")
Back to Journals » Journal of Multidisciplinary Healthcare » Volume 9
Alpers–Huttenlocher syndrome: the role of a multidisciplinary health care team
Authors Saneto R
Received 18 April 2016
Accepted for publication 10 May 2016
Published 26 July 2016 Volume 2016:9 Pages 323—333
DOI https://doi.org/10.2147/JMDH.S84900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Russell P Saneto1,2
1Department of Neurology, University of Washington, 2Division of Pediatric Neurology, Seattle Children’s Hospital, Seattle, WA, USA
Abstract: Alpers–Huttenlocher syndrome (AHS) is a mitochondrial DNA-depletion syndrome. Age of onset is bimodal: early onset at 2–4 years and later adolescent onset at 17–24 years of age. Early development is usually normal, with epilepsy heralding the disorder in ~50% of patients. The onset of seizures is coupled with progressive cognitive decline. Hepatopathy is variable, and when present is a progressive dysfunction leading to liver failure in many cases. These features of seizures, cognitive degeneration, and hepatopathy represent the “classic triad” of AHS. However, most patients develop other system involvement. Therefore, although AHS is ultimately a lethal disorder, medical care is required for sustained quality of life. Frequently, additional organ systems – gastrointestinal, respiratory, nutritional, and psychiatric – abnormalities appear and need treatment. Rarely, cardiovascular dysfunction and even pregnancy complicate medical treatment. Optimal care requires a team of physicians and caretakers to make sure quality of life is optimized. The care team, together with the family and palliative care specialists, need to be in communication as the disease progresses and medical changes occur. Although the unpredictable losses of function challenge medical care, the team approach can foster the individual quality-of-life care needed for the patient and family.
Keywords: Mitochondrial depletion syndrome, polymerase gamma 1, clinical care, mitochondria
Introduction
Alpers–Huttenlocher syndrome (AHS) is a mitochondrial disease that is part of a group of mitochondrial DNA (mtDNA)-maintenance syndromes. This group of mitochondrial disorders arises from disruption of mtDNA replication, which reduces the amount of mtDNA within the mitochondrion. The depletion of mtDNA ultimately compromises the energy-producing capabilities of the organelle and gives rise to cellular failure. Over the past few decades, the description of neuropathological, clinical presentation, biochemical, and genetic abnormalities has defined AHS as a distinct syndrome among the mtDNA-maintenance syndromes and other mitochondrial diseases.
Early findings defining Alpers–Huttenlocher syndrome
The eponym of Alpers disease was coined by Blackwood et al, based on the review of five new cases and the clinical and neuropathological findings of case reports by Alpers and Morse.1–3 In the Blackwood et al report, two siblings of the five reported cases also had liver cirrhosis with episodes of epilepsia partialis continua (EPC).1,3 In a larger series of neuropathological and clinical findings reported by Harding, the close homogeneity of findings involving the cerebral cortex and patchy thinning with striking predilection for the striate cortex was confirmed to resemble the two siblings in the case series by Blackwood et al.1,4 In this latter series of 32 patients, clinically the early development was normal, as were the two patients reported by Blackwood et al.1 Seizures often heralded the disorder, but in some they occurred with the insidious onset of cognitive and motor delays, difficult weight gain, unexplained bouts of emesis, and pronounced hypotonia. In all, with seizure onset the course of the disease rapidly progressed. Onset of liver involvement varied, with some preceding seizure onset, while in others it was only seen at the end of the clinical course and some had no overt liver findings.
Blackwood et al reported two cases within a single family who had the neuropathological features of AHS and liver involvement of portal proliferation and fibrosis.1 Huttenlocher et al described eight patients with a distinct form of hepatocerebral degeneration, with neuropathological findings demonstrating the degeneration of the cerebral gray matter of AHS.5 The liver findings were described as cirrhosis or subacute hepatitis with superimposed fatty infiltration of hepatocytes.5 Huttenlocher et al also described increased cerebral spinal fluid protein levels and possible autosomal-recessive inheritance. Although patient findings were not first described by Huttenlocher, the series he reported eventually tied his name to the syndrome.
The first suggestion of a possible mitochondrial abnormality was described in 1972. Sandbank and Lerman described large disorganized perinuclear mitochondria within neurons of a 15-month old who died from an episode status epilepticus.6 This young girl was a previously healthy 3 month old who developed a upper respiratory infection and subsequent progressive epilepsy with cognitive involvement.6 It took over 2 decades for the biochemical and genetic underpinnings of AHS to be fully described. In 1996, the mtDNA replicase POLG was cloned.7,8 The initial biochemical evidence that POLG dysfunction leading to mtDNA depletion caused AHS was described in 1999.9 This description was of a young boy who developed ataxia at 19 months of age with a progressive but episodic course of decline that finally ended with liver failure and death. He was found to have both mtDNA depletion less than 30% of control levels in muscle and liver of mtDNA and POLG activity that was not detectable by assay.9 Then, in 2004 Naviaux and Nguyen described pathological mutations in POLG responsible for the phenotype of AHS.10 These latter studies closed the loop of neuropathological, clinical phenotype, biochemical abnormality, and genetic etiology of AHS.
