")
Back to Journals » OncoTargets and Therapy » Volume 15
ALK-Positive Anaplastic Large-Cell Lymphoma in a Patient with Chronic Lymphocytic Leukemia: A Case Report and Literature Review
Authors Yu Q, Zhao Z, Wang H , Wang L
Received 12 August 2022
Accepted for publication 6 October 2022
Published 19 October 2022 Volume 2022:15 Pages 1245—1253
DOI https://doi.org/10.2147/OTT.S383779
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Gaetano Romano
Qinchuan Yu,* Zhiqiang Zhao,* He Wang, Lieyang Wang
Department of Hematology, Shanxi Province Cancer Hospital/Shanxi Hospital Affiliated to Cancer Hospital, Chinese Academy of Medical Sciences/Cancer Hospital Affiliated to Shanxi Medical University, Taiyuan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lieyang Wang, Department of Hematology, Shanxi Province Cancer Hospital/Shanxi Hospital Affiliated to Cancer Hospital, Chinese Academy of Medical Sciences/Cancer Hospital Affiliated to Shanxi Medical University, No. 3 Zhigongxin Street, Taiyuan, Shanxi, People’s Republic of China, Tel +86 15235123213, Email [email protected]
Abstract: Chronic lymphocytic leukemia (CLL) can experience histological transformation to a more aggressive lymphoma called “Richter’s transformation”. This transformation usually leads to diffuse large B cell lymphoma, though cases of T cell lymphoma have been identified. Here we report an extremely rare case of ALK (anaplastic lymphoma kinase) positive anaplastic large cell lymphoma. We performed detailed examination for this patient and reviewed related literatures to understand the transformation. Despite our best effort, this patient passed away shortly. Literature review shows no consensus on treatment and poor prognosis of this condition. We intend to report this case and explore novel treatment possibilities for these patients.
Keywords: CLL, ALCL, Richter’s transformation, Ig rearrangement
Introduction
Chronic lymphocytic leukemia (CLL) is a disease derived from mature lymphocytes. It is the most common leukemia in Western countries, but the incidence is lower in ethnic Asian population.1 This disease tends to develop in senior adults; however, increasingly more cases of early-stage CLL have been detected in younger patients. CLL mainly affects bone marrow, peripheral blood, lymph nodes, spleen and liver. The tumor cells are monoclonal mature B cells, with a few antigens expressed differently to normal B cells. For example, CD 20, CD22, and CD 79b can be downregulated while CD5, CD23, CD43 are positive.2 Despite similar clinical manifestation, CLL displays remarkably heterogeneous disease progression. Some patients may experience years of slow disease progression and do not need treatment, while others may have a rapid progressive course that leads to death 1–2 years after diagnosis.3 The underlying reason for this, thanks to the development of karyotype and sequencing techniques, has partially been discovered. CLL-IPI is a scoring system used for predicting risk of CLL patients. It integrates clinical characteristics such as age, staging and β2-MG and gene signature (ie, TP53 and IGHV mutation) to estimate prognosis. TP53-mutated and IGHV-unmutated status imply a dismal outcome, between which, TP53 mutation plays a more important role. Patients harbor TP53 mutation have a significant shorter survival, even with low variant allele frequency (VAF).4
Richter’s transformation refers to histological transformation that occurs in CLL patients, often sometime after diagnosis, to a more progressive malignancy. These transformations usually go to diffuse large B cell lymphoma (DLBCL) or Hodgkin’s lymphoma. In rare cases, T cell lymphoma can also happen.5–12 We present a patient with CLL undergoing Richter’s transformation to ALK+ ALCL (anaplastic large cell lymphoma). Literature review was performed to analyze disease characteristics.
