")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Albumin-Fusion Recombinant FIX in the Management of People with Hemophilia B: An Evidence-Based Review
Received 23 April 2022
Accepted for publication 13 September 2022
Published 15 September 2022 Volume 2022:16 Pages 3109—3116
DOI https://doi.org/10.2147/DDDT.S236788
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Samantha Pasca,1,2 Ezio Zanon3
1Biomedical Sciences Department (DSB) - Padua University Hospital, Padua, Italy; 2Medicine Department (DIMED) - Padua University Hospital, Padua, Italy; 3Hemophilia Center, General Medicine - Padua University Hospital, Padua, Italy
Correspondence: Samantha Pasca, Medicine Department (DIMED) – Padua University Hospital, Via Giustiniani, 3, Padua, 35128, Italy, Tel +39-339-6552395, Email [email protected]
Abstract: Albutrepenonacog-alfa (Idelvion®, CSL Behring) is a recombinant fusion protein in which the recombinant FIX (rFIX) links a recombinant human albumin, extending the half-life of rFIX even beyond 100 hours. In 2016, this drug was approved worldwide for the treatment of pediatric and adult persons with hemophilia B (PWH-B). Its efficacy and safety were described in the PROLONG-9FP program and subsequently confirmed in the real-world practice, even if to date there are not many manuscripts that extensively and completely deal with the use of albutrepenonacog-alfa in daily practice, also evaluating its impact on the quality of life of patients treated with this drug; this review therefore aims to analyze all the publications currently available regarding the real-world use of this extended half-life concentrate, also noting which topics need further study and research.
Keywords: hemophilia B, recombinant FIX, albutrepenonacog-alfa, albumin-fusion proteins, extended half-life concentrates
Introduction
Hemophilia B (HB), also known as Christmas disease from the name of the patient in which it was first identified in 1952,1 is an inherited recessive X-linked hemorrhagic disorder, due to clotting factor IX (FIX) deficiency, spread all over the world with an estimated prevalence of about 1 in 40,000 live-born males.2 Being a disorder linked to a mutation of the X chromosome, normally only males are affected, while females are carriers. However, the latter could themselves be affected in the rare case they received mutated alleles from both parents or through the process of lyonization, which also inactivates the other X chromosome.3 The most famous carrier of HB in history is undoubtedly Queen Victoria of England, who through her descendants passed on the mutated gene in many European courts (Russian, German, Spanish); therefore, this bleeding disorder was given the title of “royal disease”.4
Usually, HB are considered clinically less severe than hemophilia A,5 and some studies have in fact reported the low number of orthopedic surgeries in severe adult PWH-B than compared with those with severe hemophilia A,6 with a lower hemorrhagic score7 of severity disease obtained joining different topics such as annualized bleeding rate (ABR), orthopedic status and concentrate factor consumption. These differences could be assigned to a high presence of missense variants8,9 in PWH-B, which guarantee a minimal production of FIX able to mitigate the hemorrhagic phenotype, thus limiting bleeding. The typical hemorrhagic manifestations in persons with hemophilia depend on the FVIII or FIX plasma level. In mild subjects (FVIII/FIX 0.05–0.40 IU/mL), bleeding occurs especially after major surgeries or traumas, while spontaneous hemorrhages are rare, this means that the hemophilia diagnosis can sometimes be delayed. In moderate ones (FVIII/FIX 0.01–0.05 IU/mL), hemorrhagic events occur also in case of minor traumas or surgeries, while the severe patients (FVIII/FIX <0.01 IU/mL) have recurrent joint bleeds and hematomas, also spontaneous.2
Prophylactic treatment started at an early age in severe subjects is the gold standard to prevent bleeding. Today, several FIX concentrates are available to clinicians for the treatment of HB: products of plasma or recombinant origin with standard half-life (SHL) or products of recombinant origin with extended half-life (EHL) to which a molecule capable of increasing its permanence in the circulation were added.10 These new drugs have led to a reduction in the infusion number, a previous unmet need expressed by hemophilic patients,11 with an improvement in their quality of life.
Albutrepenonacog-alfa (Idelvion®, CSL Behring) is a recombinant fusion protein (FP) in which the recombinant FIX (rFIX) links a recombinant human albumin, extending the half-life of rFIX even beyond 100h.12 In 2016, this rFIX-FP was approved worldwide for the treatment of pediatric and adult PWH-B.
This review will analyze all available real-world data relating to the efficacy and safety of rFIX-FP, with a final look at the perceived quality of life of patients treated with this drug and the future role of the concentrate in the face of new ongoing treatments and gene therapy soon available to clinicians and patients.