Alpers–Huttenlocher syndrome and the phenotypes of POLG diseases
To date, there have been more than 200 mutations documented in POLG-producing mitochondrial diseases that vary in age of onset, inheritance pattern, and range of organ involvement.11–13 POLG-related disorders are a heterogeneous group of mitochondrial diseases that evolve over time and have overlapping spectra of signs and symptoms. POLG disease can be inherited in an autosomal-recessive or autosomal-dominant manner. The autosomal-dominant disorders always present in the adult years.14 Phenotypically, the dominant diseases always express external ophthalmoplegia, and were the first disorders proven to be due to mutations in POLG.15 Autosomal-recessive disorders present throughout the life span, and compromise the predominant phenotypes of POLG disease (Table 1). The phenotypic expression and timing of the recessive disorders are quite variable at onset and progression of disease unpredictable. However, given a tincture of time for full expression of the phenotype, there are a collection of findings that are fairly consistent and fall into distinct syndromes.12,13,16–26 However, many patients have a range of phenotypic expression that cannot be classified as one of the POLG syndromes (Table 1) or have been diagnosed early in the course of their disease and have been classified with ataxic neuropathy spectrum or mitochondrial-recessive ataxic syndrome.16,27 Some authors have used the term “POLG-related diseases” to describe the range of POLG disorders.28
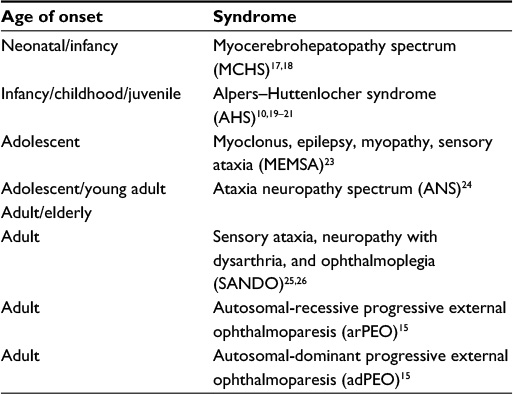 | Table 1 Major POLG clinical syndromes |
Two of the early onset POLG syndromes – myocerebrohepatopathy spectrum (MCHS) disorders and AHS – have overlapping symptoms but distinct differences. MCHS disorders are the rarest of POLG disorders. Onset is early, mostly during the first few months of life with a median of 1 year, and progression to death is rapid.12,16 The primary features are encephalopathy, pronounced myopathy, and liver dysfunction. Liver involvement is often the most devastating in MCHS.17 The pathological liver findings are distinct from that described in AHS.29 Gastrointestinal (GI) dysmotility is often seen earlier than other POLG and mitochondrial diseases. Other features of MCHS are seizures (variably present), myoclonus, neuropathy, renal tubular dysfunction, sensorineural hearing loss, pancreatitis, and respiratory chain defects.12,16,17,30 What distinguishes MCHS from AHS is the earlier onset of symptoms, pathological liver findings, more severe myopathy, and milder seizures and myoclonus.16,17,29 The progression of AHS is more prolonged, with a stepwise progression of disease involvement.9,31
Pathophysiology of Alpers–Huttenlocher syndrome
AHS is an autosomal-recessive disorder induced by mutation in the sole mtDNA polymerase – POLG. To date, homozygous dominantly inherited mutations have not been reported, nor have compound heterozygotes containing one dominant and one recessive inherited mutation been described in POLG disease(s). Since both copies of POLG are expressed in mammalian cells, the latter mutation combinations are likely embryonically lethal with a single wild-type POLG allele sufficient to avoid disease.32 Mutations in AHS can be found as homozygous recessive or as compound heterozygotes in trans. Homozygous recessive mutations most often produce the milder and latter onset phenotype, while patients with compound heterozygote mutations present earlier and have severe and more progressive disease.33,34 However, there are exceptions where identical homozygous mutations can lead to severe disease presenting early in life in one patient and a milder disease in another patient.16,20 Similarly, identical heterozygous mutations can lead to mild disease in one patient and severe disease in another.16,20 The reasons for the difference in presentation onset and severity of disease are unclear.
POLG is responsible for mtDNA replication and repair.35 The protein is part of the mitochondrial trimer replicase composed of the 140 kDa catalytic (α) subunit of the mtDNA POLG and two 55 kDa accessory (β) subunits (POLG2). Both of the proteins are synthesized from nuclear genes, transcribed in the cytoplasm, and transported into the mitochondrion, and associate with other nuclear-encoded proteins along the inner mitochondrial membrane to form the replisome. The POLG protein has three functional domains (Figure 1). The amino terminal contains the 3′–5′ proofreading exonuclease. The middle third region is the linker domain and the binding site for one POLG2 protein. When bound to POLG, the POLG2 protein enhances the DNA-binding affinity of the holoenzyme.36 The carboxyl-terminal end of the protein contains the 5′–3′-polymerase activity and 5′-deoxyribose phosphate lyase activity. The second binding site of the POLG2 protein makes limited contact with POLG at the distal accessory site and enhances the polymerization rate of the holoenzyme.36,37 There are three highly conserved sequence motifs within the exonuclease region (I, II, and III) and three within the polymerase region (A, B, and C), which are essential for full enzyme activity.14,17 There are two conserved sequence motifs within the linker region: one is the POLG2-binding site, and the second site an intrinsic processivity domain. Both motifs are required for full processivity (Figure 1; AID [accessory-interacting determinant] and IP [intrinsic processivity]). Processivity is the average number of nucleotides added by the enzyme per association/disassociation with the template DNA.37 Interestingly, even with the intimate relationship of POLG and POLG2 needed for optimal activity, to date no mutation in POLG2 has been shown to induce AHS. The replisome complex is also made up of a helicase PEO1 (Twinkle) single-stranded DNA-binding protein and an ill-defined number of accessory proteins and transcription factors.14
 | Figure 1 Functional and structural domains of the polymerase-γ protein. Notes: The N-terminal domain (TD) comprises the targeting sequence for the protein to be transported into the mitochondrion. There are two thumb regions (T) that separate the endonuclease and polymerase regions from the linker region. The exonuclease domain has three essential motifs (I, II, III) needed for full activity. The polymerase region contains one thumb region (T) and the palm (two motifs) and finger, which contain regions A, B, and C. These motifs are needed for complete and optimal functioning of the protein. The arrows point to the amino acid that begins the subdomains. Abbreviations: AID, accessory-interacting determinant; IP, intrinsic processivity. |
Although AHS is associated with mutations in POLG, there are no universal mutations that exclusively give rise to disease. Over 60 mutations in POLG are known to cause AHS, both homozygous and compound heterozygous mutations.11 One would assume that compound heterozygous mutations would compromise POLG function more than homozygous mutations, due to synergic depression of function. Indeed, certain mutation combinations induce worse disease with earlier onset of symptoms.38 The most common finding is a patient with compound mutations involving the linker region p.A467T/p.W748S and a mutation in a polymerase region. Mutations found in the latter region are associated with decreased survival and increased incidence of liver failure, compared to homozygous mutations p.A467T and p.W748S.19,39,40 Unexpectedly, some compound mutations are found in the milder juvenile-onset form of AHS, while homozygous mutations in the linker region can be found in the more severe childhood onset of AHS. Therefore, although the combinations of mutations within distinct regions of POLG likely dictate a part of the phenotypic expression, other unknown mechanisms are also involved.