Case
A 59-year-old female patient presented with lymphadenopathy in bilateral inguinal lymph nodes and underwent incisional biopsy in August 2017. The complete blood counts of the patient were WBC (White blood cell) 5.56 × 109/L, lymphocyte 4.60 × 109/L, HGB (hemoglobin) 134 g/L, PLT 278 × 109/L. β2-MG was 3.11 mg/L. The histology revealed a dense infiltrate of monomorphic small lymphoid cells that were immunopositive for CD5, CD23, CD20, and BCL-2 but immunonegative for CD10, CD3, and Cyclin D1, with 15%+ Ki67 (Figure 1). Bone marrow biopsy showed infiltration by monoclonal B lymphocytes, and a diagnosis of small lymphocytic lymphoma (SLL)/chronic lymphocytic leukemia (CLL) was made. FISH (Fluorescence in situ hybridization) for TP53 was 1.8% (negative). A recommendation to watch and wait was made due to no indication of treatment. After 20 months, the patient revisited with low fever and night sweats, and the left axillary lymph nodes increased in size to 4×6 cm. A computed tomography (CT) scan showed multiple enlarged lymph nodes in the bilateral parotid glands, axillary, mediastinum, retroperitoneum and bilateral inguinal area, with the largest one being approximately 7.4×4.5 cm. The complete blood counts at this point were WBC 7.44 × 109/L, lymphocyte 6.21 × 109/L, HGB 120 g/L, PLT 152 × 109/L. β2-MG was 4.77 mg/L. Chromosome analysis (G banding) showed a complex karyotype: 45, XX, −13, del(14)(q 24), add(15)(p 13)[2]/46, XX, t(2; 10)(q 21; q 26), del(14)(q 24)[2]/46, XX[15]. FISH results: ATM (1%) negative, P53 (11%) positive. Next-generation sequencing indicated TP53, LRP1B mutated and IGHV unmutated. TP53 mutation: site1, exon8, c.G818A:p.R273H 48.12%; site2, exon8, c.G856A:p.E286K 16.97%, with an SNP exon4:c.C215G:p.P72R rs1042522 99.57%. LRP1B mutation: exon51:c.A8150G:p.D2717G 53.13%. The patient was determined to be stage I for Rai staging and stage B for Binet staging. CLL-IPI scoring was 9, indicating high risk.
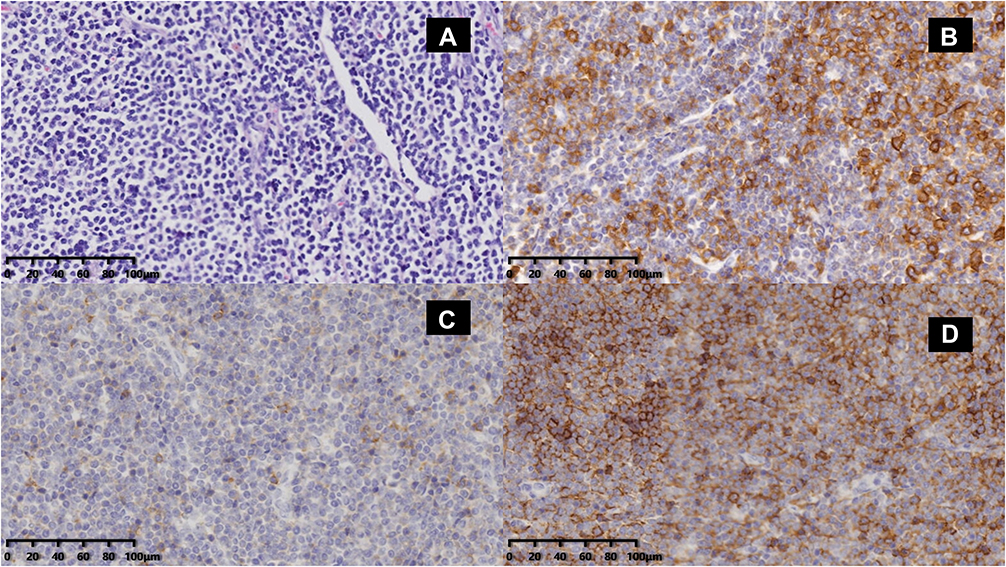 |
Figure 1 Infiltration of monomorphic small lymphoid cells in an inguinal lymph node biopsied at the first onset of disease. (A) H&E staining image (20×) showing that the tumor cells had high nucleus-to-cytoplasm ratios and condensed chromatin. Immunohistochemistry images showing that the abnormal cells were positive for CD20 (B), CD5 (C), and CD23 (D). |
Ibrutinib (420 mg once a day, administered orally) was prescribed. Subsequently, the enlarged lymph nodes gradually subsided, and lymphocyte counts also returned to normal. CT examination in June 2020 showed major regression of lymph nodes, with the largest one being 2.6×1.1cm. Liver or spleen enlargement was not observed. According to the response criteria of iwCLL, the patient had achieved PR (partial remission), and nearly achieved CR (complete remission). However, in September 2020, 1 year after treatment initiation, the