALA antioxidant effects remain largely unknown. Therefore, we hypothesized that ALA attenuated colonic visceral hypersensitivity through suppression of NaV1.7 and NaV1.8 expressions and functions in the DRG neurons innervating the colon in diabetic rats. To test this hypothesis, we chronically treated diabetic rats with ALA to examine expressions of NaV1.7 and NaV1.8 protein in DRGs. In the present study, we showed that ALA treatment significantly attenuated the colonic visceral hypersensitivity in diabetic rats. ALA treatment also suppressed VGSC expression, reduced channel current density, and reversed neuronal hyperexcitability of DRG neurons innervating the rat colon.
Materials and methods
Generation of STZ-induced diabetes
All animal experiments were approved by the Institutional Animal Care and Use Committee at the Soochow University and were in accordance with the guidelines of the International Association for the Study of Pain. Adult female Sprague–Dawley rats (weighing 160~180 g) were housed four per cage in a temperature-controlled (25°C±1°C) room and subjected to 12 hour-12 hour light-dark cycle. All rats were allowed access to tap water and standard laboratory chow ad libitum. Diabetes was induced by a single intraperitoneal (IP) injection of STZ (65 mg/kg, Sigma Chemicals, St. Louis, MO, USA), which was freshly dissolved in citrate buffer (10 mmol/L, Na citrate, pH 4.3~4.4), as described previously.24,25 The control (CON) rats only received citrate buffer in the same volume. One week later, fasting blood glucose concentration obtained from the tail vein was measured using a glucometer (Johnson & Johnson, New Brunswick, NJ, USA). Only rats with blood glucose concentration higher than 15.0 mmol/L (270 mg/dL) were included in the present experiments and data analyses.
Measurement of colonic sensitivity
All animals were habituated to the test environment for 1 week before measurement. Colonic sensitivity was measured by recording the response to colorectal distention (CRD) in rats, as described in detail previously.26,27 In short, these rats were anesthetized lightly with 1% Brevital (25 mg/kg, IP). A balloon made from a surgical glove finger and attached to tygon tubing was inserted 8 cm into the descending colon and rectum via the anus. Rats were allowed to adapt for 30 minutes. CRD was performed by rapidly inflating the balloon to a constant pressure measured using a sphygmomanometer connected to a pressure transducer. The balloon was inflated to various pressures: 20, 40, 60 and 80 mmHg, for a 20 second stimulation period followed by a 2 minutes rest. Behavioral responses to CRD were measured by visual observation of the abdominal withdrawal reflex by a blinded observer, and distention threshold (DT) was recorded in this study. DT was the minimal distention pressure to evoke abdominal visceromotor response. It was recorded in mmHg by giving a steady increase in distention pressure by sphygmomanometer.28 Each measurement was performed twice. All behavioral studies were performed in a blinded manner.
Rotarod test
Rotarod test was performed as described in detail previously.29 Briefly, rats were first placed on the Rotarod at a given speed (from 5 rpm to 15 rpm) for 1 day or 2 consecutive days for training before the beginning of the experiment. After this training, most rats step voluntarily from the operator’s hand onto the rod. The length of time that each rat is able to stay on the rod at a given rotation speed (15 rpm) was recorded before and after administration of ALA.
Cell retrograde labeling
As described previously,27,30 DRG neurons innervating the colon were labeled with a fluorescent dye DiI (Invitrogen, Carlsbad, CA, USA), which was injected into the colon wall. Rats were anesthetized with chloral hydrate (0.36 g/kg, IP), and their abdomen was opened and the colon was exposed. DiI (20 mg in 0.5 mL methanol) was injected in a 0.1 μL volume at 10 sites on the descending colon. The colon was washed and swabbed with normal saline (NS) before closing of the abdomen. One week later, rats were given STZ or citrate buffer injection IP. Three weeks after STZ injection, rats were treated with ALA or the same volume of NS once a day for consecutive 7 days. Then DRG neurons (T13-L2) innervating the colon were dissected out for patch-clamp recordings.
Acute dissociation of DRG neurons
As described previously,27,30 DRGs (T13-L2) were bilaterally dissected out from anesthetized rats and the connective tissue were removed. DRGs were quickly moved to an ice-cold, oxygenated fresh dissecting solution containing (in mM): 130 NaCl, 5 KCl, 2 KH2PO4, 1.5 CaCl2, 6 MgSO4, 10 glucose, and 10 HEPES and set to pH 7.2 (osmolarity: 305 mOsm). Then the ganglia were incubated in a 5-mL dissecting solution containing collagenase D (2.0–2.2 mg/mL; Roche, Indianapolis, IN, USA) and trypsin (1.5 mg/mL; Sigma, St. Louis, MO, USA) for 1.5 hours at 34.5°C. After the digestion, DRGs were washed. A single cell suspension was transferred onto glass cover slips for 30 minutes before recording.
Whole-cell patch-clamp recordings
The excitability of DRG neurons innervating the colon was measured using whole-cell patch-clamp recording as described previously.31 Small- and medium-sized DRG neurons in red were recorded. The external solution contained (in mM) 130 NaCl, 5 KCl, 2 KH2PO4, 2.5 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose (pH=7.2, adjusted with NaOH, osmolarity: 295–300 mOsm/kg·H2O). The pipette solution contained (in mM) 140 potassium gluconate, 10 NaCl, 10 HEPES, 10 glucose, 5 EGTA, and 1 CaCl2 (pH=7.25, adjusted with KOH; osmolarity: 292 mOsm/kg·H2O). Whole-cell current and voltage were recorded with an HEKA EPC10 patch-clamp amplifier. The data were acquired and stored in a computer and analyzed using Fit Master from HEKA. Cell input resistance (Rin), cell membrane capacitance (Cm), rheobase, resting membrane potential (RP), action potential (AP) threshold, AP overshoot, and duration elicited by current stimulation were characterized. The frequency of APs stimulated by 2 times (2×) and 3 times (3×) rheobase and ramp current stimulation was recorded under current clamp mode.