AHS is classified as an mtDNA-depletion syndrome; however, the actual mtDNA copy number in liver and muscle is in the normal range in more than 70% of the patients at the first onset of symptoms. As the disease progresses, mtDNA depletion becomes pronounced, with levels <35% of normal in liver and brain and eventually muscle.41 In contrast, the enzyme activity of POLG is less than 10% of control values in most tissues throughout life.34 This basis for early onset of symptoms in the presence of normal mtDNA levels and the tissue bias in the presence of universally decreased enzyme activity is not known. Early on in the course of the disease, involvement of the respiratory chain is not specific, and even later in the course of the disease the respiratory chain complexes may have normal activity or variable deficiencies, ranging from a single complex to multiple complexes.18,29,34,42 The insensitivity of mtDNA depletion and respiratory chain assays make early diagnoses based on clinical suspicion, constellation of symptoms, and genetic testing.
Clinical features of Alpers–Huttenlocher syndrome
The overall prevalence of AHS is estimated to be approximately 1:100,000 live births. However, due to the varying degrees of phenotypic expression, inconsistent times of onset, unpredictable onset of progression of first to second symptom, and environmental influence of disease onset, an accurate estimate of prevalence cannot be made. Clinically, it is the constellations of findings, not the sequence of signs and symptoms, that dictate the diagnosis of AHS.
The hallmark clinical features of AHS include the triad of seizures, developmental delay, and liver dysfunction, and when combined with two of eleven other clinical or laboratory findings, constitutes the clinical diagnosis (Table 2). The most variable clinical findings are the age of liver involvement and age of death.9,10,20,42 Unfortunately, what is invariant is the fatal outcome. The age of onset is bimodal, with peaks at 2–4 years (range 3 months to 8 years) and 17–24 years (range 10–27 years).4,10,20–22,30,43 Infants, children, and young adults are healthy at birth and mostly develop normally until disease onset.
 | Table 2 Diagnostic criteria for the clinical diagnosis of Alpers–Huttenlocher syndrome |
Environmental factors, such as infections, can influence the presentation of AHS. In one series, 12 of 15 children who had confirmed POLG mutations presented with their first symptoms within 3–10 days of an intercurrent infection.29 The frequency of infections is not greater in patients with AHS, but the sequelae can be, although not always more severe. Viral infections seem to be more exacerbating of disease than bacterial infections. This may be connected to the evolving central role that mitochondria play in innate immunity.44 The infection does not cause AHS, but can uncover or trigger the disease in a child/young adult with POLG mutations who was previously healthy or exacerbate the stepwise progression of this disorder.
The typical age of onset in childhood is 2–4 years of age. Children usually have uneventful pregnancy and delivery, and normal early development until the onset of their illness. A minority of children may have varying degrees of “developmental delay” prior to onset of symptoms, but it is not clear if the delay is part of the POLG defect. Typically, survival ranges from a few months to 10–12 years after onset of symptoms.41 The younger the onset, the shorter the life span, with few children diagnosed at 2–5 years living beyond their teens.
The typical age of juvenile AHS is 17–24 years of age, and as with childhood onset, early development is normal. In the approximately 30 patients described in the literature, none had an onset associated with an illness.19–22,43,45–47 Survival was much longer in this group, with some patients living into the fifth decade.19
Multidisciplinary approaches to care
AHS is a multisystem disease that usually progresses in a stepwise fashion, with indeterminate time between periods of degeneration of organ function. It is not clear why brain, liver, and musculoskeletal systems are preferentially involved, but these are high-energy-demanding systems. However, in the Seattle cohort and literature, many patients have other organ involvement, eg, psychiatric, GI, cardiac, and pregnancy/delivery (Table 3). Due to such a diversity of potential medical problems, having one overseer in the medical care team is essential. Unfortunately, treatment of AHS is limited to symptom management and supportive care. It is essential to emphasize and address quality-of-life issues and the intensity of treatment modalities. As this disease is relentless in progression, the family needs to accept the diagnosis, which varies between families. Once this happens, the family/caregivers need to be involved in the option for supportive care, which can be offered or withheld.