patient presented with a new left neck lymphadenopathy, including multiple skin nodules that grew rapidly with redness on the skin surface, indicating a more aggressive disease course. CT scan showed multiple bilateral nodules in the neck, clavicular region, armpit, and groin. Massive abdominal and pelvic lymphadenopathies were observed. Multiple skin lumps were palpable on the bilateral breast and both sides of the trunk. A core needle biopsy was performed on the left neck mass. Histological examination with light microscopy showed the diffuse distribution of abnormal large cells and partial nuclear deviation. Immunohistochemical studies revealed that the tumor cells expressed CD3, CD4, CD5, CD30, ALK, CD8 (individual +), perforin (scattered +), and granzymeB (a few +). However, AE1/AE3, CD20, CD23, CD38, desmin, ERG, CD34, CD19, PAX-5, CD79a, and TIA were not expressed. Ki-67 showed 80% proliferation. These findings were consistent with ALK-positive anaplastic large-cell lymphoma (ALCL; Figure 2). T-cell receptor (TCR) clonality studies were carried out due to the unusual presentation. TCR gene rearrangement analysis showed TCRB Vβ+Jβ 2, Dβ+Jβ 1/2, and all of TCRG V as long as J genes monoclonal rearrangement. Flow cytometric analysis of the bone marrow showed CD 19+, CD 5+ B cells accounted for 64.3% of all nucleated cells. CD 19, CD 5, CD 20 dim, CD 23, CD 200, CXCR 4, and CD 22 were expressed, while CD 10, CD 7, Kappa, Lambda, CD 38, CD 34, CD 33, CD 138, CD 123, CD 15, CD 13, CD 25, CD 11 c, FMC 7, CD 79 b, CD 30, and CD 71 were not expressed, which are abnormal phenotypic mature B lymphocytes. These findings were consistent with CLL. Consequently, the diagnosis of ALK-positive ALCL combined with CLL was made. As the second tumor infiltrated lymph nodes over both sides of diaphragm as well as skin and breast, it was determined to be stage IVB.
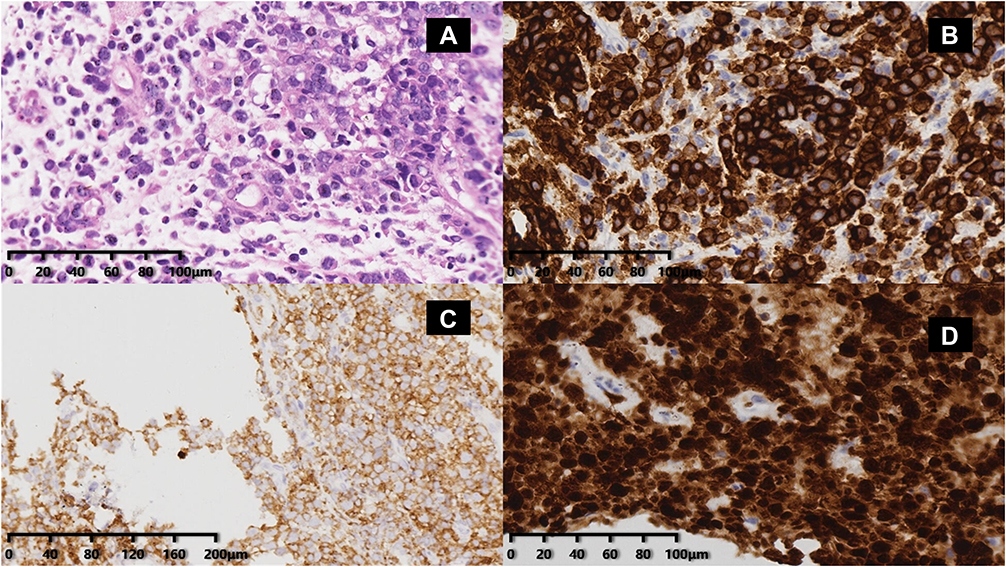 |
Figure 2 Distribution of abnormal large cells with partial nuclear deviation based on the core needle biopsy of the enlarged left neck mass 1 year after first diagnosis. (A) H&E staining image (20×) showing that these cells were pleomorphic with bent nuclei, pale chromatin, and prominent nucleoli. Immunohistochemistry images showing that the abnormal cells were positive for CD30 (B), CD4 (C), and ALK1 (D). |
The patient was prescribed ibrutinib (420 mg per day) together with the CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) regimen. The lymph nodes regressed remarkably after chemotherapy, and the abnormal color of the surface skin faded. The patient was in good spirits and had an appetite. Two weeks after the second chemotherapy cycle, the patient experienced fatigue, anorexia, fever, and cough. She took anti-cold medicine at home and then suddenly convulsed into a coma. She was taken to a local hospital for treatment but died the same night.
