")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 7
Akt/PKB activation and insulin signaling: a novel insulin signaling pathway in the treatment of type 2 diabetes
Authors Mackenzie R, Elliott B
Received 22 October 2013
Accepted for publication 11 December 2013
Published 13 February 2014 Volume 2014:7 Pages 55—64
DOI https://doi.org/10.2147/DMSO.S48260
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Richard WA Mackenzie, Bradley T Elliott
Department of Human and Health Sciences, Facility of Science and Technology, University of Westminster, London, UK
Abstract: Type 2 diabetes is a metabolic disease categorized primarily by reduced insulin sensitivity, β-cell dysfunction, and elevated hepatic glucose production. Treatments reducing hyperglycemia and the secondary complications that result from these dysfunctions are being sought after. Two distinct pathways encourage glucose transport activity in skeletal muscle, ie, the contraction-stimulated pathway reliant on Ca2+/5′-monophosphate-activated protein kinase (AMPK)-dependent mechanisms and an insulin-dependent pathway activated via upregulation of serine/threonine protein kinase Akt/PKB. Metformin is an established treatment for type 2 diabetes due to its ability to increase peripheral glucose uptake while reducing hepatic glucose production in an AMPK-dependent manner. Peripheral insulin action is reduced in type 2 diabetics whereas AMPK signaling remains largely intact. This paper firstly reviews AMPK and its role in glucose uptake and then focuses on a novel mechanism known to operate via an insulin-dependent pathway. Inositol hexakisphosphate (IP6) kinase 1 (IP6K1) produces a pyrophosphate group at the position of IP6 to generate a further inositol pyrophosphate, ie, diphosphoinositol pentakisphosphate (IP7). IP7 binds with Akt/PKB at its pleckstrin homology domain, preventing interaction with phosphatidylinositol 3,4,5-trisphosphate, and therefore reducing Akt/PKB membrane translocation and insulin-stimulated glucose uptake. Novel evidence suggesting a reduction in IP7 production via IP6K1 inhibition represents an exciting therapeutic avenue in the treatment of insulin resistance. Metformin-induced activation of AMPK is a key current intervention in the management of type 2 diabetes. However, this treatment does not seem to improve peripheral insulin resistance. In light of this evidence, we suggest that inhibition of IP6K1 may increase insulin sensitivity and provide a novel research direction in the treatment of insulin resistance.
Keywords: type 2 diabetes, insulin resistance, Akt/PKB, 5′-monophosphate-activated protein kinase
Metabolic dysfunction in type 2 diabetes
Type 2 diabetes is a multifactorial metabolic disease characterized by defects in β-cell function and insulin action and increased hepatic glucose production.1 Metabolic dysfunction in type 2 diabetes is also the product of reduced glucose effectiveness or the ability of glucose to transport itself by a mass action effect.2 Central to this metabolic condition is altered glucose and lipid metabolism resulting from the combined effects of insulin resistance in skeletal muscle, hepatic, renal, and adipose tissue. The resulting hyperglycemia is the primary cause of the secondary complications associated with type 2 diabetes. Thus, treatments that target glucose uptake while reducing gluconeogenesis are key in the management of type 2 diabetes.
Faced with an increased prevalence, which is predicted to reach 6.1% of the world’s population by 2025,1 advances in the treatment of type 2 diabetes are clearly being sought after. There are three main approaches to treatment of diabetes, ie, lifestyle modifications requiring patient-led metabolic responsibility, gastric bypass surgery, and pharmaceutical interventions. A current and effective target for the pharmaceutical approach to treatment of type 2 diabetes centers around upregulation of the metabolic master switch, ie, 5′-monophosphate-activated protein kinase (AMPK),3 with the use of metformin.