 | Table 3 Clinical features encountered in Alpers–Huttenlocher syndrome |
Childhood onset
Seizures are the initial clinical manifestation in approximately 50% of affected patients. There is no uniform seizure semiology that predominates, but the most common are focal seizures that may or may not be generalized and are parietal-occipital in location on the electroencephalography (EEG).20,47 As the disease progresses, most will develop simple sensory focal status epilepticus, EPC, and generalized motor status epilepticus.20,46,47 EPC is a motor seizure that involves only one part of the body, such as the hand, face, or arm, with a constant and repetitive clonic movement that may continue for hours to days with or without loss of sensorium or level of consciousness. A mistake can be made by the clinician by not considering they may be nonepileptic in origin. Clinical acumen is often needed when symptoms are more subtle. This can be sorted out with video-EEG studies. Most simple sensory status epilepticus manifestations are visual field changes.47 However, other types of sensory abnormalities can occur: in the Seattle cohort, the author had a 3-year-old who had ictal hallucinations of seeing a spider on the walls that lasted for hours.20
As the disease progresses, the findings on EEG change in location and can become more generalized in distribution.20 Early in the course of the disease, seizures may be well controlled with medication management; we found lamotrigine to be efficacious in seizure management, but other sodium-channel blockers may also be good choices.19,20 Overtime, some benefit can be seen using sodium channel-blocking medication with a benzodiazepine.47 However, as the disorder progresses, most patients become intractable to medical management. There is variability, as some patients present with status epilepticus and remain resistant to treatment from the onset. We have found that during episodes of status epilepticus, most patients need to be placed in a medically induced coma and slowly brought out of the coma based on EEG findings. As the disorder progresses and other organs become compromised, decisions about treatment for status epilepticus need to be fully discussed with the family, palliative care team, and providers. Myoclonus and myoclonic seizures become predominant in many patients, as well as “minor motor” seizures. Quality-of-life issues concerning persistent myoclonus treatment may include botulinum toxin (Botox) injections.
It is tempting for the treating neurologist to use valproic acid as the seizure events become resistant to other medications. For unclear reasons, exposure to valproic acid induces liver dysfunction, and in the majority of patients liver failure can occur within 6–16 weeks of starting therapy.10,16,19,20,27,29,46,47 Valproic acid should be avoided in this population, and if AHS is suspected, POLG sequencing should be obtained.20
Juvenile onset
In the 13 defined juvenile AHS patients, seven presented with migraine-like headache with visual changes.20–22,43,48 Not clearly defined as juvenile AHS, another eight of 22 presented with headache.17,19,24 Seizure was the presenting sign in eleven of 35 patients.17,19,24 Similarly to childhood onset, seizures can be explosive in onset, status epilepticus, or more easily controlled seizures with onset. Findings on the EEG are similar to childhood AHS; parieto-occipital lobe predilection during the early periods of the disorder.20,47 Over time, seizures become the most obvious symptom of juvenile AHS, as with childhood AHS. Treatment remains similar between both childhood and juvenile onset.20,47 Congruently with childhood AHS, the onset of seizures may herald a progressive decline, with death occurring within an average of 8 years.47 In another study, Kaplan–Meier survival curves indicated that some patients, even with seizures, can live into the fifth decade, showing the variability of this disorder.19
Common childhood and juvenile Alpers–Huttenlocher syndrome symptoms
Central and peripheral nervous systems
Most patients are initially healthy without symptoms before disease onset, but some can demonstrate nonspecific neurologic features of clumsiness, headache, progressive ataxia, hypoglycemia episodes, or mild medically controlled seizures before explosive onset.19–21,24 Headaches are common. Most headaches would be considered migraines with visual alterations: visual field changes, flashing light or flickering, and actual visual hallucinations. The visual changes reflect the early visual cortex involvement.20,47 Treatment should not include long-term use of acetaminophen, as this medication is a strong oxidant and removes reduced glutathione from cells, such as in the liver. Whether this could actually play a part in liver dysfunction has not been proven, but certainly based on mechanism of action could be worrisome.
The gray-matter involvement found in AHS also affects the deep gray structures, leading to abnormal involuntary movements. Myoclonus and choreoathetosis are the most common movements found in AHS. Choreoathetosis is a writhing movement involving small or large joints. Myoclonus are rapid single jerking movements that are not correlated with EEG changes, and can be difficult to distinguish from motor tics or EPC.49 Both of these types of movements are difficult to treat. Myoclonus can be so frequent that it is disabling and alters quality of life.
Severe neuronal cell loss in the calcarine and striate cortices eventually manifests as cortical blindness. Visual loss may be initially transitory, but as the disease progresses, if the patient lives long enough, it becomes complete. Visually evoked potentials are typically abnormal, with normal retinal exams.24,50 Optic atrophy is very common in AHS, but may be difficult to validate due to ongoing dementia.
Ataxia is very common if the disease process is not too rapid or the corticospinal tract involvement masks the evaluation for ataxia.19,20,23 Sensory neuropathy is extremely common, with one study of older patients with POLG disease demonstrating 100% of patients had abnormal nerve conduction.28 In the younger child, clinical neuropathy will present as distal weakness and loss of muscle-stretch reflexes, with evolution to larger proximal muscle and tendon reflexes as the disease progresses. Sensory neuropathy first presents as loss of position sensation, which may appear as limb ataxia. The distinguishing finding between sensory neuropathy-induced ataxia and cerebellar ataxia is position sense, which is very difficult to test in young children. Formal nerve-conduction testing is often not performed due to age. However, given the abnormal nerve conduction findings in the adult population, it is likely that if formally tested, most all children would eventually have similar abnormalities in conduction.