Methods
This review has been created following the “Preferred reporting items for systematic reviews and meta-analyses” (PRISMA) model.13 Literature search included clinical studies, case reports, reviews, abstracts, and all scientific articles concerning PWH-B treated with rFIX-FP in the real-world and published on PubMed until March 2022. The key terms “haemophilia B”, “hemophilia B” “albutrepenonacog (-alfa)”, “rFIX-FP”, “albumin-fusion rFIX”, “IdelvionTM” were used for the search, linked with the boolean operator AND terms such as “safety, efficacy, bleeding, annualized bleeding rate, inhibitors, alloantibodies, and anaphylactic reactions”. In the query box, these terms were searched only within the titles and/or abstracts of the articles. Reports that met the following inclusion criteria were included in this review: 1) articles/abstracts concerning PWH-B of any age; 2) articles/abstracts that described the use of rFIX-FP in the PWH-B; 3) articles/abstracts written in English.
Pivotal studies were excluded from the review and could only be cited as a term of comparison with the real-world available data.
A total of fourteen articles and abstracts dealing with efficacy, safety, and quality of life related to real-world use of rFIX-FP were finally considered for this review. The whole list of analyzed reports is described in Table 1.
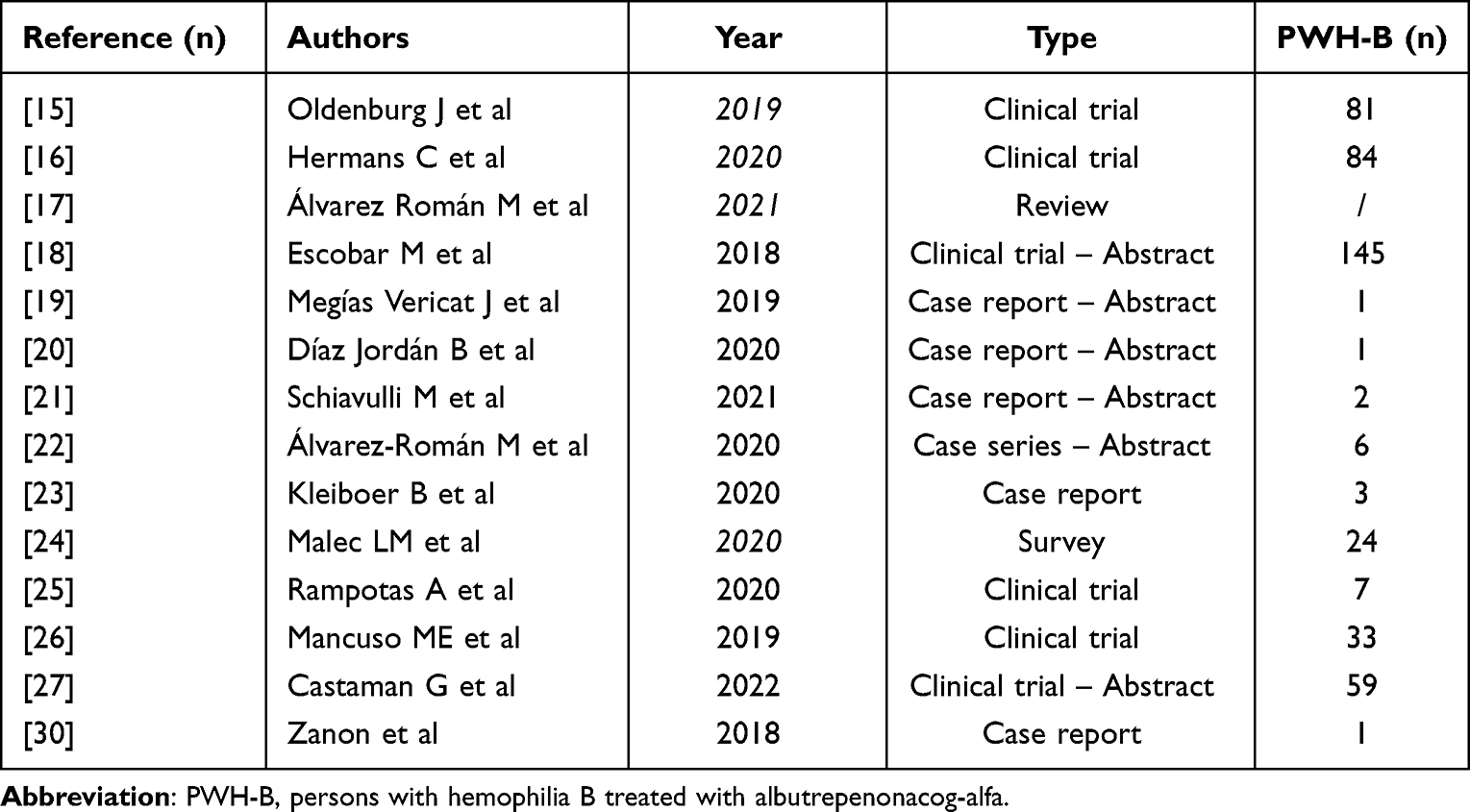 |
Table 1 All Real-World rFIX-FP Reports Included in This Review |
Efficacy
The efficacy of albutrepenonacog-alfa was described in the PROLONG-9FP program and subsequently confirmed in the real-world practice. During the Phase III studies, the median spontaneous ABR (AsBR) obtained was 0.0 for patients who received any prophylactic regimen with rFIX-FP, with similar results for total ABR and joint ABR (AjBR). The efficacy of albumin-fusion rFIX has also been described in some PWH-B at different prophylaxis regimens: up to 21 days in patients ≥18 years and up to 14 days in those <12 years.14
Oldenburg et al15 reported the anonymized patient chart data obtained from twenty-four German institutions treating PWH-B. Only subjects who have received rFIX-FP for at least 8 weeks were included in the study. Eighty-one patients were finally considered, 67 adults and 14 children. The mean duration of treatment with rFIX-FP was 39 weeks. The fifty-two weeks, before the switch, were analyzed in the case of 74 patients previously treated with another FIX concentrate. At the time of the analysis, 72 patients were on prophylactic treatment with albutrepenocog-alfa, including 59 who were previously on prophylaxis with another FIX product (prophylaxis-to-prophylaxis group). Data regarding hemorrhagic events were available only for 51 subjects, 42 of them included in the prophylaxis-to-prophylaxis group. 