Isolation of NaV currents
Voltage-gated sodium currents were examined by patch-clamp recordings under voltage-clamped conditions, as described previously.15 The sodium currents were isolated by blocking the K+ and Ca2+ currents with appropriate ion replacements and channel blockers.32–34 The NaV currents were recorded in response to every stimulation potential from –70 to +50 mV in 10 mV increments. The peak sodium current was measured as the peak of the current at every given voltage, and the current densities (pA/pF) was measured by dividing the current amplitude by Cm. The equation for the calculation of membrane conductance (G) is G=I/(Vm–Vrev), where I is the peak sodium current measured at different test potentials, Vm is command voltage, and Vrev is reversal membrane potential, which is calculated to be +78 mV for sodium based on experimental solutions. Normalized average conductances (G/Gmax) were plotted against the given membrane potentials. Data were then fit with the following modified Boltzmann equation: G/Gmax=1/{1+exp [–(V–V1/2)/k], where V is membrane potential, V1/2 is the membrane voltage at which the current was half-maximally activated, and k is the slope factor.15 Inactivation curves for sodium currents were measured using a two-pulse protocol: an 80 ms prepulse between −90 and +30 mV, followed by a 30-ms test pulse to +20 mV. The peak current amplitude was normalized to that recorded at a 90 mV conditioning step (Imax). Data were plotted as a function of conditioning step potentials and fit with the following negative Boltzmann equation: I/Imax=1/{1+exp[–(V1/2–Vm)/k]}, where I is the current, Imax is the maximal current, V1/2 is the membrane potential for half-activation, Vm is the command potential, and k is the slope factor.
Western blotting analysis
Expressions of NaV1.7 and NaV1.8 in colonic DRGs (T13-L2 DRGs) from diabetic and CON rats were determined using western blotting analysis, as previously described in detail.15 The primary antibodies used to probe the target proteins included rabbit anti-NaV1.7 or anti-NaV1.8 (1:200, Alomone Labs, Jerusalem, Israel), rabbit anti-GAPDH (1:1000, Biotechnology Co., CHN), and mouse anti-actin (1:1000; Chemicon, Temecula, CA, USA). The secondary antibodies included anti-rabbit peroxidase-conjugated secondary antibody (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-mouse horseradish peroxidase-conjugated secondary antibody (1:4000; Chemicon).
Statistical analysis
All data are expressed as means ± standard error of mean. Statistical analysis was conducted using software OriginPro 8 (OriginLab, Northampton, MA, USA) and Matlab (Mathworks, Natick, MA, USA). Normality was checked for all data before analysis. Significance was determined using Dunn’s post hoc test following Friedman analysis of variance (ANOVA), two-sample t-test, paired-sample t-test, Mann–Whitney test, one-way repeated-measures ANOVA, or two-way repeated-measures ANOVA followed by Tukey post hoc test, as appropriate. Results were considered statistically significant when a p-value was less than 0.05.
Results
ALA attenuated colonic visceral hypersensitivity in rats with diabetes
After a single IP injection of STZ, blood glucose level and colonic hypersensitivity were measured every 2 weeks until 8 weeks after the injection. As we have showed in the previous study,15 majority of the rats developed hyperglycemia compared with CON rats and also developed colonic hypersensitivity, as measured by DT in response to CRD. Four weeks after STZ treatment, a total of 24 diabetic rats were IP injected with ALA in different doses (30, 60, and 120 mg/kg). The DT to CRD was measured from diabetic rats 30 minutes after ALA treatment. NS had no significant effect on the DT. However, ALA at 30, 60 and 120 mg/kg all resulted in a dramatic increase in the DT to CRD (Figure 1A, n=6 for each group, **p<0.01, compared with Pre, two-way repeated- measures ANOVA followed by Tukey post hoc test). Then, we used ALA at 60 mg/kg to determine the time course of ALA effects. As shown in Figure 1B, the ALA-induced analgesic effect lasted for ~24 hours (Figure 1B, n=6 for each group, *p<0.05, compared with Pre, one-way repeated-measures ANOVA followed by Tukey post hoc test). ALA at 60 mg/kg did not produce significant effect on the DT in age- and sex-matched healthy CON rats (Figure 1C, n=6 for each group, p>0.05, compared with Pre, two-sample t-Test). In addition, ALA at 60 mg/kg did not affect motion on Rotarod in diabetic rats (Figure 1D, n=6, p>0.05, compared with Pre, paired-sample t-test).