AMPK: a current target in management of type 2 diabetes
AMPK is a key regulatory protein ubiquitously expressed in cells throughout the human body. In mammalian tissues, AMPK is a heterotrimer consisting of a catalytic α subunit with two further regulatory subunits, β and γ. Each of these subunits has two or more isoforms.4,5 The γ subunit consists of 4-cystathionine-b-synthase domains, which are required for binding of adenosine triphosphate (ATP) or adenosine monophosphate (AMP). The binding of two AMPs to the γ subunit leads to conformational changes in the complex that result in activation of AMPK through exposure and phosphorylation of the Thr172 binding site on the catalytic α subunit.6,7 Binding of ATP to the γ subunit operates in an opposing manner by decreasing AMPK activity, while the binding of adenosine diphosphate (ADP) to the regulatory γ subunit protects AMPK against dephosphorylation.8
A working hypothesis is that upon stimulation, AMPK mediates contraction-stimulated GLUT-4 translocation and glucose uptake in skeletal muscle via phosphorylation of the Rab guanosine triphosphatase (GTPase)-activating protein (GAP), AS160, in a manner similar to insulin.9–11AS160 is a proposed downstream target of AMPK and it is suggested that phosphorylation of AS160 by AMPK results in decreased Rab-GTPase activity, leading to an increase in GTP loading on intracellular GLUT-4 storage sites, promoting GLUT-4 translocation and cell membrane fusion while stimulating glucose uptake in adipocytes and skeletal muscle.12–14
Using an in vivo electroporation/gene delivery technique that prevents phosphorylation on four regulatory phospho-Akt substrate sites on AS160, Kramer et al9 demonstrated that both insulin-stimulated and contraction-stimulated glucose transport was impaired in skeletal muscle (tibialis anterior). In the same study, overexpression of mutant AS160 with abolished Rab-GAP activity showed normal elevations in contraction-stimulated glucose uptake.9 Furthermore, transgenic mouse models lacking AMPKα2 activity in muscle show complete inhibition of AS160 phosphorylation and glucose transport on stimulation with the AMPK activator, 5-aminoimidazole-4-carboxamide ribonucleoside. Taken together, these data suggest a prominent role of both AMPK and AS160 in glucose transport, uptake, and utilization.15
Metformin is extensively used in the treatment and management of type 2 diabetes. Metformin improves glycemic control primarily via suppression of hepatic glucose production, and to a lesser extent, but still metabolically important, increased peripheral glucose uptake.16 This pharmaceutical agent activates hepatocyte-specific AMPK, resulting in reduced acetyl-CoA carboxylase activity, increased fatty acid oxidation, and suppression of lipogenic enzyme expression.16 However, recent work suggests that inhibition of gluconeogenesis by metformin acts independently of the AMPK pathway because hepatic glucose production remains blunted in AMPK-depleted hepatocytes despite treatment with metformin.17
Metformin is also known to activate AMPK and stimulate glucose uptake in isolated rodent skeletal muscle, presumably via the ability of metformin to increase the intracellular AMP/ATP ratio.18 In addition, administration of metformin is known to increase AMPK activity in human skeletal muscle, promote GLUT-4 membrane translocation, and stimulate insulin-independent glucose uptake,18 in a manner similar to muscle contraction. Although inhibition of hepatic glucose production by metformin is considered to be the primary mechanism by which AMPK lowers hyperglycemia, its ability to increase glucose uptake in peripheral tissue is no less important. This action is of particular relevance given that peripheral insulin resistance is considered to occur prior to hepatic insulin resistance.19 However, it has been noted in more recent work using a high-fat mouse model that insulin resistance in the liver precedes reduced insulin action in skeletal muscle.20 Recent work carried out in type 2 diabetic humans found that administration of metformin 2,550 mg/day for 3–4 months increased glucose disposal but did not alter insulin receptor substrate-1, class IA phosphatidylinositol-3 (PI3) kinase, or Akt/protein kinase B (PKB) activity in skeletal muscle.21 These results suggest that metformin can stimulate glucose transport activity in type 2 diabetics, but does so via a mechanism that is independent of the traditional insulin signaling pathway in skeletal muscle.
Pathways in type 2 diabetes stimulated by insulin or contraction
When forming an understanding of the progression of this disease, it is important to recognize two points. First, peripheral glucose uptake into skeletal muscle (the main disposal site for glucose) can be promoted via two distinct pathways, ie, insulin-dependent mechanisms resulting in recruitment and activation of Akt/PKB and contraction-mediated stimulation22 or hypoxia-mediated stimulation23 of AMPK (Figure 1). It has been consistently shown that PI3 kinase is necessary for insulin-stimulated but not for contraction-stimulated glucose uptake,24–27 while Akt2 knockout mice demonstrate normal basal and contraction-stimulated glucose uptake.28 Indeed, glucose transport is additive when either hypoxia or contractile activity are coupled with insulin, whereas hypoxia and contractile activity are not.29,30 In support of this is the observation that wortmannin, a selective inhibitor of PI3 kinase, completely blocks insulin-stimulated glucose transport but has no effect on contraction-mediated or hypoxia-mediated 2-deoxy-D-glucose uptake in rat skeletal muscle.25
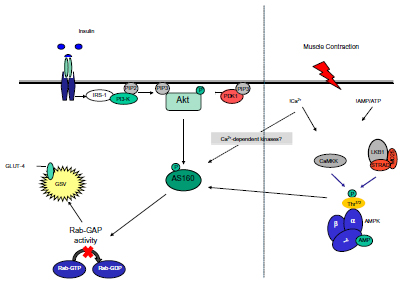 | Figure 1 Insulin and contraction signaling pathways during GLUT-4 recruitment and translocation. |
Thus, insulin-stimulated glucose transport is thought to occur independently of AMPK activity,31,32 and when defects in the Akt/PKB pathway occur, the likely outcome is peripheral insulin resistance.33 In addition, it is worth noting that Akt/PKB and target proteins sitting upstream and downstream thereof are defective in type 2 diabetes whereas AMPK activation remains largely intact.23,34 This latter point is important because peripheral insulin resistance is a catalyst for β-cell dysfunction, increased hepatic glucose production, and progression of the disease,35 so any intervention that improves peripheral insulin resistance may conceivably reverse β-cell function and reduce hepatic glucose production.