76% of PWH-B of this cohort were severe, 19% moderate and 5% mild. Overall, the mean ABR during prophylaxis with rFIX-FP was 0.3 ± 0.6, while it was 2.6 ± 2.9 during prophylaxis with prior FIX products, and similar data have been obtained in the adult patients (≥12 years), while in the subgroup 0–11 years, the results were significantly lower, but data of only four children were available. 81% of PWH-B on albutrepenonacog-alfa presenting zero bleeds. Among the 72 subjects on prophylaxis with this drug, 68% were infused once a week, while the remaining ones were infused every 9 or more days. This increased efficacy was also obtained with a reduced consumption of drug; in fact, the resulted mean weekly dose of rFIX-FP was 44.2 IU/kg, compared with the mean weekly dose of nonacog-alfa of 85.5 IU/kg, almost two-times higher, and with the mean weekly dose of all plasma-derived FIX (pdFIX) products of 74.5 IU/kg. The data were also confirmed in the two different subgroups: adults (≥12 years) and children (0–11 years). Indeed, in adults, the mean weekly dose of rFIX-FP was 44.1 IU/kg; the mean weekly dose of nonacog-alfa was 84.3 IU/kg and the mean weekly dose of all pdFIX concentrates was 74.0 IU/kg; whereas in children the mean weekly dose of rFIX-FP was 45.0 IU/kg, the mean weekly dose of nonacog-alfa was 94.2 IU/kg and the mean weekly dose of all pdFIX products was 83.5 IU/kg. Similarly, Hermans et al16 conducted a retrospective review of anonymized data collected from twenty-three hemophilia treatment centers in Italy (n = 13), the United Kingdom (n = 7) and Belgium (n = 3). Eighty-four PWH-B treated with rFIX-FP for at least 8 weeks between May and September 2018 were included in this analysis. Overall, 92.8% had severe hemophilia B, while 73/84 were previously in prophylaxis with other FIX products. In this prophylaxis-to-prophylaxis cohort, the mean ABR reductions with rFIX-FP were 94.3% in Italy, 93.9% in Belgium, and 67.7% in the United Kingdom, respectively. Zero bleeds were noted in the 84.1% of Italian subjects, in the 71.4% of Belgians and in the 36.4% of British, reached to 97.7%, 100% and 63.6%, respectively, in case of zero spontaneous bleeds. As reported in the German study,15 this great efficacy was attributed to a low frequency of infusions; in fact, 61/73 PWH-B received rFIX-FP every 7–12 days, 9/73 once every two weeks and 2/73 every 15 days; and to a reduction in the drug consumption of 71%, 59% and 54% in Belgium, the United Kingdom and Italy, respectively, if compared with previous prophylaxis with the different FIX products.