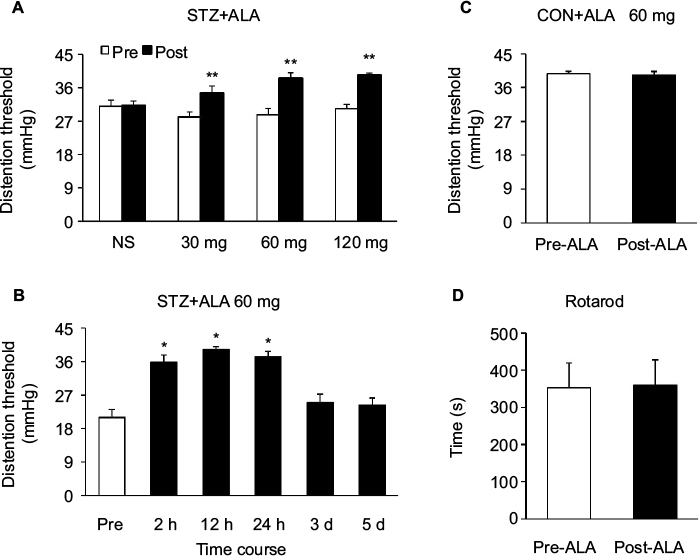 | Figure 1 ALA attenuated colonic hypersensitivity in rats with diabetes. Notes: (A) ALA at 30, 60, and 120 mg/kg resulted in a dramatic increase in the DT to CRD, and NS had no significant effect on the DT (n=6 for each group, **p<0.01, compared with Pre, two-way repeated-measures ANOVA followed by Tukey post hoc test). (B) Time course of ALA-induced analgesic effect. ALA at 60 mg/kg significantly increased the DT at 2 hours, which lasted for ~24 hours (n=6 for each group, *p<0.05, compared with Pre, one-way repeated-measures ANOVA followed by Tukey post hoc test). (C) ALA at 60 mg/kg had no significant effect on the DT in age- and sex-matched healthy CON rats. (n=6 for each group, p>0.05, compared with Pre, two-sample t-test). (D) ALA at 60 mg/kg did not affect motion on Rotarod of diabetic rats. (n=6, p>0.05, compared with Pre, paired-sample t-test). Abbreviations: ALA, α-lipoic acid; ANOVA, analysis of variance; CON, control; CRD, colorectal distension; DT, distention threshold; NS, normal saline. |
ALA reversed hyperexcitability of DRG neurons
Fluorescent dye DiI was injected into the colon wall, which helped trace, in a retrograde manner, DRG neurons innervating the colon (Figure 2A). One week later, rats were given STZ or citrate buffer injection IP. Three weeks after STZ injection, ALA at 60 mg/kg or NS in the same volume was injected (IP) once a day for consecutive 7 days. T13-L2 DRGs were dissociated acutely, and small and medium-sized DRG neurons in red were recorded (Figure 2B, arrow), as we have shown in a previous study.15 ALA treatment significantly reduced excitability of colonic DRG neurons, compared with NS. The resulted showed that ALA treatment reduced RP of colonic DRG neurons from diabetic rats (Figure 3A, **p<0.01, compared with NS, Mann–Whitney test). AP threshold was depolarized in ALA group compared with NS group (Figure 3B, **p<0.01, compared with NS, two-sample t-test). Rheobase was much higher in ALA group than in NS group (Figure 3C, **p<0.01, compared with NS, Mann–Whitney test). ALA at 60 mg/kg and NS did not produce significant effect on the frequency of APs in response to 2 times (2×) rheobase and three times (3×) rheobase current stimulation in DRG neurons from diabetic rats (Figure 3D–G, p>0.05, Mann–Whitney test and two-sample t-test).
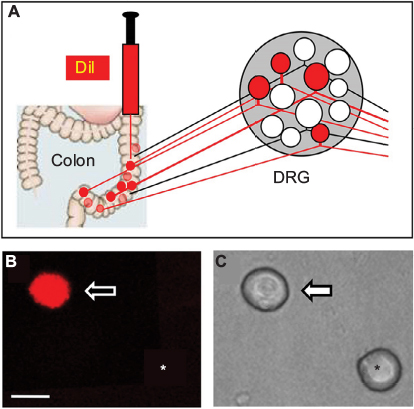 | Figure 2 Retrograde labeling of DRG neurons innervating the colon. Notes: (A) Fluorescent dye DiI was injected into the colon wall, which helped trace, in a retrograde manner, DRG neurons innervating the colon. (B) Example of a Dil-labeled DRG neuron (arrow). Asterisk indicates the place where a neuron is not labeled by DiI. (C) Phase image of the same DRG neuron labeled by DiI is shown on the left (arrow) and the neuron not labeled by DiI is shown on the right (*). Scale bar: 25.0 μm. Abbreviation: DRG, dorsal root ganglion. |
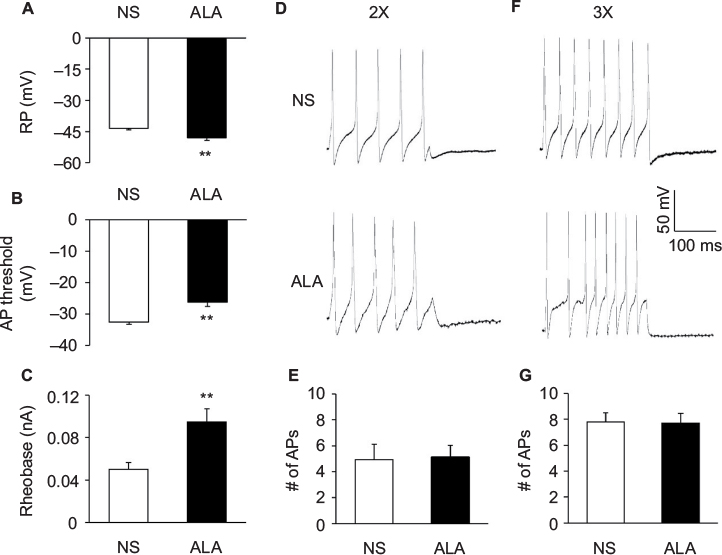 | Figure 3 ALA treatment changed membrane properties of DRG neurons. Notes: (A) ALA treatment hyperpolarized the RP in DRG neurons (n=24 for each group, **p<0.01, compared with NS, Mann–Whitney test). (B) ALA treatment remarkably depolarized AP threshold (n=24 for each group, **p<0.01, compared with NS, two-sample t-test). (C) ALA treatment resulted in a marked increase of rheobase (n=24 for each group, **p<0.01, compared with NS, Mann–Whitney test). (D, F) Examples of AP trances evoked by 2× (left) and 3× (right) rheobase current stimulation of DRG neurons from NS- (top) and ALA-treatment rats (bottom). (E, G) Bar graphs showed that ALA treatment did not change frequencies of APs evoked by 2 times (2×) and 3 times (3×) rheobase current stimulation (n=24 for each group, p>0.05, Mann–Whitney test and two-sample t-test). Abbreviations: ALA, α-lipoic acid; AP, action potential; DRG, dorsal root ganglion; NS, normal saline; RP, resting membrane potential. |
In addition, ALA treatment reduced the frequencies of APs in response to 0.1, 0.3, and 0.5 nA ramp current stimulation in diabetic rats, compared with NS (Figure 4A–C, *p<0.05, **p<0.01, compared with NS, Friedman ANOVA). Furthermore, at the 0.1, 0.3, and 0.5 nA stimulation, the time to first spike (TTFS) was significantly long in ALA group than in NS group (Figure 4E–G, *p<0.05, **p<0.01, Friedman ANOVA). But at the 1.0 nA stimulation, the frequencies of APs and TTFS did not alter after ALA treatment (Figure 4D and H, p>0.05, Friedman ANOVA).