Hypoxia and contractile activity are known stimulators of AMPK, and upon activation, AMPK is widely accepted to increase glucose transport activity and GLUT translocation via an insulin-independent mechanism. Azevedo et al30 demonstrated that insulin stimulation of 2-deoxyglucose transport is impaired in insulin-resistant tissue. These investigators further demonstrated no differences in hypoxia-stimulated glucose transport rates between lean and obese diabetic groups, suggesting that this insulin-independent (or AMPK-dependent) pathway is intact in insulin-resistant muscle tissue.30 The muscle protein content of AMPKα1, α2, and γ3 is similar in type 2 diabetics when compared with healthy controls.22 In addition, no difference was found in basal phosphorylated AMPK activity when the same comparison was made between insulin-resistant and lean muscle tissue.22 Furthermore, the basal activity of AMPKα1, α2, and Thr172 AMPK-α phosphorylation are all normal in type 2 diabetes.35,36 In support of this, normal AMPK activity was found despite decreased insulin-stimulated glucose transport when measured during euglycemic-hyperinsulinemic clamp in the same population.36
The data reported by Højlund et al,36 along with the finding that type 2 diabetics have normal AMPK activation during exercise35 while showing metformin-induced increases in AMPK phosphorylation,34 suggest again that AMPK function is intact in type 2 diabetes. The conclusion drawn from the work of both Musi et al34 and Højlund et al36 is that normal AMPK activity and reduced insulin sensitivity are metabolic aspects of insulin resistance, indirectly supporting the notion that two separate pathways operate to facilitate glucose transport. Further, Højlund et al36 also demonstrated that AMPK activity under basal conditions in human skeletal muscle is not regulated by normal physiological concentrations of insulin. These findings taken together may suggest that future therapeutic targets should focus not on AMPK specifically, but also on prominent proteins associated with the insulin-signaling pathway for the treatment of insulin resistance. However, the targeting of AMPK for insulin resistance cannot be completely disregarded, with McBride et al37 proposing a novel role for the AMPKβ subunit in glycogen sensing and synthesis. Further, AMPKα2 knockout mice show whole body insulin resistance, suggesting that AMPK may play a role in modulating insulin sensitivity.38 Insulin sensitivity is not altered in a dominant-negative mutant AMPK rodent model (Tg-KD-AMPKα2),39 although it is more susceptible to high-fat diet-induced insulin resistance,40 suggesting that AMPK is not an important mediator of insulin sensitivity in skeletal muscle.41 The potential role of AMPK in insulin-stimulated glucose transport may be complicated by the recent findings from Sakamoto’s laboratory.42 5-aminoimidazole-4-carboxamide ribonucleoside-induced activation of AMPK in knockin mice expressing a glucose-6-phosphate-insensitive glycogen synthase mutant demonstrates elevated glycogen accumulation through allosteric activation of glycogen synthase, caused by elevated glucose uptake and intracellular glucose-6-phosphate levels. This evidence suggests that AMPK may be able to promote glycogen synthesis in skeletal muscle independent of the traditional insulin-signaling nexus.42 More work is clearly needed to identify how, or even if, AMPK affects insulin-induced glucose uptake, given that a direct mechanism via which AMPK is activated by insulin is yet to be determined.
AMPK is clearly a major target for the treatment of type 2 diabetes. However, AMPK activation is known to be largely normal in this population22,43 and provides a rationale for the prescription of exercise in the treatment of type 2 diabetes. Although AMPK activation produces a number of desirable effects in metabolically responsive tissue, it seems to bypass the problem, at least in skeletal muscle. It therefore seems logical to focus future research on the activation/inhibition of signaling mechanisms known to be defective in insulin-resistant tissue in the pursuit of future pharmaceutical treatment.
Akt/PKB isoforms
Akt, also known as PKB, is a serine/threonine-specific protein kinase that plays a key role in multiple cellular processes, including cell growth, survival, proliferation, and metabolism.44 Three homologous isoforms have been identified, ie, Akt1/PKBα, Akt2/PKBβ, and Akt3/PKBγ.45 Research conducted over the past decade has identified distinct roles for each isoform, with Akt1/PKBα linked to cell survival, Akt2/PKBβ with cell-substrate metabolism,46 and Akt3/PKBγ with brain development.47
Akt1/PKBα is known to inhibit apoptosis via activation of NF-ºB and regulation of IºB kinase, resulting in upregulation of prosurvival genes.48 The same isoform is also implicated in stimulating skeletal muscle hypertrophy by promoting protein synthesis and inhibiting protein degradation pathways, providing evidence for its roles in muscle homeostasis.49
Hyperglycemia and reduced glucose transport in muscle is present in knockout Akt2/PKBβ mice,50 but is not apparent with deletion of the Akt1/PKBα and Akt3/PKBγ isoforms. Involvement of the Akt3/PKBγ isoform in brain development comes from evidence examining Akt3 knockout mice, which shows reduced cerebral cell numbers and seemingly increased protein degradation and reduced synthesis. Further, the same Akt3/PKBγ knockout mice show diminished mTOR (mammalian target of rapamycin) signaling,51 highlighting its role in cell death and growth inhibition.