There are several abstracts presented at the World Congresses in recent years have described real-world data, as highlighted by Álvarez Román et al17 in their review. In the first, dated 2018, Escobar et al18 reported the data on 145 PWH-B treated with different rFIX concentrates and obtained from the US healthcare providers (hematologists and Hemophilia Treatment Centers). In the comparison between rFIX-FP and rFIX-Fc, the observed ABR was 0.7 ± 1.0 and 8.9 ± 9.6, respectively, while in the comparison between rFIX-FP and nonacog-alfa, this was 1.5 ± 5.8 and 4.5 ± 5.9, respectively. This highlights the efficacy of albutrepenonacog-alfa in reducing bleeding compared to both extended half-life and standard half-life concentrates, even with lower drug consumption. A first Spanish clinical, pharmacokinetic, and economic evaluation was described by Megías Vericat et al.19 Zero bleeds, reduced drug consumption and costs were reported in a young male switched to rFIX-FP from a standard half-life rFIX. Efficacy and safety of albumin-fusion rFIX were further showed in two other reports describing overall three different pediatric cases,20,21 and in a Spanish case series22 of six PWH-B treated for two years in which the total ABR was 0.50, while spontaneous ABR was 0.25.
Unlike previously reported, two different papers described excessive breakthrough bleeding23 and poor response24 in PWH-B switched to albutrepenonacog-alfa. In the first report, three different adult patients with severe HB, previously treated with rFIX twice a week (dose range 35–50 IU/kg) without bleeding, experienced unexpected joint bleeds after switch to rFIX-FP 60–65 IU/kg every fourteen days and in presence of a trough level over 0.10 IU/mL. No subject had an inhibitor, so a possible explanation for the poor response was a relatively low volume of drug distribution. The reasons for the bleeding are therefore not clear, clinicians must therefore consider this possibility in case of switching from another concentrate, and this clinical event will need to be further investigated. The second one is a letter published in 2020 reporting a US survey on seventy-one patients with severe HB enrolled in different hemophilia centers, among these fifty-five were treated with EHL-rFIX. Albutrepenonacog-alfa was used in a total of twenty-four PWH-B, rFIX-Fc in thirty-one subjects, while the remaining sixteen were in prophylaxis with standard half-life FIX. Bleeding was described in thirteen patients, all in treatment with rFIX-FP; again the rationale for this poor response to therapy was attributed to low volume of drug distribution in the extravascular space, which however needs further investigation. In this case, no data on dosage or infusion times were reported.
Rampotas et al25 reported the safety and the efficacy of two different EHL-rFIX used at lower dosages than recommended. 7/20 of evaluated PWH-B were treated with a median dose rFIX-FP of 20.2 IU/kg/week, reaching a median trough level of 0.08 IU/mL, three of them experienced bleeding, with a reported median ABR of 2, halved compared to that obtained with rFIX-Fc, despite the latter being used at a higher dosage (31.5 IU/kg/week).
A high adherence to therapy yet remarked in the pivotal studies was reported by Mancuso et al26 in the real-world experience. In the three different hemophilia centers, the adherence was 100% (14/14 patients), 100% (15/15 patients) and 57.1% (4/7 patients), respectively. The recorded non-adherence in three patients of the last center was linked to insurance-related and parental issues.
Preliminary data from the Phase IV observational IDEAL study were presented at the ISTH 2022 congress by Castaman et al.27 Overall, 59 male subjects with moderate or severe hemophilia B, treated for at least 6 months with rFIX-FP, were enrolled in this study and followed for two years. The comparison with the previous treatment showed an increase in patients with ABR zero, from 66% to 78%, with the achievement of a median trough level of 0.117IU/mL, against the previous 0.038 IU/mL.
Surgery
The high hemorrhagic risk always makes the surgical intervention in PWH critical; therefore, an accurate evaluation of the type of intervention, the duration and the prophylactic treatment to be implemented is necessary. The efficacy and safety of rFIX-FP in the surgical setting have been described in pivotal trials. Curtin et al28 evaluated thirty surgeries (8 minor and 22 major) performed in twenty-one patients of any age, as a sub-analysis of PROLONG-9FP studies. A single pre-operative bolus of rFIX-FP was sufficient in 29/30 surgeries to achieve a mean plasma FIX level of 1.084 IU/mL, without needing any additional intra-operative infusion. 62.5% of patients undergoing minor surgery did not require post-operative infusion of albutrepenonacog-alfa and resumed their usual prophylaxis 72h after intervention. Red blood cell transfusions were required for six arthroplasties in five patients, but this significant blood loss was predicted prior to surgery. Overall, hemostatic efficacy was considered as excellent or good in 87.5% (7/8) of minor surgeries and in 95.5% (21/22) of major surgeries. No serious adverse events were reported during the peri-operative time.