Spastic paraparesis evolves over months to years as the disease progresses, due to destruction of the cerebral cortex. Both physical and occupational therapy can be very helpful in managing spasticity. To help with ambulation, proper wheelchair management is vital for families. The use of antispasticity medication may be helpful, and discussions with the team that manages spasticity would be of great help for quality-of-life issues.
The onset of cognitive impairment is variable, but seems to be parallel to seizures and seizure severity. A rapid loss of cognition often occurs with infection, and clinically can include persistent somnolence and loss of receptive and expressive language and normal emotional responses, as well as increased irritability. The increased use of seizure medications can compound the losses if infection also exacerbates seizures.
Anxiety and depression can occur at any stage of the disorder, and in one study were found in >40%.28 In the Seattle cohort, one juvenile AHS patient required psychiatric care and eventually medication for her anxiety. She also developed what was eventually diagnosed as paroxysmal nonepileptic events during her anxiety attacks, based on video-EEG studies. This points out the need to validate actual seizures, and in some cases video-EEG studies are needed to sort out true seizures. In our patient, she continued to get increased seizure medications when she came to our emergency department, and it was not until we captured her events on video EEG that we realized the events were nonepileptic.
Gastrointestinal system
Liver involvement
Severe liver dysfunction has been a defining feature of AHS.1,4,5,10,20,29 However, the use of valproic acid has influenced the modern-day conception of AHS, as almost all patients exposed to valproic acid exposure develop liver dysfunction, leading to liver failure and ultimate demise of the patient.10,20,29,46,51 Before valproic acid was available, autopsy studies identified the liver changes that accompanied many but not all patients with AHS. Still, hepatotoxicity was defining and present, even without frank liver failure or valproic acid exposure.1,5 Liver pathology remains identical in those patients exposed to and those not exposed to valproic acid.4,29,52,53 Nguyen et al developed criteria for liver involvement that is independent of valproic acid exposure (Table 4).29 In patients who do not have AHS, the pathological features differ from other chemically induced or toxic exposure.52,53 In the author’s personal experience, even patients with MCHS do not meet the Nguyen et al criteria for liver involvement. One patient with MCHS passed away from liver failure at 1 year of age. He had p.A143/p.R227W mutations, and biopsy of the liver showed steatosis and dense eosinophilic cytoplasm, and thus did not meet the Nguyen et al AHS criteria.
 | Table 4 Specific histological features of Alpers–Huttenlocher syndrome |
Hepatopathy is one of the clinical triad in diagnosis of AHS. However, hepatopathy may not be seen early, and varies in onset during the disease process, and if the child/adolescent dies early in the course of the disease, then proper diagnoses may be missed. A common early cause of death is status epilepticus.19,47 Utilization of the Nguyen et al criteria (Tables 2 and 4) or neuropathological investigation at autopsy should be used to confirm the diagnosis when there is no suggestion of liver involvement.
Fasting hypoglycemia can occur early in the course of AHS or at the end stage of AHS. Onset as early as the first year of life has been described with normal liver biopsy, as well as later in early life.20,54 It is not clear why these events resolve over time, but this may reflect secondary impairment of fatty oxidation and inadequate gluconeogenesis induced by liver immaturity.
Coagulopathies can be a problem as liver dysfunction becomes more prominent. Vitamin K-dependent clotting factor II, VII, IX, and X activities become compromised as hepatic biosynthesis declines. Bleeding issues during hospitalization need to be monitored closely. During intensive care visits, hepatic biosynthesis of anticoagulant proteins C and S or anti-thrombin III may produce prothrombosis of central line placement. Hematology consultations can be of value to control these issues.
Gastrointestinal manifestations
GI dysfunction is a common finding in AHS. As the disease progresses, patients develop dysphagia, delayed gastric emptying, and intestinal dysmotility. Due to the need for proper nutrition, as symptoms develop, critical assessments by the nutritionist and gastroenterologist are needed. Dysphagia is very common, and due to poor muscle strength and bulbar reflexes, protection of the airway can be compromised. Swallow studies are needed to assess possible aspiration. Children will often require placement of a gastric tube for nutrition, with initial bolus feedings, and eventually require continuous feeding due to poor gastric and GI motility. The degree of GI involvement can be severe and even mimic the mtDNA depletion of mitochondrial neurogastrointestinal encephalopathy. Both disorders can demonstrate complete loss of the longitudinal muscularis propria external muscle layer.31,55 If the patient lives long enough, total parenteral nutrition maybe required.
Pancreatitis may also occur in AHS. A confounding factor is that valproic acid exposure can also give rise to pancreatitis.56 However, Harding described pancreatitis in patients without exposure to valproic acid, demonstrating AHS-induced organ dysfunction.4 A child with AHS with upper abdominal pain, emesis, fever, and very tender abdomen needs careful investigation for this possibility. The mechanism of AHS-induced pancreatitis remains unknown.
Respiratory support
Both nutritional and respiratory support are the most important issues for the provider as neurological function degenerates. There is a wide range of ethical positions regarding the intensity of respiratory interventions in the US. Airway maintenance ranges from full ventilator support to less invasive partial external treatments, eg, bilevel-positive airway pressure. As the myopathy progresses, the pharynx and chest wall weakens, which alters the ability to clear secretions and properly remove carbon dioxide. Acutely, respiratory infections can increase the workload of the chest-wall muscles, and respiratory function can spiral downward. Short-term ventilator support until the infections resolve may be needed. Less invasive modalities, such as bilevel-positive airway pressure, or respiration vests, such as the biphasic cuirass ventilation system, or other devices can be considered instead of tracheal intubation with mechanical ventilation. The use of such devices allows the respiratory muscles to rest during the infection process. We need to learn more about how conserving mitochondrial overwork can limit further dysfunction and improve quality and quantity of life. As the disease progresses, the loss of central respiratory drive due to cortical injury and progression of myopathy can combine or separately induce obstructive and/or central apnea. Apnea can result in hypoventilation and hypercarbia, with resulting encephalopathy and sleep awakenings that can worsen inattention, increase irritability and aggravate memory loss. Referral to a sleep specialist for polysomnography and options for intervention can be helpful. Ethically, we as providers want what increases quality of life without artificially extending intolerable aspects of dying. As the need for intervention becomes more invasive, an open conversation with palliative care, hospice, physicians, and family needs to occur and likely be repeated as the disease worsens.