Discussion
CLL is a chronic hematopoietic malignancy involving B cells. It is believed that overt CLL is precedented by a stage called monoclonal B lymphocytosis (MBL), in which the count of clonal B cells is fewer than 5 × 109/L. In that stage, patients may experience monoclonal B lymphocytosis, albeit without symptoms. In the case we report, the patient’s monoclonal lymphocyte count did not reach the CLL criteria at first, suggesting that her CLL was at an early stage. However, the malignancy eventually developed to overt CLL 2 years after first presentation.
CLL is indeed a heterogeneous disease. Lots of work has been done to identify high-risk patients, and to give them appropriate treatment. Among various identified biomarkers, TP53 mutation is one of the most effective indicators for distinguishing patient risk groups. Patients harboring TP53 mutations, 17p deletion or both have shorter time-to-first treatment (TTFT), progression-free survival (PFS) and overall survival (OS).13 In addition, TP53 mutations are associated with more chances of Richter’s transformation.14 These patients are resistant to traditional chemoimmunotherapy (CIT).15 Besides, CIT may make clonal selection of TP53-mutated subclones. As in the case of the reported patient, TP53 aberration was only detected in later reexamination. Two TP53 mutations, c.G818A and c.G856A, were found in the patient. TP53 mutation c.818G>A leads to a single amino acid change (R273H) in the p53 protein. This mutation was proposed to promote oncogenic gain-of-function activity by destabilizing p53 tetramer formation.16 R273H mutation can also drive AKT signaling and suppress BMF expression, resulting in enhanced cell survivability and anoikis resistance.17 Additionally, R273H mutation can promote colorectal cancer stem cells, which leads to enhanced cancer self-renewal, tumor propagation and chemoresisitance.18 Despite being relatively well-characterized in other cancers, little is known about the effect of this mutation in CLL.19 Much less is known about the TP53 mutation c.G856A that also leads to a single amino acid mutation (E286K) in the p53 protein. This mutation has been reported to decrease transactivation activity in cultured cells,20 and was predicted to abolish p53 protein activity. C.C215G is an SNP of TP53, which leads to single amino acid change of P72R. P72R is a common polymorphism. It does not impair p53 function, but could be associated with cancer risk.21
The presence of these mutations indicates that the patient was in high risk and requires novel treatment to control the disease progression. Ibrutinib is a BTK (Bruton tyrosine kinase) inhibitor, which can irreversibly bind to BTK and inhibit downstream activities, such as BCR (B cell receptor) and NF-κB signaling pathways.22 Ibrutinib offers superior disease control compared to CIT in CLL patients bearing TP53 mutations, with an estimated PFS of more than 30 months.23
IGHV mutation is an essential step during B cell maturation in germinal center (GC). Consequently, IGHV mutations may suggest the origin of B cells. It is believed that IGHV-mutated CLL derives from antigen-experienced B cells, while IGHV unmutated CLL derives from GC-independent or pre-GC B cells.24 In traditional CIT settings, IGHV-unmutated status correlates with worse clinical outcome.3 With novel compounds such as BTK and Bcl-2 inhibitors, however, the adverse effect of unmutated IGHV is eliminated.25 Since the reported patient had TP53-mutated and IGHV-unmutated CLL, ibrutinib treatment was prescribed.