No data is currently available regarding the efficacy of rFIX-FP in real-world surgery, therefore reports describing the use of the drug in this area and in everyday life would be necessary.
Target Joints Resolution
A target joint is defined as a joint where at least three or more bleeds occur in six months.29 The control of these hemorrhages is undoubtedly one of the main goals of the hemophilia treatment, in order to reduce the risk of the onset or the progression of hemophilic arthropathy. Santagostino et al14 described the data obtained in the phase III study PROLONG-9FP, in which 100% resolution of the target joints was reported in previously treated subjects switched from on-demand to prophylaxis treatment with albutrepenonacog-alfa (p < 0.0001).
The recommended dosage of Idelvion is 35–50 IU/kg/week, which can in some cases be raised to 75 IU/kg every 10–14 days, following the results obtained in the pivotal studies. Sometimes, however, especially in the real-world studies, the dosages may be different from those recommended, chosen by clinicians based on their experience, but this can lead to a reduced hemostatic efficacy of the drug. An example of this comes from the report by Rampotas et al,25 where the median dosage of 20.2 IU/kg, more than halved compared to what is recommended, resulted in an ABR of 2.
Despite the dosages and infusion frequency used in the phase IV observational IDEAL study27 have not yet been reported, the efficacy of rFIX-FP was proven by the reduction of patients with target joints (8.0% vs 30.0%) and chronic joint pain (16.0% vs 44.0%).
Further real-world data on the effect of this drug in reducing target joints are not currently available; therefore, it is hoped that in the future studies and registries will also consider this aspect in order to better evaluate its anti-hemorrhagic efficacy.
Safety
Inhibitor Development
The safety of rFIX-FP was widely valued on the pivotal trials. No inhibitor development in the previously treated PWH-B was reported in Phase I–III studies of the PROLONG-9FP program.14 Similar data were reported in the real-world, where only one case of inhibitor development in an adult male with severe hemophilia B switched to albutrepenonacog-alfa was described.30 In this case report, the patient, previously in prophylactic treatment with rFIX, was put on prophylaxis with IdelvionTM after a pharmacokinetic (PK) assessment to guarantee a higher trough level (0.057 IU/mL on the fifteenth day) and a lower number of infusions, he developed a low-titer inhibitor (0.9 BU/mL) after eleven exposure days (ED) in the presence of an ileo-psoas hematoma, confirmed by CT-scan. The inhibitor was rapidly eradicated following a first infusion of albutrepenonacog-alfa 50 IU/kg, which resulted in a FIX recovery of 13.4% and an incremental in vivo recovery (IVR) of 0.19, and subsequent infusions of rFIX (30 IU/kg/bid), due to the patient’s refusal to continue the same drug that had caused the inhibitor development.
It can therefore be stated, based on the results of clinical studies and real-world data, that rFIX-FP is a safe product, in which the risk of developing inhibitors following its use is minimal.
Anaphylactic Reaction
Anaphylactic reaction in PWH-B could be associated with inhibitor development. The mechanism of action is still unclear, and some hypotheses were formulated, such as an IgE-mediated hypersensitivity response or a complement activation by IgG1 antibody formation. This adverse event has been described by some articles published in the literature. Mauro et al31 reported three different cases of anaphylaxis in children during the immune tolerance induction (ITI) regimen performed using high-dose recombinant plasma-derived FIX concentrates, results similar to those illustrated by Castaman et al32 in their Italian experience.
The only patient who developed inhibitors30 after switch to rFIX-FP had no anaphylactic reaction, and no other allergic responses to this drug were described in the pivotal trials or in the real-world experiences. Only one hypersensitivity reaction was reported by a patient included in a phase III study, but without any consequences.14
Albumin-fusion rFIX can therefore be considered a safe product, with a negligible risk of developing anaphylaxis.