Akt/PKB activation
PI3 kinase, following upstream stimulation by the insulin receptor substrate, causes recruitment of phosphatidylinositol (4,5)-bisphosphate (PIP2) and production of the second lipid messenger, phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 then binds to the pleckstrin homology domain of Akt/PKB, allowing for its translocation and binding to the cell membrane. Following this membrane translocation, Akt/PKB can then be phosphorylated and activated52 by two further kinases, PDK1 and mTOR complex 2, within the T-loop of the catalytic domain (Thr308) and the carboxyl terminal hydrophobic domain (Ser473), respectively, resulting in phosphorylation of many downstream targets involved in cellular growth and metabolism.44
Akt/PKB and insulin signaling
It is clear that Akt signaling plays a central role in insulin-stimulated glucose uptake in both muscle and adipose tissue while inhibiting glucose release from hepatocytes.5 The effect of insulin on glucose uptake in peripheral tissue via Akt/PKB is through its ability to translocate GLUTs to the cell membrane, thereby facilitating glucose uptake. Binding of insulin to its cell surface protein receptors causes subsequent tyrosine phosphorylation, resulting in phosphorylation of insulin receptor substrates on specific tyrosine residues and activation and recruitment of PI3 kinase and its downstream target Akt/PKB.52 The Akt substrate of 160 kDa (AS160), also known as TBC1D4, is an established candidate in Akt/PKB-induced GLUT-4 translocation in skeletal muscle. AS160 functionally maintains Rab-GTPase(s) in an inactive form via guanosine-50-diphosphate-loading, thereby retaining GLUT-4 within GLUT storage vesicles.14 Upon activation, Akt/PKB phosphorylates AS160, leading to a reduction in Rab-GAP activity, promoting GLUT-4 translocation and glucose uptake.14,53,54 Thus, any defects in the PI3 kinase/Akt/AS160 transduction pathway would ultimately reduce glucose uptake in skeletal muscle. Similarly, hepatic deletion of the Akt1 and Akt2 isoform causes glucose intolerance, insulin resistance, and a defective insulin transcriptional response to feeding in hepatocytes.55
Akt/PKB and insulin resistance
Evidence from both human and animal work links insulin resistance with defects to both upstream and downstream targets of Akt/PKB in the form of dephosphorylation of protein side chains on insulin receptor substrates as well as complete loss of insulin receptor substrates from the cell surface membrane,56 reduced PI3 kinase activity57 and impaired phosphorylation of the Akt/PKB substrate, AS160, in type 2 diabetic skeletal muscle.58 In addition, knockdown or depletion of the Akt2/PKBβ isoform in mice causes insulin resistance and diabetic-like symptoms, with Akt2/PKBβ knockout rodents also demonstrating hepatic insulin resistance.59 This work has been extended to humans, with the finding that a mutation in the gene encoding Akt2/PKBβ results in severe insulin resistance,60 establishing Akt2/PKBβ as a key protein in the maintenance of euglycemia, given that defects in this important mediator would presumably result in reduced AS160-induced GLUT translocation.
In substituting the AS160-Thr649 phosphorylation/14-3-3 binding residue with a nonphosphorylatable alanine, Chen et al were able to show that insulin-stimulated AS160 binding to 14-3-4 is completely abolished.11 The same work also showed that insulin-stimulated glucose uptake and cell surface GLUT-4 content were reduced in this mutated knockin model.11 These results demonstrate the key role played by Akt/PKB and its downstream targets in insulin sensitivity and normal glucose tolerance, and may therefore represent an argument for its targeting as an intervention in the treatment of insulin resistance.
Inositol pyrophosphate and Akt/PKB signaling: targeting peripheral insulin resistance
Research in the field highlights Akt/PKB as a major signaling intermediate in insulin-stimulated glucose transport in type 2 diabetes characterized by reduced insulin signaling of Akt/PKB.45,58 Akt/PKB is known to upregulate S6 kinase 1 (S6K1) via activation of mTOR complex 1, leading to phosphorylation of the serine residues on insulin receptor substrate-1 and resulting in Akt/PKB inhibition and a reduction in insulin-stimulated glucose uptake.61 PDK1-mediated phosphorylation of Akt at Thr308 is dramatically increased by binding of PIP3 to the pleckstrin homology domain on Akt.62 Recent work from Chakraborty et al33 suggests a separate and seemingly novel feedback mechanism in the regulation of insulin signaling, whereby specific inositol pyrophosphates can also have an inhibitory effect on insulin stimulation of Akt/PKB by competing with PIP3 at the pleckstrin homology domain.