LRP1B is a putative tumor suppressor gene, and its mutation status has been found to be associated with enhanced cell proliferation and colony formation in various cancers.26 In CLL, whole exome analysis showed that LRP1B mutation affected 5% of the patients, but its role is not fully understood.27
CLL can undergo histological transformation during the course of the disease, also known as Richter’s transformation.28 Typically, the transformation leads to highly malignant lymphomas such as diffuse large B-cell lymphoma or Hodgkin’s lymphoma, with an associated incidence rate of 3–10%.2 However, transformation to ALCL is very rare and, to our knowledge, has not been reported in China. To date, only 16 cases of post-CLL transformed ALK-positive or ALK-negative ALCL have been reported worldwide. Here we review the literature on the development of ALCL after CLL (Table 1). Patients with this kind of transformation have a median age of 61 (43–86 years) and a male-to-female ratio of 9:8. The median time from CLL to the development of ALCL was 3.5 years (0–16 years). Despite radiotherapy, chemotherapy, and immunotherapy of CD 30 monoclonal antibody, the median survival time was only 36 months (0–79 months). ALCL can appear in the same place as the CLL or in a different place. It can occur at the same time as CLL8,9 or at some point after CLL.5,7,9–12 Additionally, ALCL can even appear in combination with other types of lymphomas such as Hodgkin’s lymphoma.5 ALCL after CLL commonly involves lymph nodes (13 out of 17 cases). Extra nodal involvement such as bone marrow, testis, and skin may also occur simultaneously or individually. Eight out of the 17 patients were ALK-positive, eight were ALK-negative, and one had unknown ALK status.
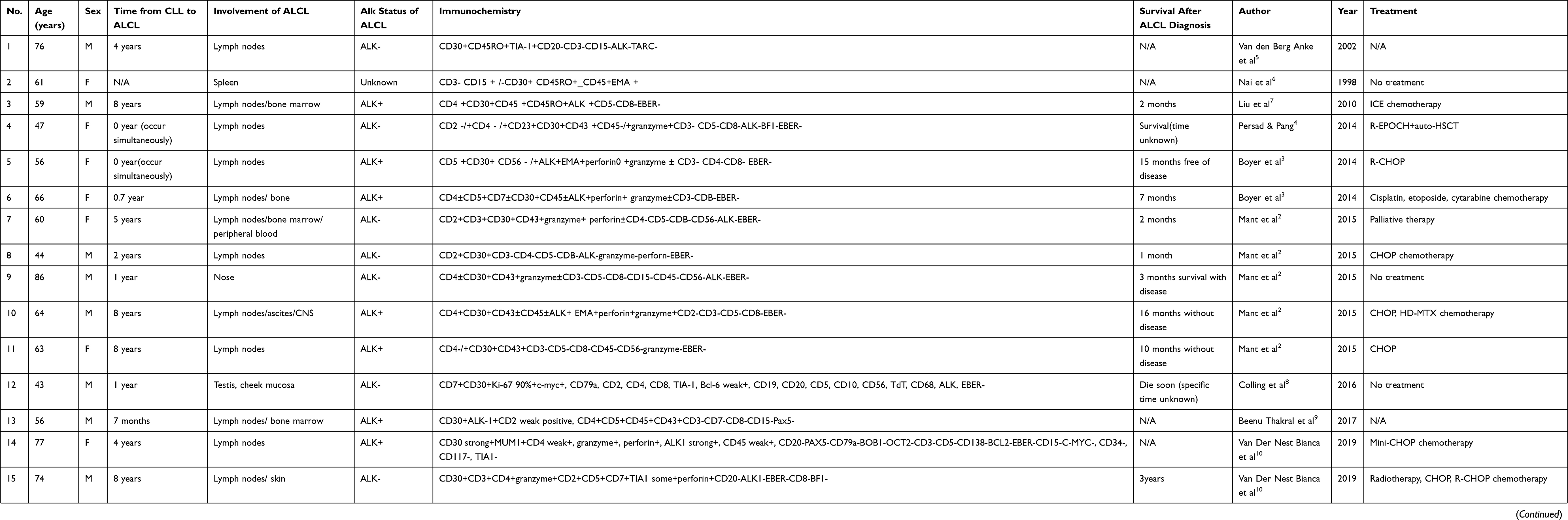 |
Table 1 Summary of Patients Experiencing ALCL in the Presence of CLL |
Whether CLL and ALCL develop independently or share a common cellular origin remains unclear. Clonality studies with Ig rearrangement analysis on CLL and subsequent ALCL specimens indicated that the two tumors may share the same origin, although they displayed completely different morphological and histochemical characteristics.5 Mature B lymphocytes have natural clonal markers. Under antigenic stimulation, each B lymphocyte forms a specific IgH VDJ sequence by binding with numerous V, D, and J fragments. This specific VDJ rearrangement can be inherited in all the descendants of that B cell. Therefore, it is possible to describe intra-tumor heterogeneity and infer clonal evolutionary pathways by comparing the VDJ and IGHV patterns of tumor specimens. A retrospective analysis of specimens from our patient revealed a clonal IGH sequence (TGTGCGAGACATAGGAGGTATTACGATTTTTGGAGTGGTTATTATCTGCCTGGTGCTTTTGATATCTGG) in