Quality of Life (QoL)
In 2017, von Mackensen et al11 described the unmet needs of PWH, where the reduction in the number of infusions came first, followed by the efficacy and safety of the product. 15.5% of the study participants had hemophilia B. Since then, two different EHL-rFIX concentrates were placed on the market and some trials on non-replacement drugs began. Patients were then able to assess whether these new products met their needs or not. Unlike what happens for hemophilia A, there are currently no real-world data regarding the quality of life perceived by PWH-B following treatments with extended half-life concentrates, such as IdelvionTM. To date, the only report dealing with this topic, and always written by von Mackensen et al,33 is based on the QoL data of pediatric patients enrolled in the PROLONG-9FP trial. Overall, twenty children aged 4–12 years were included in this sub-analysis, and all the scores used to assess their QoL (eg, Haemo-QoL and Hemo-Sat) resulted improved after treatment with rFIX.FP. Although the results obtained in this analysis are very interesting, real-world quality of life data is needed to better understand the impact of rFIX-FP on PWH-B.
Future Role of Albumin-Fusion rFIX
Albumin-fusion rFIX has certainly brought great benefits to PWH-B. An extended half-life, sometimes over 100 hours, and the possibility of infusing the drug once every 7-10-14 and even 21 days have changed the therapy and everything that revolves around it.8,20 Today, new subcutaneous therapies, such as fitusiran and concizumab,34,35 are being studied, while gene therapy36 is about to be made available worldwide. Will these new therapies make the use of concentrates obsolete? It is difficult to say what will happen in a few years, but today concentrates are still needed in the management of the PWH-B. In case of breakthrough bleeding and surgery, these new drugs are not sufficient, and their monitoring also results in difficult, if not impossible, with assays currently available.37 The risk of thrombosis reported in clinical trials of products that rebalance the coagulation cascade, when improperly associated with factor concentrates, caused the suspension of the studies, the resumption of which was conditioned by the reduction of the dosage of the study drug.38,39 The risk of a rise in transaminases, to be treated with corticosteroids, and the use of viral vectors are currently a critical point for patients to use gene therapy,40 while the exclusion from treatment of children ≤12 years places further limits on the use of this therapeutic approach. There are still many things to clarify regarding the efficacy, and above all safety, of the novel therapies for hemophilia, which still guarantee a wide use of factor concentrates, especially in PWH-B.
Although rFIX-FP infusion frequency is infrequent, it is not comparable to the monthly or even bimonthly frequency of fitusiran, but certainly less administrative burden compared to the daily administration of concizumab, while a trough level adequate to the needs of patients can be maintained with low costs. Considering this, “the sunset boulevard” for the albumin-fusion rFIX is still far away.
Conclusion
The extended half-life rFIX concentrates have contributed significantly to modifying the therapy of HB. Albutrepenonacog-alfa is a purified protein produced by recombinant DNA technology, generated by the genetic fusion of recombinant albumin with rFIX, it presents a median half-life of about 100 h, a low clearance, and a small volume of distribution.41 This pharmacokinetic profile allows a reduction in the number of infusions, with a consequent increase in therapeutic adherence. Pivotal trials have shown the efficacy and safety of rFIX-FP with reduced total, spontaneous or joint ABR in both the adult and pediatric population, no inhibitor development and absence of other serious adverse reactions.14 However, the available data from the real-world are not many, and if, on the one hand, they confirm what is highlighted in the PROLONG-9FP program, we can say nothing about the quality of life with this drug or its efficacy in case of surgery.
The poor response highlighted in some reports,23,24 attributed to the low extravascular volume of distribution of rFIX-FP, should also be further investigated to give clear answers to clinicians and patients.
In the next few years, both new non-replacement drugs and gene therapy for the treatment of hemophilia B will be widely available, but this will certainly not lead to the permanent storage of factor concentrates. The possibility of using them at very long intervals (from 7 to 21 days) makes them like subcutaneous drugs, and the predictability of the pharmacokinetic response and the practically absent thromboembolic risk also makes them easy to prescribe to anyone.
Disclosure
S. Pasca and E. Zanon have received fees from Bayer, Kedrion, Roche, Baxalta, Novo Nordisk, CSL Behring, Pfizer, Sobi, and Takeda. The authors report no other conflicts of interest in this work.