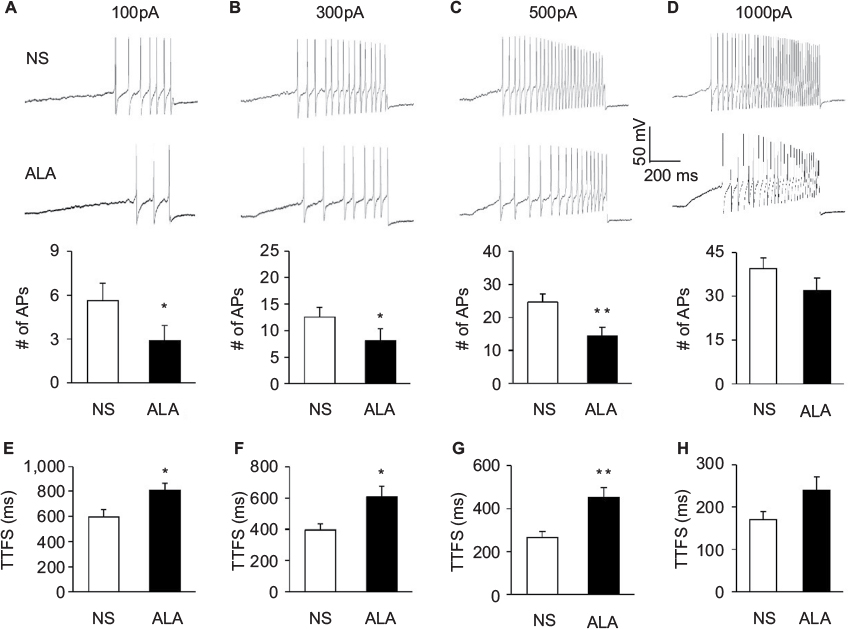 | Figure 4 ALA treatment decreased frequencies of APs evoked by ramp current stimulation. Notes: (A–D) Examples of APs by 0.1, 0.3, 0.5, and 1.0 nA ramp current injection from NS (above) and ALA (below) rats. (A–D) Bar graphs showed a significant decrease in frequencies of APs evoked by 0.1, 0.3, and 0.5 nA ramp current stimulation in NS- and ALA-treated diabetic rats (n=17 for NS group, n=18 for ALA group, *p<0.05, **p<0.01, compared with NS, Friedman ANOVA). (E–H) Bar graphs showed the mean TTFS in response to 0.1, 0.3, 0.5, and 1.0 nA ramp current stimulation from NS- or ALA-treated diabetic rats (n=17 for NS group, n=18 for ALA group, *p<0.05, **p<0.01, Friedman ANOVA). Abbreviations: ALA, α-lipoic acid; ANOVA, analysis of variance; AP, action potential; NS, normal saline; TTFS, time to first spike. |
The other membrane properties are shown in Table 1. There was no significant difference in cell size, Cm, Rin, AP overshoot, and AP duration (at 0 mV) of DRG neurons innervating the colon in ALA group compared with NS group.
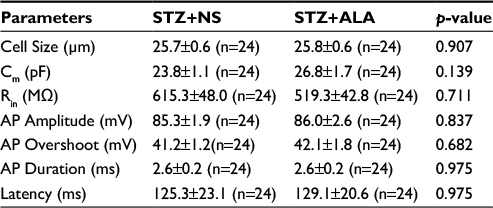 | Table 1 Changes of membrane properties of colonic DRG neurons after ALA treatment Notes: Values are mean ± SEM, with sample size in parenthesis. P-values were determined by two-sample t-test or Mann–Whitney test. Abbreviations: ALA, α-lipoic acid; AP, action potential; Cm, membrane capacitance; DRG, dorsal root ganglion; NS, normal saline; Rin, input resistance; STZ, streptozoin. |
ALA decreased sodium current densities of DRG neurons
Figure 5A shows examples of VGSCs recorded from NS (left) and ALA-treated diabetic rats (right). The membrane potential was held at –60 mV, and depolarization steps from –70 mV to +50 mV in 10 mV increments with duration of 80 ms were used to activate NaV channels. The amplitude of sodium current decreased in ALA-treated diabetic rats. The peak current–voltage (I–V) curves are shown in Figure 5B. ALA injection significantly decreased the average of peak sodium current densities in DiI-labeled neurons compared with the NS group (Figure 5C, NS: –75.82±7.31 pA/pF, n=10; ALA: –47.23±6.39 pA/pF, n=13, **p<0.01, Mann–Whitney test).