an inguinal lymph node biopsy specimen taken upon the first diagnosis of CLL. Another clonal IGK sequence was also found: TGTCAGCAGTATGGTAGCTCACCTCCGCTCACTTTC. Unfortunately, due to the insufficient puncture tissue from the left neck mass, this test could not be performed to determine whether the ALCL tissue had the same clonal origin as the CLL tissue. However, the IGH clonal rearrangements in the bone marrow specimens detected at the onset of ALCL were identical to those observed at the time of CLL diagnosis, indicating that the tumor cells in the bone marrow of this patient were the same population as the original CLL cells. Karyotype analysis of bone marrow showed t(2; 10)(q 21; q 26) translocation in our patient. Given that the Alk gene is located on chromosome 2, this translocation may lead to the upregulation of Alk and potentially leads to the occurrence of ALCL.
Treatment plan of second malignancies in patients with CLL should be carefully designed according to the patient’s disease control and organ function. In settings of DLBCL transformation, it is recommended to test clonal relationship of CLL and DLBCL.14 If they are clonally unrelated (~15% cases), treatment as de novo DLBCL is preferred. If they are clonally related (~85% cases), response to treatment is usually poor. However, in the ALCL setting, there is no such consensus due to the rarity of this condition. ALK+ ALCL itself is a T cell lymphoma with favorable prognosis (5-year OS 92.3% in a single-center study),29 but patients’ survival become dismal in the CLL background. This may partly due to compromised immunity, especially undermined T cell function in CLL.30 Shared genetic aberrations with CLL may also alter clinical and biological characterization of ALCL, similar to the clonally related DLBCL.31 Due to unavailability of ibrutinib, patients reviewed did not receive it in CLL treatment. After ALCL transformation, most of them received CHOP regimen chemotherapy, which led to dismal outcomes. Remarkably, one patient received high-dose chemotherapy followed by autologous hematopoietic transplantation (auto-HSCT) and achieved complete remission that lasted at least until the publication of the case.8 However, high-dose chemotherapy plus auto-HSCT brings greater risk of severe infection, especially in older subjects, which limits its application in CLL patients. Brentuximab vedotin plus CHP chemotherapy is currently the front-line therapy of ALK+ ALCL according to NCCN guidelines. One patient received vincristine/gemcitabine and brentuximab vedotin (BV) chemotherapy.5 His skin lesions got better but bone disease progressed. The patient survived for 4 years after the diagnosis of ALCL. This case suggests that the addition of BV may bring some benefits.