References
1. Graham JB, Brinkhous KM. Christmas disease. Br Med J. 1953;2(4827):97. doi:10.1136/bmj.2.4827.97-a
2. Alshaikli A, Rokkam VR. Hemophilia B. StatPearls Publishing; 2022.
3. Garagiola I, Mortarino M, Siboni SM, et al. X Chromosome inactivation: a modifier of factor VIII and IX plasma levels and bleeding phenotype in haemophilia carriers. Eur J Hum Genet. 2021;29(2):241–249. doi:10.1038/s41431-020-00742-4
4. Rogaev EI, Grigorenko AP, Faskhutdinova G, et al. Genotype analysis identifies the cause of the ”royal disease”. Science. 2009;326(5954):817. doi:10.1126/science.1180660
5. Lowe GD, Ludlam CA. Less severe bleeding in hemophilia B than in hemophilia A. J Thromb Haemost. 2008;6(11):1982–1983. doi:10.1111/j.1538-7836.2008.03126.x
6. Lambert T, Benson G, Dolan G, et al. Practical aspects of extended half-life products for the treatment of haemophilia. Ther Adv Hematol. 2018;9(9):295. doi:10.1177/2040620718796429
7. Tagariello G, Iorio A, Santagostino E, et al.; Italian Association Hemophilia Centres. Comparison of the rates of joint arthroplasty in patients with severe factor VIII and IX deficiency: an index of different severity of the 2 coagulation disorders. Blood. 114;2009:747–755. doi:10.1182/blood-2009-01-195313
8. Schulman S, Eelde A, Holmstrom M, et al. Validation of a composite score for clinical severity of hemophilia. J Thromb Haemost. 2008;6:1113–1121. doi:10.1111/j.1538-7836.2008.03001.x
9. Belvini D, Salviato R, Radossi P, et al.; AICE HB Study Group. Molecular genotyping of the Italian cohort of patients with hemophilia B. Haematologica. 2005;90:635–642.
10. Rallapalli PM, Kemball-Cook G, Tuddenham EG, et al. An interactive mutation database for human coagulation factor IX provides novel insights into the phenotypes and genetics of hemophilia B. J Thromb Haemost. 2013;11(7):1329–1340. doi:10.1111/jth.12276
11. von Mackensen S, Kalnins W, Krucker J, et al. Hemophilia patients’ unmet needs and their expectations of the new extended half-life factor concentrates. Hemophilia. 2017;23(4):566–574. doi:10.1111/hae.13221
12. Chia J, Louber J, Glauser I, et al. Half-life-extended recombinant coagulation factor IX-albumin fusion protein is recycled via the FcRn-mediated pathway. J Biol Chem. 2018;293:6363–6373. doi:10.1074/jbc.M117.817064
13. Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339(1):b2535. doi:10.1136/bmj.b2535
14. Santagostino E, Martinowitz U, Lissitchkov T, et al.; PROLONG-9FP Investigators Study Group. Long-acting recombinant coagulation factor IX albumin fusion protein (rIX-FP) in hemophilia B: results of a Phase 3 trial. Blood. 2016;127(14):1761–1769. doi:10.1182/blood-2015-09-669234
15. Oldenburg J, Yan S, Maro G, et al. Assessing bleeding rates, related clinical impact and factor utilization in German hemophilia B patients treated with extended half-life rIX-FP compared to prior drug therapy. Curr Med Res Opin. 2019;36:9–15. doi:10.1080/03007995.2019.1662675
16. Hermans C, Marino R, Lambert C, et al. Real-world utilisation and bleed rates in patients with haemophilia B who switched to recombinant factor IX fusion protein (rIX-FP): a retrospective international analysis. Adv Ther. 2020;37:2988–2998. doi:10.1007/s12325-020-01300-6
17. Álvarez Román MT, Benítez O, Canaro MI, et al. Expert opinion paper on the treatment of hemophilia B with albutrepenonacog-alfa alfa. Expert Opin Biol Ther. 2021;21(9):1165–1171. doi:10.1080/14712598.2021.1932811
18. Escobar M, Leissinger C, Yan S, et al. Comparison of rFIX utilization and bleed rates in US hemophilia B patients on rIX- FP and their prior rFIX drug. Res Pract Thromb Haemost. 2018;2(S1):69–70, PB150. doi:10.1002/rth2.12053
19. Megías Vericat J, Rodríguez López M, Poveda J, et al. Clinical, pharmacokinetic, and economic analysis of the first switch to an extended half-life factor IX (albutrepenonacog-alfa alfa) in Spain. Res Pract Thromb Haemost. 2019;3(S1):440, PB1436.
20. Díaz Jordán B, Valverde Templado A, Cebanu T, et al. Real-life experience using rIX-FP in a patient with hemophilia B on prophylaxis after 100 days of switch to extended life factor IX.
21. Schiavulli M, Lupone MR, Pollio B, et al. Real-world experience of rIX-FP in two previously untreated paediatric patients with severe haemophilia B in Italy. Haemophilia. 2021;21(S2):118, ABS179.
22. Álvarez-Román M, López M, Martínez M, et al. Retrospective analysis of the use, effectiveness, and quality of life in patients with hemophilia B in prophylaxis treatment with albumin-fused recombinant FIX (rIX-FP) in Spain.