Cardiac support
Cardiac myopathy is not well reported to occur in AHS, although in a review article cardiomyopathy was stated as occurring in 10%–20% of AHS patients, but late in the course of the disease.41 Typically, a dilated cardiomyopathy is found.41 However, cardiomyopathy must be rare early in the disease. In three large reviews, cardiomyopathy was found in only two of 97 patients.16,39,49 In the Seattle cohort, the author cared for a young girl with AHS due to compound heterozygote mutations p.W788S and p.R852C. She presented at age 3 years with focal seizures and noncompaction dilated cardiomyopathy. The seizures were initially controlled by seizure medications, but later became intractable to multiple medications. Her dilated cardiomyopathy was progressive, and within several years developed a sinus-node dysfunction with junctional escape rate in the 40 to 45 heart beats per minute range and severe ventricular function with left-bundle branch block. In consultation with our cardiologist, a cardiac resynchronization defibrillator pacemaker was implanted. She responded well to the pacemaker and her quality of life improved, with abilities to play and interact with her friends. Unfortunately, her seizures became more intractable, and during her last episode of status epilepticus, she developed severe sinus and ventricular tachycardia and her pacemaker induced pain with defibrillation. At that point, her parents withdrew care support; this patient was 7 years of age.
The author’s patient represents the variability in the range of onset of clinical findings, as well as the breadth of organ involvement. The combination of status epilepticus and cardiac rhythm abnormalities induced her demise. On autopsy, the liver expressed only microvesicular steatosis. We think that her early death in the course of her disease progression was likely the reason for the lack of full liver involvement. However, she did meet the Nguyen et al criteria for AHS.
Pregnancy management
Pregnancy and delivery have not been described in the literature. In the Seattle cohort, the author had one patient with juvenile AHS who became pregnant unexpectedly at age 19 years. She was referred to a “high-risk” pregnancy clinic, as the stress of pregnancy could alter her disease course as well as create metabolic imbalance for fetal development. Prior to pregnancy, her seizures were controlled on dual therapy of lamotrigine and levetiracetam. Postpregnancy, we followed medication levels every 4 weeks and increased dosing to maintain prepregnancy levels of lamotrigine.57 Postpartum decrease in lamotrigine clearance is known, and we empirically reduced doses on days 3, 7, and 10 with a return to prepregnancy dosing.58 A single possible seizure occurred during the second trimester, but it was uncertain if a dose was missed. We obtained liver-function results (prothrombin time/international normalized ratio, partial thromboplastin time, ammonia) and fasting glucose levels to follow possible liver involvement (which she did not have prior to pregnancy). The high-risk pregnancy service followed her during the pregnancy with repeated ultrasounds, routine clinical visits, and physiological fetal testing. Delivery was at 39 weeks; she received an epidural for anesthesia and delivered without problems. The baby was born with Apgar scores of 8 and 9 at 5 and 10 minutes respectively, and did not have any problems postdelivery. She and the baby left the hospital within 2 days of delivery. Genetic testing was performed on the father during the pregnancy, and he was found not to carry POLG pathological mutations.
Palliative care/hospice care
Involvement of palliative care/hospice care services is essential. Early in the course of the disorder, rehabilitation medicine and GI and respiratory services are important for care and treatment of GI tubes, ventilator support, and therapies. However, a global perspective of care to maximize quality of life should involve palliative care early in the course of the disorder. “Quality of life” varies between ethnic, religious, personal, and community standards, and is thus very subjective. Open discussion concerning the issues and feelings of the family needs to be addressed by the caring “team”, together with the family, palliative/hospice team, and if needed an ethicist.
Surveillance
The varying presentation and range of possible organ involvement demands individualized patient assessment of disease progression. A coordinated team approach to care is needed, and requires a “medical home” involving the primary care physician and mitochondrial neurologist/mitochondrial medicine specialist. Communication among the various providers is essential for proper care of the patient. If possible, other physicians may need to be involved, such as emergency room physicians at local hospitals, hospitalists who work at local and tertiary hospitals, and anesthesiologists for designated procedures. In particular, management of the major disease-triad disorders needs to be monitored closely for ongoing progression of disease. However, monitoring of other system involvement is critical to overall care and to maximize quality of life.