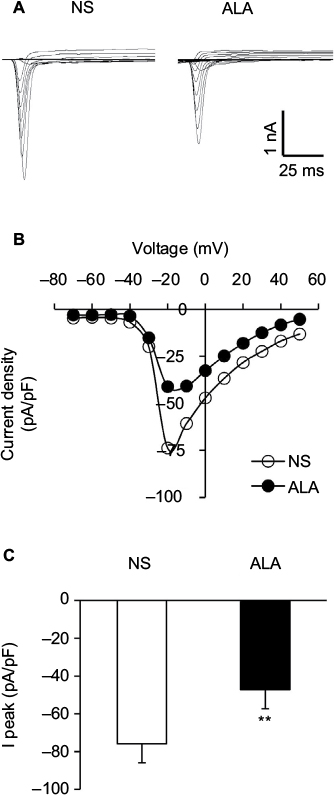 | Figure 5 ALA decreased sodium current densities of DRG neurons. Notes: (A) Examples of voltage-gated sodium currents recorded from NS- (left) and ALA-treated rats (right). Membrane potential was held at –60 mV and voltage steps were from –70 to +50 mV with 10 mV increments and 80 ms duration. (B) I–V curves for sodium currents of colon DRG neurons from NS- (n=10) and ALA-treated rats (n=13). (C) Bar graph showing the mean peak sodium current densities. ALA treatment significantly reduced the peak sodium currents densities (**p<0.01, compared with NS, Mann–Whitney test). Abbreviations: ALA, α-lipoic acid; DRG, dorsal root ganglion; I–V, current–voltage; NS, normal saline. |
We also analyzed the dynamic modification of activation and inactivation responses of sodium currents. Activation of the I–V relationship was constructed from I–V curves of neurons from NS- and ALA-treated diabetic rats (Figure 6A). V1/2 did not change significantly after ALA treatment (Figure 6B, n=10 for each group, p>0.05, compared with NS group, Mann–Whitney test), whereas k changed significantly after ALA treatment (Figure 6C, n=10 for each group, *p<0.05, compared with NS group, Mann–Whitney test). Furthermore, we determined the effect of ALA on the voltage dependence of steady-state inactivation of sodium channels of colon-innervating DRG neurons. The membrane potential was held at –60 mV, and voltage steps were from –90 mV to +30 mV with 10 mV increments and 80 ms duration. For steady-state inactivation curve, the peak current amplitude was normalized to that recorded at a 90 mV conditioning step (I max). ALA treatment did not alter the inactivation curve compared with NS group (Figure 6D). Both V1/2 and k did not alter after ALA treatment (Figure 6E and F, n=9 for each group p>0.05, compared with NS, Mann–Whitney test).
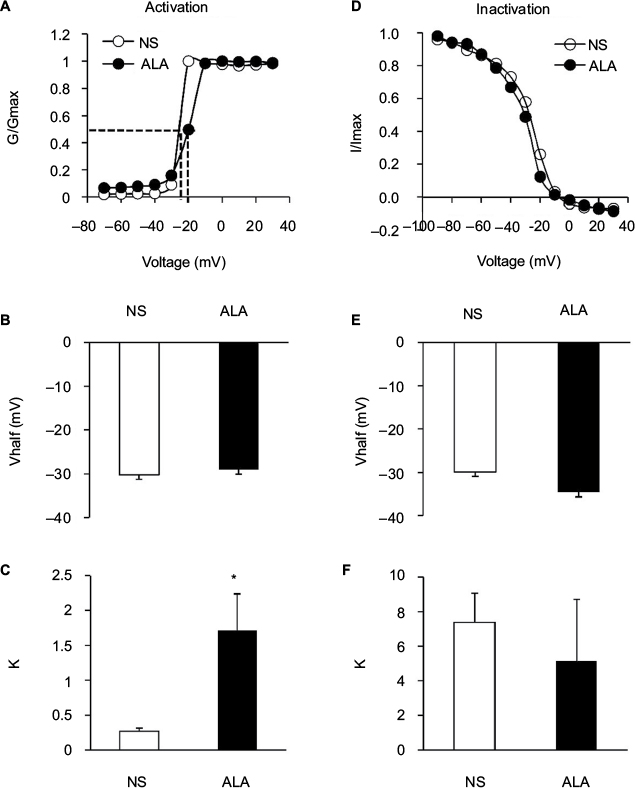 | Figure 6 Changes of activation and inactivation curves of sodium channel currents. Notes: (A) Activation curves of sodium currents were generated by stimulation steps from –70 to +30 mV in 10 mV increment in DRG neuron from NS- and ALA-treated rat. (B) ALA treatment did not change the V1/2 (n=10 for each group, p>0.05, compared with NS- group, Mann–Whitney test). (C) The slope factor (k) in activation of sodium current changed significantly after ALA treatment (n=10 for each group, *p<0.05, compared with NS group, Mann–Whitney test). (D) Steady-state inactivation curves of sodium currents evoked by conditional steps of various voltages from –90 to +30 mV with 10 mV increment in DRG neuron from NS- and ALA-treated rat. (E, F) ALA treatment did not change the V1/2 and k in inactivation of sodium current (n=9 for each group, p>0.05, compared with NS, Mann–Whitney test). Abbreviations: ALA, α-lipoic acid; DRG, dorsal root ganglion; k, slope factor; NS, normal saline; V1/2, half-maximal activation potential. |
ALA downregulated NaV1.7 and NaV1.8 expression
Three weeks after STZ treatment, ALA at 60 mg/kg or NS in the same volume was IP injected once a day for consecutive 7 days in diabetic rats. ALA treatment significantly suppressed NaV1.7 and NaV1.8 expression at protein levels in diabetic rats, compared with NS. Anti-NaV1.7 antibody (Figure 7A) and anti-NaV1.8 antibody (Figure 7B) labeled a 226-kDa and 220-kDa molecular mass protein, respectively. The expressions of NaV1.7 and NaV1.8 were decreased significantly after ALA treatment (Figure 7A and 7B, *p<0.05, **p<0.01, compared with NS, two-sample t-test). The relative density for NaV1.7 was 0.67±0.08 (n=4) and 0.32±0.04 (n=4) in NS and ALA, respectively. The relative density for NaV1.8 was 0.99±0.09 (n=5) and 0.62±0.07 (n=4) in NS and ALA, respectively.