23. Kleiboer B, Nielsen B, Ma AD, et al. Excessive breakthrough bleeding in haemophilia B patients on factor IX-albumin fusion protein prophylactic therapy: a single centre case series. Haemophilia. 2020;26(1):e23–e25. doi:10.1111/hae.13896
24. Malec LM, Croteau SE, Callaghan MU, et al. Spontaneous bleeding and poor bleeding response with extended half-life factor IX products: a survey of select US haemophilia treatment centres. Haemophilia. 2020;26(3):e128–e129. doi:10.1111/hae.13943
25. Rampotas A, Desborough MJR, Raza-Burton S, et al. A single centre retrospective study of low dose prophylaxis with extended half-life factor IX for severe haemophilia B. Haemophilia. 2020;26(2):278–281. doi:10.1111/hae.13936
26. Mancuso ME, Oldenburg J, Boggio L, et al. High adherence to prophylaxis regimens in haemophilia B patients receiving rIX-FP: evidence from clinical trials and real-world practice. Haemophilia. 2020;26(4):637–642. doi:10.1111/hae.14018
27. Castaman G, Tagliaferri A, Molinari AC, et al. IDEAL study: a real-world assessment of pattern of use and clinical outcomes with recombinant factor IX albumin fusion protein (rIX-FP) in patients with haemophilia B in Italy. ISTH 2022 Congress, VPB1165; 2022.
28. Curtin J, Santagostino E, Faraizah AK, et al. Simplifying surgery in haemophilia B: low factor IX consumption and infrequent infusions in surgical procedures with rIX-FP. Thromb Res. 2020;188:85–89. doi:10.1016/j.thromres.2020.02.011
29. Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935–1939. doi:10.1111/jth.12672
30. Zanon E, Pasca S, Simioni P. The sudden and unexpected appearance of inhibitors in a previously treated severe haemophilia B patient after the switch to albutrepenonacog-alfa alpha. Haemophilia. 2018;24(5):e372–e375. doi:10.1111/hae.13590
31. Mauro M, Bonetti E, Balter R, et al. Recurrent episodes of anaphylaxis in a patient with haemophilia B: a case report. Blood Transfus. 2016;14(6):582–584. doi:10.2450/2016.0297-15
32. Castaman G, Bonetti E, Messina M, et al. Inhibitors in haemophilia B: the Italian experience. Haemophilia. 2013;19(5):686–690. doi:10.1111/hae.12158
33. von Mackensen S, Shah J, Seifert W, Kenet G. Health-related quality of life in paediatric haemophilia B patients treated with rIX-F. Haemophilia. 2019;25(1):45–53. doi:10.1111/hae.13624
34. Shapiro AD. Concizumab: a novel anti-TFPI therapeutic for hemophilia. Blood Adv. 2021;5(1):279. doi:10.1182/bloodadvances.2019001140
35. Machin N, Ragni MV. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med. 2018;9:135–140. doi:10.2147/JBM.S159297
36. Rodríguez-Merchán EC, De Pablo-Moreno JA, Liras A. Gene therapy in hemophilia: recent advances. Int J Mol Sci. 2021;22(14):7647. doi:10.3390/ijms22147647
37. Ellsworth P, Ma A. Factor-mimetic and rebalancing therapies in hemophilia A and B: the end of factor concentrates? Hematology. 2021;2021(1):219–225. doi:10.1182/hematology.2021000253
38. Young G, Srivastava A, Kaan K, et al. Efficacy and safety of fitusiran prophylaxis, an siRNA therapeutic, in a multicenter Phase 3 study (ATLAS-INH) in people with hemophilia A or B, with inhibitors (PwHI). Blood. 2021;138(S1):4. doi:10.1182/blood-2021-150273
39. Chowdary P, Eichler H, Matsushita T, et al. Dose optimisation and risk mitigation during concizumab prophylaxis in patients with haemophilia A/B with and without inhibitors in phase 3 clinical trials. Haemophilia. 2022;28(S1):
40. Miesbach W, Sawyer EK. Practical implications of factor IX gene transfer for individuals with hemophilia B: a clinical perspective. Hum Gene Ther Clin Dev. 2018;29(2):80–89. doi:10.1089/humc.2017.253
41. Morfini M. Pharmacokinetic drug evaluation of albutrepenonacog-alfa alfa (CSL654) for the treatment of hemophilia. Expert Opin Drug Metab Toxicol. 2016;12(11):1359–1365. doi:10.1080/17425255.2016.1240168
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.