Diphosphoinositol polyphosphates, also known as inositol pyrophosphates, are a family of water-soluble inositol phosphates.63 The most notable of these is diphosphoinositol pentakisphosphate (IP7),64 which has been implicated in insulin secretion65 and peripheral insulin signaling (Figure 2).45 Inositol pyrophosphates were first discovered in 1993 and have received a great deal of attention since due to their “high energy” pyrophosphate bond, which can be synthesized by a family of three inositol hexakisphosphate (IP6) kinases (IP6Ks).66 The inositol hexakisphosphate kinase-1 (IP6K1) isoform produces a pyrophosphate group at the position of IP6 to generate a further inositol pyrophosphate, IP7. Production of IP7 results in its binding to the pleckstrin homology domain of Akt/PKB, preventing its translocation to the cell membrane and reducing its subsequent phosphorylation by PDK1. The evidence for this comes from the finding that IP7 fails to prevent PDK1 Thr308 phosphorylation of Akt/PKB lacking a pleckstrin homology domain.33 The consequence of this is a reduction in insulin (Akt/PKB)-stimulated glucose uptake in muscle and adipose tissue (Figure 2), with a potential increase in accumulation of hepatic fat.33 Unpublished data from the authors’ laboratory shows that IP6K1 is detectable in human skeletal muscle and correlates with one-compartment measures of insulin sensitivity (SI) in glucose-intolerant individuals. It is well known that calorie restriction in humans improves insulin sensitivity,67 while starvation reduces IP7 formation in wild-type embryonic fibroblasts, again highlighting a potential mechanism whereby IP7 may play a role in insulin resistance.33
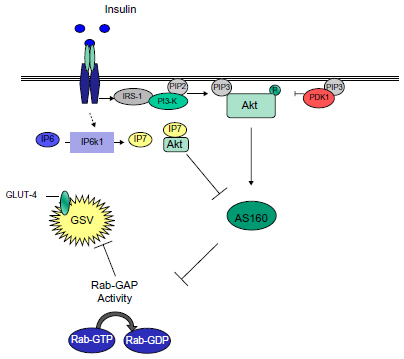 | Figure 2 Inositol phosphates and Akt/PKB inhibition, a potential Akt/PKB feedback mechanism. |
Does inhibition of IP7 formation restore normal Akt? Implications for obesity and insulin resistance
It seems that depletion or chemical inhibition of IP6K1 may improve peripheral insulin action and hepatic function. Specific IP6K1 inhibitors have been shown, at least in cell culture, to reduce the conversion of IP6 to IP7, and so increase PIP3 binding to the pleckstrin homology domain of Akt/PKB.33,68 Padmanabhan et al68 showed that the selective inhibitor, N2-(m-(trifluoromethyl)benzyl) N6-(p-nitrobenzyl)purine (TNP), reduces IP6K1 activity in a dose-dependent manner in vitro while reducing IP7 synthesis in vivo, providing the potential for decreased IP7-Akt/PKB interaction and an increase in Akt/PKB membrane translocation. At least in the HeLa cell line, IP7 formation is returned to near baseline concentrations 2 hours post removal of TNP, suggesting that the action of TNP on IP6K1 activity is both rapid and reversible.68 Using IP6K1 knockout hepatocytes,33 a 60% reduction in IP7 and elevations in Akt/PKB and GSK3β are seen in response to insulin stimulation. Similarly, these same hepatocytes show enhanced age-dependent phosphorylation of Akt at Thr308,33 suggesting a role for IP7 in age-related insulin resistance, with either depletion or knockout resulting in improved glucose tolerance by a presumable decrease in hepatic glucose production. The work on IP6K1 knockout and improved hepatic insulin resistance is a novel research topic with very little current published work, so provides an exciting avenue for future research.
This finding has been extended in IP6K1 knockout mouse models which have elevated Akt/PKB activity in response to insulin stimulation, accompanied by increased glucose transport rates in skeletal muscle.33 IP6K1 mice also display diminished circulating insulin levels, suggesting increased insulin sensitivity or efficiency.69 These data are of note because IP6K1 knockout mice have a two-fold increase in insulin sensitivity when compared with their wild-type counterparts, and phosphorylated Akt/PKB at sites Thr308 and Ser473 were both increased while phosphorylated GSK3β was reduced in skeletal muscle.33 This is an important discovery because phosphorylated GSK3β Ser9 renders glycogen synthesis active, resulting in the potential for increased glucose uptake and glycogenesis. These data were accompanied by a significant elevation in muscle glycogen content in IP6K1 knockout models versus wild-type mice,33 with low muscle glycogen content being an inherent metabolic characteristic of type 2 diabetes.70 These data are important because they suggest that IP6K1 deletion may result in insulin hypersensitivity, a known physiological consequence of elevated Akt/PKB activity.71 Further, a reduction in IP7 by 10 μM TNP increased phosphorylation of Akt/PKB at Thr308 without affecting phosphorylation of Ser473.33 This latter finding is of clinical significance given that it is not likely that future pharmaceutical intervention acting via IP7 inhibition will negatively affect muscle growth and development through inhibition of mTOR signaling.
Interestingly, IP6K1 knockout mice also exhibit a lean phenotype,72 presumably as a result of increased fatty acid metabolism via β oxidation45 and reduced adipogenesis.73 Akt has been referred to as lipogenic, with double knockout of the Akt1 and Akt2 isoforms displaying reduced fat mass.74 Thus, it seems that IP7 formation, as synthesized by IP6K1 in mice, is implicated in obesity and insulin resistance via inhibition of Akt and increased GSK3β activity.33 Indeed, increased Akt activity and resistance to obesity are defined characteristics in genetic models of insulin hypersensitivity (ie, protein phosphatase 1B, S6K1, and JNK mutants).33,75
IPK61 inhibition and β-cell function
Decreased β-cell function is critical to the development of overt type 2 diabetes.1 IP6K1 knockout mice were found to have lower fasting serum insulin when compared with their wild-type counterparts, which the authors suggested represents a loss of β-cell function.69 Depletion of IP6K1 seems to inhibit insulin exocytosis in β-cells,69 while maintenance of IP7 levels in the same cell type may enhance insulin secretion.76 Indeed, IP6K1 knockout rodents demonstrate a 65%–70% reduction in circulating insulin concentrations,69 suggesting that mice lacking IP6K1 in their pancreatic β-cells have reduced insulin release. However, the mutant model used by Bhandari et al69 also showed signs of improved insulin-stimulated glucose uptake. The reduction in circulating insulin levels may therefore be a product of the hyperbolic relationship that exists between insulin sensitivity and insulin secretion,77 ie, a lower insulin requirement due to a more efficient rate of glucose disposal. Further, IP6K1 knockout mice with low physiological insulin concentrations do not demonstrate diabetic characteristics, with normal fasting glucose and glycosylated hemoglobin levels, and show reduced body weight over a 10-week period69 and resistance to obesity.33 These findings are particularly exciting in light of the positive effect that inhibition of IP6K1 might have on the sensitivity of peripheral tissue to insulin via increased Akt/PKB activity. Inhibition of IP6K1 in β-cells requires more research because incubation of pancreatic Min6 cells with TNP results in direct inhibition of insulin release,68 and more importantly, this reduction in insulin release is not a product of increased peripheral insulin action.