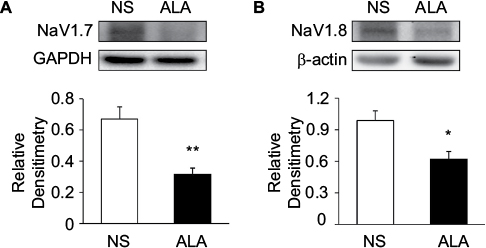 | Figure 7 ALA downregulated NaV1.7 and NaV1.8 expression. Notes: (A) Western blots for NaV1.7 of T13-L2 DRGs from NS- and ALA-treatment rats. Bar graph showed mean density relative to GAPHD for NaV1.7. ALA treatment greatly reduced expressions of NaV1.7 (n=4 for each group, **p<0.01, compared with NS, two-sample t-test). (B) Western blots for NaV1.8 of T13-L2 DRGs from NS- and ALA-treatment rats. Bar graph showed mean density relative to β-actin for NaV1.8. ALA treatment greatly reduced expressions of NaV1.8 (n=5 for NS group, n=4 for ALA group, *p<0.05, compared with NS, two-sample t-test). Abbreviations: ALA, α-lipoic acid; DRG, dorsal root ganglion; NS, normal saline.
|
Discussion
GI dysfunction and complications are very common in DM. More than 75% of diabetic patients show one or more GI symptoms such as early satiety, postprandial fullness, bloating, nausea, vomiting, constipation, diarrhea, abdominal pain, and fecal incontinence.35 It was reported that STZ injection led to gastric and colonic hypersensitivity,25,36 in addition to peripheral sensorimotor neuropathy in rats.24,37 Recently, we have reported that diabetic rats display colonic visceral hypersensitivity in response to CRD.15 In the present study, we provided direct evidence for the first time to demonstrate that systematical application of ALA significantly attenuated the colonic visceral hypersensitivity in rats with diabetes. The current findings are significant because ALA possesses beneficial effects in the treatment of diabetes not only for the nephropathy, retinopathy, and vascular diseases but also for the GI complications. However, the mechanisms underlying the ALA antinociceptive effect in GI tract remain unknown. We have previously demonstrated that DM rats displayed hyperexcitability of DRG neurons innervating the colon.15 Moreover, the sodium current density and the expression of NaV1.7 and NaV1.8 were remarkably enhanced in colon DRGs of rats with diabetes,15 indicating that both NaV1.7 and NaV1.8 play an important role in generation of diabetic colon hypersensitivity. This is consistent with previous reports that both NaV1.7 and NaV1.8 are involved in chronic pain conditions.38,39 Since ALA attenuated the colonic visceral hypersensitivity, it is reasonable to hypothesize that ALA treatment attenuated the colonic visceral hypersensitivity through suppressing NaV1.7 and NaV1.8 expressions and sodium current density in colon DRG neurons of rats with diabetes. Indeed, we showed that ALA treatment reversed neuronal hyperexcitability of colon-specific DRGs in rats with diabetes. This conclusion is mainly based on the observations that ALA hyperpolarized the RPs, depolarized the AP threshold, increased rheobase, and decreased numbers of APs evoked by ramp current stimulation. Although the reason why ALA treatment did reduce the numbers of APs evoked by 2× and 3× rheobase current stimulation, our electrophysiological data support the behavioral findings that ALA dramatically reduced the DT in diabetic rats. Very importantly, we also showed that ALA greatly suppressed the expression of NaV1.7 and NaV1.8 and remarkably decreased sodium current density in colon DRG neurons of rats with diabetes.
How ALA affects NaV1.7 and NaV1.8 expression and function has not been investigated. It is possible that ALA effect is through antioxidant mechanisms since it is recognized as a strong antioxidant. Diabetic complications are closely associated with enhanced oxidative stress.40 Oxidative stress is an increase in concentration of ROS in cells and tissue, which leads to lipid, protein and DNA damage due to peroxidation and ultimately to cell death.41 In addition, ROS affects ion channel expressions and functions directly and indirectly. For example, ROS can be the underlying cause of increased the calcium-activated K+ channel activity in the microvasculature.33 It was also reported that the activity of brain voltage-dependent Ca2+ channels, which are the key factors in the genesis and procession of brain aging and neuronal diseases, is enhanced by oxidative conditions such as toxic fragments or direct oxidation with H2O2.16 In neurodegenerative process, ROS may play a crucial role in modulation of voltage Na+ channels and cause changes in the properties of voltage sensitivity of Na+ channels that may play a relevant role in the regulation of membrane excitability.33 Moreover, ion channel modulation by ROS may occur in several different ways, including posttranslational modification of channel proteins, alterations in the gene expression and activity of channels, and alterations in trafficking or turnover of channel proteins. An example is the activation of the MAPK pathways by ROS or oxygen radical activation of NF-κB.42 Therefore, ROS may impact ion channel function via both direct oxidation and indirect dysregulation of their signaling pathways. ALA increased glutathione level in vivo and in vitro,43 which is an important endogenous antioxidant. They can significantly reduce ROS production. We hypothesized that ALA regulates the expression of NaV1.7 and NaV1.8 through reducing ROS activity.
In addition, ALA has significant anti-inflammatory effects. It was reported that ALA downregulated the expression of redox-sensitive proinflammatory proteins including TNF and inducible NO synthase.22,43 ALA has also been reported to inhibit TNF-α-induced NF-κB activation through a different mechanism from its antioxidant effect.44 On the other hand, inflammation is one of the important mechanisms of diabetic neuropathy and plays an important role in the regulation of ion channel expression and function. Clinic studies have shown that blood plasma TNF-α levels are higher in painful diabetic patients than normal CONs and that the severity of pain is positively correlated with the plasma TNF-α levels.45 The increased TNF-α upregulated the expression of NaV1.7 in DRG neurons.45 It was also reported that peripheral inflammation enhanced NaV1.846 and TRPV1 function47 in DRG neurons. All those changes induced by inflammation contributed to the neuronal hypersensitivity and persistent pain. Therefore, ALA suppressed the expressions of NaV1.7 and NaV1.8, which is likely via inhibition of inflammation. However, these hypotheses need to be confirmed by further studies.
