")
Back to Journals » OncoTargets and Therapy » Volume 12
Akt/mTOR-Mediated Autophagy Confers Resistance To BET Inhibitor JQ1 In Ovarian Cancer
Authors Luan W, Pang Y, Li R, Wei X, Jiao X, Shi J , Yu J , Mao H, Liu P
Received 21 June 2019
Accepted for publication 18 September 2019
Published 3 October 2019 Volume 2019:12 Pages 8063—8074
DOI https://doi.org/10.2147/OTT.S220267
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Wenqing Luan,1 Yingxin Pang,1 Rui Li,1 Xuan Wei,1 Xiaoxiao Jiao,2 Juanjuan Shi,1 Jiangtao Yu,1 Hongluan Mao,1 Peishu Liu1
1Department of Obstetrics and Gynecology, Qilu Hospital of Shandong University, Jinan 250012, People’s Republic of China; 2Department of Gastroenterology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, People’s Republic of China
Correspondence: Peishu Liu; Hongluan Mao
Department of Obstetrics and Gynecology, Qilu Hospital of Shandong University, 107 Wenhua Xi Road, Jinan 250012, People’s Republic of China
Tel +86-531-82169570
Fax +86-531-86927544
Email [email protected]; [email protected]
Background: Bromodomain and extra-terminal domain inhibitors like JQ1 have proved to be promising epigenetic agents for the treatment of malignant ovarian carcinoma. However, the resistance of ovarian cancer cells to BET inhibitors has not been elucidated. In this study, we investigated the potential mechanisms underlying the resistance of ovarian cancer cell lines to the BET inhibitor JQ1.
Materials and methods: We evaluated the apoptotic and proliferative response of four ovarian cancer cell lines to JQ1. The cell lines were designated as resistant (A2780 and HO-8910) and sensitive groups (SKOV-3 and HEY). Further experiments detected the different levels of JQ1-induced autophagy. Anti-tumour effect of the combination of JQ1 and autophagy inhibitors was tested both in vitro and in vivo.
Results: In the JQ1-sensitive group, JQ1 effectively inhibited proliferation and apoptosis in a concentration-dependent manner. Conversely, JQ1 showed modest inhibition of proliferation and negligible apoptosis in the resistant group. We detected increased LC3-II lipidation, autophagosome formation, upregulation of Beclin-1 and ATG5, and downregulation of P62/SQSTM1 in the resistant group. Inhibition of JQ1-induced autophagy by pharmacologic inhibitors 3-MA and CQ enhanced the inhibition of proliferation and significantly increased the apoptosis in the JQ1-resistant group, which was also verified by in vivo experiments, indicating that JQ1-induced autophagy played a cytoprotective role. Inactivation of Akt (Ser473)/mTOR(Ser2448) pathway was associated with JQ1-induced autophagy in the resistant group. Overexpression of Akt1 suppressed autophagy and increased the anti-tumour effect of JQ1.
Conclusion: These findings revealed that JQ1-induced pro-survival autophagy might be a potential mechanism in the resistance of ovarian cancer cells to BET inhibition by JQ1. Combination of JQ1 and autophagy inhibitors could be an effective therapeutic strategy for overcoming BET inhibitor resistance in ovarian cancer.
Keywords: BET inhibitor, ovarian cancer, drug resistance, autophagy, Akt/mTOR pathway
A corrigendum has been published for this article
Introduction
Epithelial ovarian cancer (EOC) is the leading cause of gynaecological cancer-associated death worldwide, partly because it is detected often at an advanced stage.1 Although carboplatin and paclitaxel chemotherapies are first-line treatment and the initial response rate is high (∼80%), most patients eventually recur, and mortality occurs within 5-years,2 and patients with high-grade serous carcinoma (HGSC) have shorter survival.3
BET bromodomain protein BRD4 has recently emerged as an exciting new class of target for the treatment of cancer, as BRD4 overexpression can enhance transcription of critical oncogenes, for example, MYC.4,5 Consequently, BET bromodomain inhibitors (BETi) were developed. Recent studies have shown that inhibition of BRD4 could reduce the expression of oncogenes and result in tumour regression.6 Therefore, clinical trials of BETi are in progress for many cancers.7 Although BETi showed great promise as cancer therapeutics, the anti-cancer effects of BETi were quite variable.8 Besides, emerging evidence showed that cancer cells acquire resistance to BETi, indicating single agent targeting of BRD4 may not produce ideal therapeutic response.9
JQ1, a selective BET inhibitor that mimics the acetyl moiety and occludes the acetyl-lysine binding pocket, thus replacing BET proteins from chromatin and shown to be highly effective against epithelial ovarian cancer.10 Recently JQ1 was shown to induce cell cycle arrest and apoptosis in EOC cells.11 JQ1 also suppresses PD-L1 expression in both immune and tumour cells to promote anti-tumour immunity.12 Another study showed that JQ1 reduces homologous recombination(HR) and enhances PARP inhibitor-induced DNA damage.13 Thus, JQ1 appears to be a promising therapeutic option for targeting EOC.