Conclusion
AHS is an autosomal-recessive disorder induced by mutations in the POLG gene. There is no precise phenotype–genotype known to exist. The pathophysiology of the disorder results from the depletion of mtDNA and is influenced by multiple environmental factors, in particular viral infections and medication exposure. Our complete understanding of the expression of the complete triad of intractable seizures, psychomotor regression, and liver dysfunction in a particular patient remains obscure. Other organ involvement is usually found, which can be influenced by age of onset. Like most mitochondrial disease, expression of phenotype is not dependent on the sequence of clinical findings but the constellation of symptoms. What confounds the expression of the phenotype of AHS is the severe progression of seizures, which may be the presenting symptom as status epilepticus or appear more gradually with early medication control, but gradually becomes medically intractable. Multiple episodes of status epilepticus and/or EPC are likely to occur. Many patients do not recover in episodes of status epilepticus, which can truncate the full expression of the disease. The use of the Nguyen et al criteria can fully identify AHS in this latter population of patients.29
The range of systemic involvement necessitates the collaborative effort of multiple specialties. As the disease progresses, the depth of invasive treatment requires proper communication among the medical team, family, and palliative care. The makeup of the medical team depends on the range of systemic involvement. Open dialogue is needed to protect quality of life and family/patient wishes. Unfortunately, AHS is a progressive lethal disorder. Therefore, the balance of family wishes and extent of medical intervention will be a changing target for the medical team.
Acknowledgments
The author wishes to thank the patients and their families who allowed him to be part of the medication care team. The author received funding from the National Institutes of Health (1U54NS078059-01).
Disclosure
The author reports no conflicts of interest in this work.
References
Blackwood W, Buxton PH, Cumings JN, Roertson DJ, Tucker SM. Diffuse cerebral degeneration in infancy (Alpers’ disease). Arch Dis Child. 1963;38:193–204. | ||
Alpers BJ. Diffuse progressive degeneration of the gray matter of the cerebrum. Arch Neurol Psychiatry. 1931;25:469–505. | ||
Morse WI. Hereditary myoclonus epilepsy: two cases with pathological findings. Bull Johns Hopkins Hosp. 1949;84:116–133. | ||
Harding BN. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher syndrome): a personal review. J Child Neurol. 1990;5:273–287. | ||
Huttenlocher PR, Solitare GB, Adams G. Infantile diffuse cerebral degeneration with hepatic cirrhosis. Arch Neurol. 1976;33:186–192. | ||
Sandbank U, Lerman J. Progressive cerebral poliodystrophy – Alpers’ disease: disorganized giant neuronal mitochondria on electron microscopy. J Neurol Neurosurg Psychiatry. 1972;35:749–755. | ||
Ropp PA, Copeland WC. Cloning and characterization of the human mitochondrial DNA polymerase γ. Genomics. 1996;36:449–458. | ||
Lecrenier N, Van Der Bruggen P, Foury F. Mitochondrial DNA polymerases from yeast to man: a new family of polymerases. Gene. 1997;185:147–152. | ||
Naviaux RK, Nyhan WL, Barshop BA, et al. Mitochondrial DNA polymerase γ deficiency and mtDNA depletion in a child with Alpers’ syndrome. Ann Neurol. 1999;45:54–58. | ||
Naviaux RK, Nguyen KV. POLG mutations associated with Alpers syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55:706–712. | ||
National Institute of Environmental Health Sciences. Human DNA Polymerase gamma Mutation Database. Available from: http://tools.niehs.nih.gov/polg. Accessed May 31, 2016. | ||
Saneto RP, Naviaux RK. Polymerase gamma disease through the ages. Dev Disabil Res Rev. 2010;16:163–174. | ||
Cohen BH, Naviaux RK. The clinical diagnosis of POLG disease and other mitochondrial DNA depletion disorders. Methods. 2010;51:364–373. | ||
Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase γ in mitochondrial DNA replication and repair. Chem Rev. 2006;106:383–405. | ||
Van Goethem G, Dermaut B, Löfgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211–212. | ||
Wong LJ, Naviaux RK, Brunetti-Pierri N, et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat. 2008;29:E150–E172. | ||
Ferrari G, Lamanea E, Donati A, et al. Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-γA. Brain. 2005;128:723–731. | ||
de Vries MC, Rodenburg RJ, Morva E, et al. Multiple oxidative phosphorylation deficiencies in severe childhood multi-system disorders due to polymerase gamma (POLG1) mutations. Eur J Pediatr. 2007;166:229–234. | ||
Tzoulis C, Engelsen BA, Telstad W, et al. The spectrum of clinical disease caused by A467T and W748S POLG mutations: a study of 26 cases. Brain. 2006;129:1685–1692. | ||
Saneto RP, Lee IC, Koenig MK, et al. POLG DNA testing as an emerging standard of care before institution valproic acid therapy for pediatric seizure disorders. Seizure. 2010;19:140–146. | ||
Wiltshire E, Davidzon G, DiMauro S, et al. Juvenile Alpers disease. Arch Neurol. 2008;65:121–124. | ||
Uusimaa J, Hinttala R, Rantala H, et al. Homozygous W748S mutation in the POLG1 gene in patients with juvenile-onset Alpers syndrome. Epilepsia. 2008;49:1038–1045. | ||
Van Goetham G, Luoma P, Rantamäki M, et al. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology. 2004;63:1251–1257. | ||
Hakonen AH, Heiskanen S, Juvonen V, et al. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005;77:430–441. | ||