Conclusion
In conclusion, ALA attenuated colonic visceral hypersensitivity in rats with diabetes, which might be mediated by downregulation of NaV1.7 and NaV1.8 expression and function. ALA might be a potential therapeutic strategy for colonic visceral hypersensitivity in patients with diabetes.
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (31400947 to HHZ, 81471041 to JH, 81230024 and 81471137 to GYX) and from the Science and Technology Development Program of Suzhou (SYS201472 to JH). This work was also supported by grants from the Jiangsu Youth Medical Talents Project (QNRC2016874 to HHZ). This work was also supported by grants from the Priority Academic Program Development of Jiangsu Higher Education Institutions of China and from the preponderant clinic group project of the Second Affiliated Hospital of Soochow University (XKQ2015001 and XKQ2015008). The authors also wish to thank Shufen Hu from the Institute of Neuroscience of Soochow University for valuable advice on the sodium current recordings.
Author contributions
YS researched and analyzed data and wrote the manuscript. PPY researched, analyzed data and wrote the manuscript. ZYS researched data. YF researched data. D-MH reviewed and edited the manuscript. JH reviewed and edited the manuscript. G-YX designed the experiments and edited the manuscript. H-HZ performed, designed, and supervised the experiments and edited the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Camilleri M, Malagelada JR. Abnormal intestinal motility in diabetics with the gastroparesis syndrome. Eur J Clin Invest. 1984;14(6):420–427. | ||
Keshavarzian A, Iber FL, Vaeth J. Gastric emptying in patients with insulin- requiring diabetes mellitus. Am J Gastroenterol. 1987;82(1):29–35. | ||
Chaikomin R, Rayner CK, Jones KL, Horowitz M. Upper gastrointestinal function and glycemic control in diabetes mellitus. World J Gastroenterol. 2006;12(35):5611–5621. | ||
Domenech A, Pasquinelli G, De Giorgio R, et al. Morphofunctional changes underlying intestinal dysmotility in diabetic RIP-I/hIFNbeta transgenic mice. Int J Exp Pathol. 2011;92(6):400–412. | ||
Chandrasekharan B, Anitha M, Blatt R, et al. Colonic motor dysfunction in human diabetes is associated with enteric neuronal loss and increased oxidative stress. Neurogastroenterol Motil. 2011;23(2):131–138, e126. | ||
Miller SM, Narasimhan RA, Schmalz PF, et al. Distribution of interstitial cells of Cajal and nitrergic neurons in normal and diabetic human appendix. Neurogastroenterol Motil. 2008;20(4):349–357. | ||
Osawa T, Kato Y. Protective role of antioxidative food factors in oxidative stress caused by hyperglycemia. Ann N Y Acad Sci. 2005;1043:440–451. | ||
Aly HF, Mantawy MM. Comparative effects of zinc, selenium and vitamin E or their combination on carbohydrate metabolizing enzymes and oxidative stress in streptozotocin induced-diabetic rats. Eur Rev Med Pharmacol Sci. 2012;16(1):66–78. | ||
Vicentini J, Valentini J, Grotto D, et al. Association among microalbuminuria and oxidative stress biomarkers in patients with type 2 diabetes. J Investig Med. 2011;59(4):649–654. | ||
Maschirow L, Khalaf K, Al-Aubaidy HA, Jelinek HF. Inflammation, coagulation, endothelial dysfunction and oxidative stress in prediabetes – Biomarkers as a possible tool for early disease detection for rural screening. Clin Biochem. 2015;48(9):581–585. | ||
Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. | ||
Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82(2):291–295. | ||
McHugh JM, McHugh WB. Diabetes and peripheral sensory neurons: what we don’t know and how it can hurt us. AACN Clin Issues. 2004;15(1):136–149. | ||
Grabauskas G, Heldsinger A, Wu X, Xu D, Zhou S, Owyang C. Diabetic visceral hypersensitivity is associated with activation of mitogen-activated kinase in rat dorsal root ganglia. Diabetes. 2011;60(6):1743–1751. | ||
Hu J, Song ZY, Zhang HH, et al. Colonic Hypersensitivity and sensitization of voltage-gated sodium channels in primary sensory neurons in rats with diabetes. J Neurogastroenterol Motility. 2016;22(1):129–140. | ||
Annunziato L, Pannaccione A, Cataldi M, et al. Modulation of ion channels by reactive oxygen and nitrogen species: a pathophysiological role in brain aging? Neurobiol Aging. 2002;23(5):819–834. | ||
Naziroglu M. New molecular mechanisms on the activation of TRPM2 channels by oxidative stress and ADP-ribose. Neurochem Res. 2007;32(11):1990–2001. | ||
Shimizu S, Takahashi N, Mori Y. TRPs as chemosensors (ROS, RNS, RCS, gasotransmitters). Handb Exp Pharmacol. 2014;223:767–794. | ||
Moini H, Packer L, Saris NE. Antioxidant and prooxidant activities of alpha-lipoic acid and dihydrolipoic acid. Toxicol Appl Pharmacol. 2002;182(1):84–90. | ||
Ziegler D, Gries FA. Alpha-lipoic acid in the treatment of diabetic peripheral and cardiac autonomic neuropathy. Diabetes. 1997;46(Suppl 2): S62–S66. | ||
Maczurek A, Hager K, Kenklies M, et al. Lipoic acid as an anti-inflammatory and neuroprotective treatment for Alzheimer’s disease. Adv Drug Deliv Rev. 2008;60(13–14):1463–1470. | ||