Targeting isoforms: β-cell function, cancer, and cell growth
The work of Bhandari et al69 in a mouse model is clearly exciting, given that these authors assessed the effects of targeted depletion of the coding sequence of IP6K1 exon 6 on glucose control and insulin secretion. However, the IP6K1 mutant mice used in this study69 were smaller than their wild-type counterparts despite the same food intake, suggesting a negative consequence for protein synthesis and cell growth. This is surprising, given that a reduction in IP7 using the IP6K1 inhibitor TNP does not affect phosphorylation of Akt/PKB at Ser473.33 The work of Bhandari et al69 may be countered by the knowledge that IP6K1 depletion may increase muscle protein synthesis while reducing fat accumulation,33 decreasing adipogenesis via GSK3β,78 and increasing Akt/PKB-induced fat oxidation.79 These results offer indirect evidence that reduced animal size69 may be due to increased fat loss rather than muscle atrophy, particularly given that increased p-Akt/PKB at Ser473, as seen in IP6K1 knockout mice,33 may increase protein synthesis via mTOR activation.
Hyperactivity of Akt/PKB is a hallmark of most human forms of cancer, given that it modulates cell proliferation, growth, survival, and transit metabolism. In addition, overexpression of Akt/PKB is noted in various cancers and is possibly related to specific Akt/PKB isoforms.44 Indeed, Akt1/PKBα is linked to breast cancer,80 Akt2/PKBβ is reported in pancreatic cancer,33 and Akt3/PKBγ mutations are associated with human melanoma.81 A detailed description of Akt/PKB and its link to cancer is beyond the scope of this paper, but this area has been comprehensively reviewed by others.44 However, it is clear that blanket inhibition of IP6K1, with resulting hyperactivity of Akt/PKB in all cell types and/or in the whole body, cannot be advised at this stage.
Concluding remarks
Activation of AMPK has a clear beneficial effect in metabolically responsive tissue in type 2 diabetics. We suggest that future research and potential therapies, in parallel with continued research into the role of AMPK in glucose control, should be directed towards mechanisms known to be involved in insulin resistance. Recent published work provides exciting and novel data that may lead to a pharmaceutical agent capable of promoting insulin-stimulated glucose transport in insulin-resistant tissue via inhibition of IP6K1. Although early in development, use of IP6K1 inhibitors provides an interesting area of research in the fight against insulin resistance and type 2 diabetes. Due to the varied response of Akt/PKB in different tissue types and the resulting individual and/or combined effects this kinase may have on whole body metabolism, cell growth, and human cancers, blanket inhibition of IP6K1 and reduced production of IP7 is not recommended at this point. Indeed, while AMPK activity promotes growth of prostate cancer cells,82 use of metformin in vivo reduces the occurrence of pancreatic and hepatic malignancies.83 Further, it is important to point out that even AMPK, the current target protein, which is induced by metformin, is implicated in some cancers,82 but use of metformin is inversely correlated with other types of cancer in a dose-dependent manner.83
The introduction of Akt1/PKBα, Akt2/PKBβ, and Akt3/PKBγ knockout rodents has suggested different physiological functions for specific isoforms, but there is now some evidence pointing towards compensatory effects in a given isoform when another is knocked out. In terms of insulin resistance, it appears that future work should focus on pharmaceutical inhibition of IP6K1 in established animal models of diabetes (for example, Ob/Ob) before human trials can be considered. Likewise, the roles of IP6K1 depletion in pancreatic β-cells and in insulin production and release, warrant further investigation.
Acknowledgment
The authors are thankful to Sarah Rotundo-Fregusson for her kind help with English language copy editing.