Our patient received ibrutinib in combination with CHOP chemotherapy. After one cycle of CHOP, her enlarged lymph nodes regressed significantly. Unfortunately, she died at home 3 weeks after the second round of chemotherapy. Prior to death, the patient showed cold-like symptoms such as coughing and fever according to the relatives. No postmortem was performed, and therefore the cause of death is unknown. Taken the patient’s age and chemotherapy regimen into consideration, the death might be caused by infection after chemotherapy-induced neutropenia. Less intensive regimens, such as dose-reduction CHOP might bring some benefit, but the outcome might still be poor considering the disease courses of other reported cases. Adding BV into treatment regimen shows some promise, but further investigation is needed before any conclusion can be drawn from the single case. In other similar cases, patients typically die within a few months after diagnosis due to ineffective treatment. It is critical to learn from this experience and to develop improved treatment plan for Richter’s transformation.
Ethics Approval and Informed Consent
This study was approved by the ethics committee of Shanxi Tumor Hospital. We certify that the study was performed in accordance with the 1964 Declaration of Helsinki and later amendments. Written informed consent was obtained from the patient prior to the publication of this case report.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Acknowledgment
The authors thank Dr Hui Guo for a critical reading of the manuscript. We thank Dr Liping Su for involving in managing patient and making treatment strategies. We thank Dr Jing Li for carrying out pathological examinations and providing histological images.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors report no conflict of interests in this work.
References
1. Kawamata N, Moreilhon C, Saitoh T, et al. Genetic differences between Asian and Caucasian chronic lymphocytic leukemia. Int J Oncol. 2013;43(2):561–565. doi:10.3892/ijo.2013.1966
2. Salem DA, Stetler-Stevenson M. Clinical flow-cytometric testing in chronic lymphocytic leukemia. In: McCoy JP
3. International CLL-IPI Working Group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet Oncol. 2016;17(6):779–790. doi:10.1016/S1470-2045(16)30029-8
4. Bomben R, Rossi FM, Vit F, et al. TP53 mutations with low variant allele frequency predict short survival in chronic lymphocytic leukemia. Clin Cancer Res. 2021;27(20):5566–5575. doi:10.1158/1078-0432.CCR-21-0701
5. van den Berg A, Maggio E, Rust R, Kooistra K, Diepstra A, Poppema S. Clonal relation in a case of CLL, ALCL, and Hodgkin composite lymphoma. Blood. 2002;100(4):1425–1429. doi:10.1182/blood.V100.4.1425.h81602001425_1425_1429
6. Nai GA, Cabello-Inchausti B, Suster S. Anaplastic large cell lymphoma of the spleen. Pathol Res Pract. 1998;194(7):517–522. doi:10.1016/S0344-0338(98)80122-2
7. Liu T, Mai H, Carlson DL, Hedvat C, Teruya-Feldstein J. ALK-positive anaplastic large cell lymphoma in a patient with chronic lymphocytic leukemia. Int J Surg Pathol. 2010;18(5):424–428. doi:10.1177/1066896908324259
8. Persad P, Pang CS. Composite ALK-negative anaplastic large cell lymphoma and small lymphocytic lymphoma involving the right inguinal lymph node. Pathol Res Pract. 2014;210(2):127–129. doi:10.1016/j.prp.2013.09.006
9. Boyer DF, Lindeman NI, Harris NL, Ferry JA. Peripheral T-cell lymphomas with cytotoxic phenotype in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma. Am J Surg Pathol. 2014;38(2):279–288. doi:10.1097/PAS.0000000000000140
10. Mant S, Taylor G, Dutton D, Butler A, Browett P, Ganly P. Development of T-cell lymphomas with an activated cytotoxic immunophenotype, including anaplastic large cell lymphomas, in patients with chronic lymphocytic leukemia: a series of six cases. Leuk Lymphoma. 2015;56(3):774–778. doi:10.3109/10428194.2014.927460