Schulte C, Synofzik M, Gasser T, Schöls L. Ataxia with ophthalmoplegia or sensory neuropathy is frequently caused by POLG mutations. Neurology. 2009;73:898–900. | ||
Van Goethem G, Martin JJ, Dermaut B, et al. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygotes patients with progressive external ophthalmoplegia. Neuromuscul Disord. 2003;13:133–142. | ||
Chinnery PF, Zeviani M. 155th ENMC workshop: polymerase gamma and disorders of mitochondrial DNA synthesis, 21–23 September 2007, Naarden, the Netherlands. Neuromuscul Disord. 2008;18:259–267. | ||
Tchikviladzé M, Gillerson M, Maisonobe T, et al. A diagnostic flow chart for POLG-related diseases based on signs sensitivity and specific. J Neurol Neurosurg Psychiatry. 2015;86:646–654. | ||
Nguyen KV, Sharief FS, Chan SS, Copeland WC, Naviaux RK. Molecular diagnosis of Alpers syndrome. J Hepatol. 2006;45:108–116. | ||
de Vries MC, Rodenburg RJ, Morava E, et al. Multiple oxidative phosphorylation deficiencies in severe childhood multi-system disorders due to polymerase gamma (POLG1) mutations. Eur J Pediatr. 2007;166:229–234. | ||
Saneto RP, Cohen BH, Copeland WC, Naviaux RK. Alpers-Huttenlocher syndrome. Pediatr Neurol. 2013;48:167–178. | ||
Chan SS, Longley M, Copeland WC. The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J Biol Chem. 2005;36:31341–31346. | ||
Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase γ in mitochondrial DNA replication and repair. Chem Rev. 2006;106:383–405. | ||
Nguyen KV, Østergaard E, Ravin SH, et al. POLG mutations in Alpers syndrome. Neurology. 2005;65:1493–1495. | ||
Kaguni LS. DNA polymerase γ, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320. | ||
Lee YS, Johnson KA, Molineux IJ, Yin YW. A single mutation in human mitochondrial DNA polymerase Pol γA affects both polymerization and proofreading activities of only the holoenzyme. J Biol Chem. 2010;285:28105–28116. | ||
Lee YS, Kennedy WD, Yin YW. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell. 2009;139:312–324. | ||
Euro L, Farnum GA, Palin E, Suomalainen A, Kaguni LS. Clustering of Alpers disease mutations and catalytic defects in biochemical variants reveal new features of molecular mechanism of the human mitochondrial replicase, Pol γ. Nucleic Acids Res. 2011;39:9072–9084. | ||
Tang S, Wang J, Lee NC, et al. Mitochondrial DNA polymerase γ mutations: an ever expanding molecular and clinical spectrum. J Med Genet. 2011;48:669–681. | ||
Chan SS, Copeland WC. DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta. 2009;1787:312–319. | ||
Naviaux RK. Childhood Alpers-Huttenlocher syndrome. In: Saneto RP, Parikh S, Cohen BH, editors. Mitochondrial Case Studies: Underlying Mechanisms and Diagnosis. Amsterdam: Elsevier; 2016:135–148. | ||
Davidzon G, Mancuso M, Ferraris S, et al. POLG mutations and Alpers syndrome. Ann Neurol. 2005;57:921–923. | ||
Harding BN. Progressive neurological degeneration of childhood with liver disease (Alpers’ disease) presenting in young adults. J Neurol Neurosurg Psychiatry. 1995;58:320–325. | ||
Naviaux RK. Metabolic features of the cell danger response. Mitochondrion. 2014;16:7–17. | ||
Beatty C, Saneto RP. Juvenile Alpers-Huttenlocher syndrome. In: Saneto RP, Parikh S, Cohen BH, editors. Mitochondrial Case Studies: Underlying Mechanisms and Diagnosis. Amsterdam: Elsevier; 2016:149–158. | ||
Wolf NI, Rahman S, Schmitt B, et al. Status epilepticus in children with Alpers’ disease caused by POLG1 mutations: EEG and MRI features. Epilepsia. 2009;50:1596–1607. | ||
Engelsen BA, Tzoulis C, Karlsen B, et al. POLG1 mutations cause a syndromic epilepsy with occipital lobe predilection. Brain. 2008;131:818–828. | ||
Visser NA, Kraun KP, van den Bergh WM, et al. Juvenile-onset Alpers syndrome: interpreting MRI findings. Neurology. 2010;74:1231–1233. | ||
Horvath R, Hudson G, Ferrari G, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase γ gene. Brain. 2006;129:1674–1684. | ||
Luoma P, Melberg A, Rinne JO, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase γ mutations: clinical and molecular genetic study. Lancet. 2004;364:875–882. | ||
Bicknese AR, May W, Hickey WR, Dodson WE. Early childhood hepatocerebral degeneration misdiagnosed as valproate hepatotoxicity. Ann Neurol. 1992;32:767–775. | ||
Simonati A, Filosto M, Savio C, et al. Features of cell death in brain and liver, the target tissues of progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher disease). Acta Neuropathol. 2003;106:57–65. | ||
Boyd SG, Harden A, Egger J, Pampiglione G. Progressive neuronal degeneration of childhood with liver disease (“Alpers’ disease”): characteristic neurophysiological features. Neuropediatrics. 1986;17:75–80. | ||
Fellman V, Dotarsky H. Mitochondrial hepatopathies in the newborn period. Semin Fetal Neonatal Med. 2011;16:222–228. | ||
Giordano C, Sebastiani M, De Giorgio R, et al. Gastrointestinal dysmotility in mitochondrial neurogastrointestinal encephalomyopathy is caused by mitochondrial DNA depletion. Am J Pathol. 2008;173:1120–1128. | ||
Bai HX, Ma MH, Orabi AI, et al. Novel characterization of drug-associated pancreatitis in children. J Pediatr Gastroenterol Nutr. 2011;53:423–428. | ||
Pennell PB, Newport DJ, Stowe ZN, Helmers SL, Montogomery JQ, Henry TR. The impact of pregnancy and childbirth on the metabolism of lamotrigine. Neurology. 2004;62:292–295. | ||
de Haan GH, Edelbroek P, Segers J, et al. Gestation-induced changes in lamotrigine pharmacokinetics: a monotherapy study. Neurology. 2004;63:571–573. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.