Disclosure
The authors have nothing to declare in relation to the present work. This research did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
References
Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333–1346. | |
Basu A, Caumo A, Bettini F, et al. Impaired basal glucose effectiveness in NIDDM: contribution of defects in glucose disappearance and production, measured using an optimized minimal model independent protocol. Diabetes. 1997;46:421–432. | |
Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–855. | |
Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology (Bethesda). 2006;21:48–60. | |
Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–1078. | |
Scott J, Norman D, Hawley S, Kontogiannis L, Hardie D. Protein kinase substrate recognition studied using the recombinant catalytic domain of AMP-activated protein kinase and a model substrate. J Mol Biol. 2002;317:309–323. | |
Sakamoto K, McCarthy A, Smith D, et al. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 2005;24:1810–1820. | |
Xiao B, Sanders MJ, Underwood E, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. | |
Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem. 2006;281:31478–31485. | |
Cartee GD, Wojtaszewski JF. Role of Akt substrate of 160 kDa in insulin-stimulated and contraction-stimulated glucose transport. Appl Physiol Nutr Metab. 2007;32:557–566. | |
Chen S, Wasserman DH, MacKintosh C, Sakamoto K. Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell Metab. 2011;13:68–79. | |
Ramm G, Larance M, Guilhaus M, James DE. A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem. 2006;281:29174–29180. | |
Sano H, Kane S, Sano E, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. | |
Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. 2008;295:E29–E37. | |
Kramer HF, Witczak CA, Fujii N, et al. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes. 2006;55:2067–2076. | |
Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. | |
Foretz M, Hébrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. | |
Musi N, Hirshman MF, Nygren J, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51:2074–2081. | |
DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM. A balanced overview. Diabetes Care. 1992;15:318–368. | |
Turner N, Kowalski GM, Leslie SJ, et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia. 2013;56:1638–1648. | |
Kim YB, Ciaraldi TP, Kong A, et al. Troglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases p110beta protein levels in skeletal muscle of type 2 diabetic subjects. Diabetes. 2002;51:443–448. | |
Sriwijitkamol A, Coletta DK, Wajcberg E, et al. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. 2007;56:836–848. | |
Jing M, Cheruvu VK, Ismail-Beigi F. Stimulation of glucose transport in response to activation of distinct AMPK signaling pathways. Am J Physiol Cell Physiol. 2008;295:C1071–C1082. | |
Goodyear LJ, Giorgino F, Balon TW, Condorelli G, Smith RJ. Effects of contractile activity on tyrosine phosphoproteins and PI 3-kinase activity in rat skeletal muscle. Am J Physiol. 1995;268:E987–E995. | |
Lee AD, Hansen PA, Holloszy JO. Wortmannin inhibits insulin-stimulated but not contraction-stimulated glucose transport activity in skeletal muscle. FEBS Lett. 1995;361:51–54. | |
Lund S, Holman GD, Schmitz O, Pedersen, O. Contraction stimulates translocation of glucose transporter GLUT4 in skeletal muscle through a mechanism distinct from that of insulin. Proc Natl Acad Sci U S A. 1995;92:5817–5821. | |
Yeh JI, Gulve EA, Rameh L, Birnbaum MJ. The effects of wortmannin on rat skeletal muscle. J Biol Chem. 1995;270:2107–2111. | |
Sakamoto K, Arnolds DE, Fujii N, Kramer HF, Hirshman MF, Goodyear LJ. Role of Akt2 in contraction-stimulated cell signaling and glucose uptake in skeletal muscle. Am J Physiol Endocrinol Metab. 2006;291:E1031–E1037. | |
Cartee GD, Douen AG, Ramlal T, Klip A, Holloszy JO. Stimulation of glucose transport in skeletal muscle by hypoxia. J Appl Physiol. 1991;70:1593–1600. | |
Azevedo JL Jr, Carey JO, Pories WJ, Morris PG, Dohm GL. Hypoxia stimulates glucose transport in insulin-resistant human skeletal muscle. Diabetes. 1995;44:695–698. | |
Bergeron R, Russell RR 3rd, Young LH, et al. Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol. 1999;276:E938–E944. | |
Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998;47:1369–1373. | |
Chakraborty A, Koldobskiy MA, Bello NT, et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell. 2010;143:897–910. | |
Musi N, Fujii N, Hirshman MF, et al. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes. 2001;50:921–927. | |
DeFronzo RA. Pathogenesis of type 2 (non-insulin dependent) diabetes mellitus: a balanced overview. Diabetologia. 1992;35:389–397. | |
Højlund K, Mustard KJ, Staehr P, et al. AMPK activity and isoform protein expression are similar in muscle of obese subjects with and without type 2 diabetes. Am J Physiol Endocrinol Metab. 2004;286:E239–E244. | |
McBride A, Ghilagabe S, Nikolaev A, Hardie DG. The glycogen-binding domain on the AMPK ≤ subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009;9:23–34. | |
McBride A, Ghilagaber S, Nikolaev A, Hardie DG. Physiological role of AMP-activated protein kinase (AMPK): insights from knockout mouse models. Biochem Soc Trans. 2003;31:216–219. | |
Mu J, Brozinick JT Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–1094. | |
Fujii N, Ho RC, Manabe Y, et al. Ablation of AMP-activated protein kinase alpha2 activity exacerbates insulin resistance induced by high-fat feeding of mice. Diabetes. 2008;57:2958–2966. | |
Jensen TE, Wojtaszewski JFP, Richter EA. AMP-activated protein kinase in contraction regulation of skeletal muscle metabolism: necessary and/or sufficient? Acta Physiol (Oxf). 2009;196:155–174. | |