11. Colling R, Royston D, Soilleux E. Transformation of CLL to ALCL: the role of clonality studies in diagnostic molecular haematopathology. J Hematopathol. 2016;9(3):143–147. doi:10.1007/s12308-016-0280-9
12. Thakral B, Konoplev S. “Soccer ball” cells to “donut” cells: an unusual case of Richter syndrome. Blood. 2017;130(21):2358. doi:10.1182/blood-2017-08-802900
13. Rossi D, Rasi S, Spina V, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood. 2013;121(8):1403–1412. doi:10.1182/blood-2012-09-458265
14. Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131(25):2761–2772. doi:10.1182/blood-2018-01-791376
15. Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. doi:10.1016/j.cell.2013.01.019
16. Annor GK, Elshabassy N, Lundine D, et al. Oligomerization of mutant p53 R273H is not required for gain-of-function chromatin associated activities. Front Cell Dev Biol. 2021;9:772315. doi:10.3389/fcell.2021.772315
17. Tan BS, Tiong KH, Choo HL, et al. Mutant p53-R273H mediates cancer cell survival and anoikis resistance through AKT-dependent suppression of BCL2-modifying factor (BMF). Cell Death Dis. 2015;6(7):e1826–e1826. doi:10.1038/cddis.2015.191
18. Zhao Y, Li Y, Sheng J, et al. P53-R273H mutation enhances colorectal cancer stemness through regulating specific lncRNAs. J Exp Clin Cancer Res. 2019;38(1):379. doi:10.1186/s13046-019-1375-9
19. Ravikrishnan J, Lai TH, Muhowski E, et al. Role of mutant p53 in the progression of chronic lymphocytic leukemia. Blood. 2019;134(Supplement_1):2526. doi:10.1182/blood-2019-128574
20. Slovackova J, Grochova D, Navratilova J, Smarda J, Smardova J. Transactivation by temperature-dependent p53 mutants in yeast and human cells. Cell Cycle. 2010;9(11):2141–2148. doi:10.4161/cc.9.11.11808
21. Doffe F, Carbonnier V, Tissier M, et al. Identification and functional characterization of new missense SNPs in the coding region of the TP53 gene. Cell Death Differ. 2021;28(5):1477–1492. doi:10.1038/s41418-020-00672-0
22. Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14(4):219–232. doi:10.1038/nrc3702
23. Jones J, Mato A, Coutre S, et al. Evaluation of 230 patients with relapsed/refractory deletion 17p chronic lymphocytic leukaemia treated with ibrutinib from 3 clinical trials. Br J Haematol. 2018;182(4):504–512. doi:10.1111/bjh.15421
24. Klein U, Tu Y, Stolovitzky GA, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194(11):1625–1638. doi:10.1084/jem.194.11.1625
25. Rotbain EC, Frederiksen H, Hjalgrim H, et al. IGHV mutational status and outcome for patients with chronic lymphocytic leukemia upon treatment: a Danish nationwide population-based study. Haematologica. 2020;105(6):1621–1629. doi:10.3324/haematol.2019.220194
26. Príncipe C, Dionísio de Sousa IJ, Prazeres H, Soares P, Lima RT. LRP1B: a giant lost in cancer translation. Pharmaceuticals. 2021;14(9):836. doi:10.3390/ph14090836
27. Karim S Development of a targeted next-generation sequencing gene panel to investigate recurrent mutations in chronic lymphocytic leukaemia Thesis (PhD). University of Liverpool; 2016.
28. Wang Y, Tschautscher MA, Rabe KG, et al. Clinical characteristics and outcomes of Richter transformation: experience of 204 patients from a single center. Haematologica. 2020;105(3):765–773. doi:10.3324/haematol.2019.224121
29. Chen H, Tao Y, Zhou Y, et al. The clinical features, treatment, and prognostic factors for peripheral T‐cell lymphomas: a single‐institution analysis of 240 Chinese patients. Asia Pac J Clncl Oncol. 2022:13831. doi:10.1111/ajco.13831
30. Liu Y, Song Y, Yin Q. Effects of ibrutinib on T-cell immunity in patients with chronic lymphocytic leukemia. Front Immunol. 2022;13:962552. doi:10.3389/fimmu.2022.962552
31. Rossi D, Spina V, Deambrogi C, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117(12):3391–3401. doi:10.1182/blood-2010-09-302174
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.