Costantino M, Guaraldi C, Costantino D, De Grazia S, Unfer V. Peripheral neuropathy in obstetrics: efficacy and safety of alpha-lipoic acid supplementation. Eur Rev Med Pharmacol Sci. 2014;18(18):2766–2771. | ||
Packer L, Kraemer K, Rimbach G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition. 2001;17(10):888–895. | ||
Xu GY, Li G, Liu N, Huang LY. Mechanisms underlying purinergic P2X3 receptor-mediated mechanical allodynia induced in diabetic rats. Mol Pain. 2011;7:60. | ||
Zhang HH, Hu J, Zhou YL, et al. Promoted interaction of nuclear factor-kappaB with demethylated cystathionine-beta-synthetase gene contributes to gastric hypersensitivity in diabetic rats. J Neurosci. 2013;33(21):9028–9038. | ||
Winston J, Shenoy M, Medley D, Naniwadekar A, Pasricha PJ. The vanilloid receptor initiates and maintains colonic hypersensitivity induced by neonatal colon irritation in rats. Gastroenterology. 2007;132(2):615–627. | ||
Xu GY, Shenoy M, Winston JH, Mittal S, Pasricha PJ. P2X receptor-mediated visceral hyperalgesia in a rat model of chronic visceral hypersensitivity. Gut. 2008;57(9):1230–1237. | ||
Li L, Xie R, Hu S, et al. Upregulation of cystathionine beta-synthetase expression by nuclear factor-kappa B activation contributes to visceral hypersensitivity in adult rats with neonatal maternal deprivation. Mol Pain. 2012;8:89. | ||
Rozas G, Labandeira Garcia JL. Drug-free evaluation of rat models of parkinsonism and nigral grafts using a new automated rotarod test. Brain Res. 1997;749(2):188–199. | ||
Xu GY, Winston JH, Shenoy M, Zhou S, Chen JD, Pasricha PJ. The endogenous hydrogen sulfide producing enzyme cystathionine-beta synthase contributes to visceral hypersensitivity in a rat model of irritable bowel syndrome. Mol Pain. 2009;5:44. | ||
Wang Y, Qu R, Hu S, Xiao Y, Jiang X, Xu GY. Upregulation of cystathionine beta-synthetase expression contributes to visceral hyperalgesia induced by heterotypic intermittent stress in rats. PloS One. 2012;7(12):e53165. | ||
Singh JN, Jain G, Sharma SS. In vitro hyperglycemia enhances sodium currents in dorsal root ganglion neurons: an effect attenuated by carbamazepine. Neurosci. 2013;232:64–73. | ||
Patel R, Sesti F. Oxidation of ion channels in the aging nervous system. Brain Res. 2016;1639:174–185. | ||
Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014;2:702–714. | ||
Brock C, Softeland E, Gunterberg V, et al. Diabetic autonomic neuropathy affects symptom generation and brain-gut axis. Diabetes Care. 2013;36(11):3698–3705. | ||
Cheng KI, Wang HC, Chuang YT, et al. Persistent mechanical allodynia positively correlates with an increase in activated microglia and increased P-p38 mitogen-activated protein kinase activation in streptozotocin-induced diabetic rats. Eur J Pain. 2014;18(2):162–173. | ||
Shi L, Zhang HH, Xiao Y, Hu J, Xu GY. Electroacupuncture suppresses mechanical allodynia and nuclear factor kappa B signaling in streptozotocin-induced diabetic rats. CNS Neurosci Ther. 2013;19(2):83–90. | ||
Pan HL, Liu BL, Lin W, Zhang YQ. Modulation of Nav1.8 by lysophosphatidic acid in the induction of bone cancer pain. Neurosci Bulletin. 2016;32(5):445–454. | ||
Bierhaus A, Fleming T, Stoyanov S, et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nature Med. 2012;18(6):926–933. | ||
Asmat U, Abad K, Ismail K. Diabetes mellitus and oxidative stress-A concise review. Saudi Pharm J. 2016;24(5):547–553. | ||
Lopez-Lopez AL, Jaime HB, Escobar Villanueva Mdel C, Padilla MB, Palacios GV, Aguilar FJ. Chronic unpredictable mild stress generates oxidative stress and systemic inflammation in rats. Physiol Behav. 2016;161:15–23. | ||
Matalon S, Hardiman KM, Jain L, et al. Regulation of ion channel structure and function by reactive oxygen-nitrogen species. Am J Physiol. 2003;285(6):L1184–L1189. | ||
Vasconcelos GS, Ximenes NC, de Sousa CN, et al. Alpha-lipoic acid alone and combined with clozapine reverses schizophrenia-like symptoms induced by ketamine in mice: participation of antioxidant, nitrergic and neurotrophic mechanisms. Schizophr Res. 2015;165(2–3):163–170. | ||
Cho YS, Lee J, Lee TH, et al. Alpha-Lipoic acid inhibits airway inflammation and hyperresponsiveness in a mouse model of asthma. J Allergy Clin Immunol. 2004;114(2):429–435. | ||
Huang Y, Zang Y, Zhou L, Gui W, Liu X, Zhong Y. The role of TNF-alpha/NF-kappa B pathway on the up-regulation of voltage-gated sodium channel Nav1.7 in DRG neurons of rats with diabetic neuropathy. Neurochem Int. 2014;75:112–119. | ||
Belkouch M, Dansereau MA, Tetreault P, et al. Functional up-regulation of Nav1.8 sodium channel in Abeta afferent fibers subjected to chronic peripheral inflammation. J Neuroinflammation. 2014;11:45. | ||
Dong F, Du YR, Xie W, Strong JA, He XJ, Zhang JM. Increased function of the TRPV1 channel in small sensory neurons after local inflammation or in vitro exposure to the pro-inflammatory cytokine GRO/KC. Neurosci Bull. 2012;28(2):155–164. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.