Hunter RW, Treebak JT, Wojtaszewski JF, Sakamoto, K. Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes. 2011;60:766–774. | |
Kennedy JW, Hirshman MF, Gervino EV, et al. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes. 1999;48:1192–1197. | |
Gonzalez E, McGraw TE. Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling. Proc Natl Sci Acad U S A. 2009;106:7004–7009. | |
Manning BD. Insulin signaling: inositol phosphates get into the Akt. Cell. 2010;143(6):861–863. | |
Stein SC, Woods A, Jone NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J. 2000;345:437–443. | |
Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-[beta] is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. | |
Faissner A, Heck N, Dobbertin A, Garwood J. DSD-1-proteoglycan/phosphacan and receptor protein tyrosine phosphatase-beta isoforms during development and regeneration of neural tissues. Adv Exp Med Biol. 2006;557:25–53. | |
Lai KM, Gonzalez M, Poueymirou WT, et al. Conditional activation of AKT in adult skeletal muscle induces rapid hypertrophy. Mol Cell Biol. 2004;24:9295–9304. | |
Garofalo RS, Orena SJ, Rafidi K, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. | |
Easton RM, Cho H, Roovers K, et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. | |
Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. | |
Howlett KF, Sakamoto K, Garnham A, Cameron-Smith D, Hargreave M. Resistance exercise and insulin regulate AS160 and interaction with 14-3-3 in human skeletal muscle. Diabetes. 2007;56:1608–1614. | |
Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–31378. | |
Lu M, Wan M, Leavens KF, et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18:388–395. | |
Zick Y. Insulin resistance: a phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol. 2001;11:437–441. | |
Tremblay F, Lavigne C, Jacques H, Marette A. Defective insulin-induced GLUT4 translocation in skeletal muscle of high fat-fed rats is associated with alterations in both Akt/protein kinase B and atypical protein kinase C activities. Diabetes. 2001;50:1901–1910. | |
Karlsson HK, Zierath JR, Kane S, Krook A, Lienhard GE, Wallberg-Henriksson H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes. 2005;54:1692–1697. | |
Cho H, Mu J, Kim JK, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science. 2001;292:1728–1731. | |
George S, Rochford JJ, Wolfrum C, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–1328. | |
Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005;30:35–42. | |
Calleja V, Alcor D, Laguerre M, et al. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007;5:e95. | |
Werner JK Jr, Speed T, Bhandari R. Protein pyrophosphorylation by diphosphoinositol pentakisphosphate (InsP7). Methods Mol Biol. 2009;645:87–102. | |
Barker CJ, Illies C, Gaboardi GC, Berggren PO. Inositol pyrophosphates: structure, enzymology and function. Cell Mol Life Sci. 2009;66:3851–3871. | |
Nagata E, Luo HR, Saiardi A, Bae BI, Suzuki N, Snyder SH. Inositol hexakisphosphate kinase-2, a physiologic mediator of cell death. J Biol Chem. 2005;280:1634–1640. | |
Saiardi A, Nagata E, Luo HR, Snowman AM, Snyder SH. Identification and characterization of a novel inositol hexakisphosphate kinase. J Biol Chem. 2001;276:39179–39185. | |
Weiss EP, Racette SB, Villareal DT, et al. Improvements in glucose tolerance and insulin action induced by increasing energy expenditure or decreasing energy intake: a randomized controlled trial. Am J Clin Nutr. 2006;84:1033–1042. | |
Padmanabhan U, Dollins DE, Fridy PC, York JD, Downes CP. Characterization of a selective inhibitor of inositol hexakisphosphate kinases: use in defining biological roles and metabolic relationships of inositol pyrophosphates. J Biol Chem. 2009;284:10571–10582. | |
Bhandari R, Juluri KR, Resnick AC, Snyder SH. Gene deletion of inositol hexakisphosphate kinase 1 reveals inositol pyrophosphate regulation of insulin secretion, growth, and spermiogenesis. Proc Natl Acad Sci U S A. 2008;105:2349–2353. | |
Reynolds T 4th, Brozinick JT, Rogers MA, Cushman SW. Mechanism of hypoxia-stimulated glucose transport in rat skeletal muscle: potential role of glycogen. Am J Physiol. 1998;274:E773–E778. | |
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. | |
Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–377. | |
Kaidanovich O, Eldar-Finkelman H. The role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Expert Opin Ther Targets. 2002;6:555–561. | |
Peng XD, Xu PZ, Chen ML, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–1365. | |
Elchebly M, Payette P, Michaliszyn E, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. | |
Illies C, Gromada J, Fiume R, et al. Requirement of inositol pyrophosphates for full exocytotic capacity in pancreatic beta cells. Science. 2007;318:1299–1302. | |
Utzschneider KM, Prigeon RL, Carr DB, et al. Impact of differences in fasting glucose and glucose tolerance on the hyperbolic relationship between insulin sensitivity and insulin responses. Diabetes Care. 2006;29:356–362. | |
Ross SE, Hemati N, Longo KA, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–953. | |
Izumiya Y, Hopkins T, Morris C, et al. Fast/glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 2008;7:159–172. | |
Stål O, Pérez-Tenorio G, Akerberg L, et al. Akt kinases in breast cancer and the results of adjuvant therapy. Breast Cancer Res. 2003;5:R37–R44. | |
Stahl JM, Sharma A, Cheung M, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–7010. | |
Park HU, Suy S, Danner M, et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol Cancer Ther. 2009;8:733–741. | |
Decensi A, Puntoni M, Goodwin P, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila). 2010;3:1451–1461. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.