However, EOC cell lines exhibit different proliferative and apoptotic responses to JQ1.9 The resistance of some EOC cell lines to JQ1 cannot be explained by differences in basal expression of BRD4 or changes in the levels of c-Myc and BRD4.14,15 These findings suggested that primary resistance to JQ1 may not describe its inability to suppress c-Myc or different expression levels of basal BRD4, but could be due to compensatory mechanisms triggered by c-Myc inhibition. A recently published study suggested that resistance to BET inhibitors in EOC is mediated by adaptive kinome reprogramming,9 where activation of compensatory pro-survival kinase networks overcomes BET protein inhibition. However, the mechanisms underlying different sensitivities of EOC cell lines to JQ1 remain elusive. A better understanding of the mechanisms involved in resistance of EOC cell lines to JQ1 is needed to combat BET inhibitor resistance in EOC.
Autophagy was defined initially as a self-digestion process by which cytoplasmic contents were sequestered in autophagosomes and delivered to lysosomes for degradation.16,17 Autophagy plays a dual role both as pro-death or pro-survival in malignant tumour treatment, largely depends on the tumour type and treatment characteristics.18 Many cellular pathways like the Akt/mTOR pathway and AMPK/ULK1 pathway have been reported to be involved in autophagy initiation.19–21 Autophagy protects MDR (multi-drug resistant) cancer cells from apoptosis and promotes resistance to chemotherapy treatment. Inhibition of autophagy may sensitise MDR cells to anticancer drugs.22 It represents a new battle-line in the fight against drug resistance. Accordingly, it is vital to have a better understanding of the roles and mechanisms of autophagy and key signalling pathways involved in cancer resistance for targeting autophagy as an approach to overcome drug resistance.
In the present study, we investigated the mechanisms underlying the resistance of EOC cell lines to JQ1. We showed that JQ1-resistant A2780 and HO-8910 cell lines acquired resistance to JQ1 through induction of autophagy for the first time. Autophagy in JQ1-resistant EOC cells was mediated by inactivation of the Akt/mTOR pathway, Blocking JQ1-induced autophagy enhanced JQ1 anti-tumour effect on EOC proliferation and survival both in vitro and in vivo.
Statistical Analysis
All experiments were repeated at least three times for statistical analysis. All data were analysed using GraphPad Prism version 7.0. The tumour growth curve data were presented as mean ± standard error of the mean (SEM); other results were shown as mean ± standard deviation (SD). Unpaired Student’s t-test determined statistical differences between two independent groups. P < 0.05 was considered significant.
Results
Effect Of JQ1 On Ovarian Cancer Cell Lines
Initially, we examined the effect of JQ1 on proliferation and apoptosis of four ovarian cancer cell lines A2780, HO-8910, SKOV-3, and HEY. The four cell lines were incubated for 48 hrs, with varying concentrations of JQ1. As shown in Figure 1A, JQ1 inhibited proliferation of the four cell lines in a concentration-dependent manner. However, the sensitivity of four cell lines to JQ1 was different, since the IC50 values of A2780 and HO-8910 (6.963 and 5.18 μmol/L) were higher than SKOV-3 and HEY (1.503 and 0.503 μmol/L), suggesting A2780 and HO-8910 cell lines are more resistant to JQ1. The OC cell lines were stained with PI and Annexin V to detect the total apoptotic cell populations. As shown in Figure 1B and C, after 48 hrs incubation with JQ1, the apoptotic response to JQ1 was different in the four cell lines. In SKOV-3 and HEY cells, total apoptotic cell population increased significantly in a dose-dependent manner. While apoptotic cell death was increased modestly in A2780, and HO-8910 cell lines, this means A2780, and HO-8910 was indeed more resistant to JQ1 compared with other two OC cell lines. For analysis, four OC cell lines were classified into JQ1-sensitive (n=2) and JQ1-resistant (n=2) groups based on IC50 and the median value of apoptosis. Between the sensitive group and resistant group, the total apoptotic cell population showed a significant difference (P<0.01).
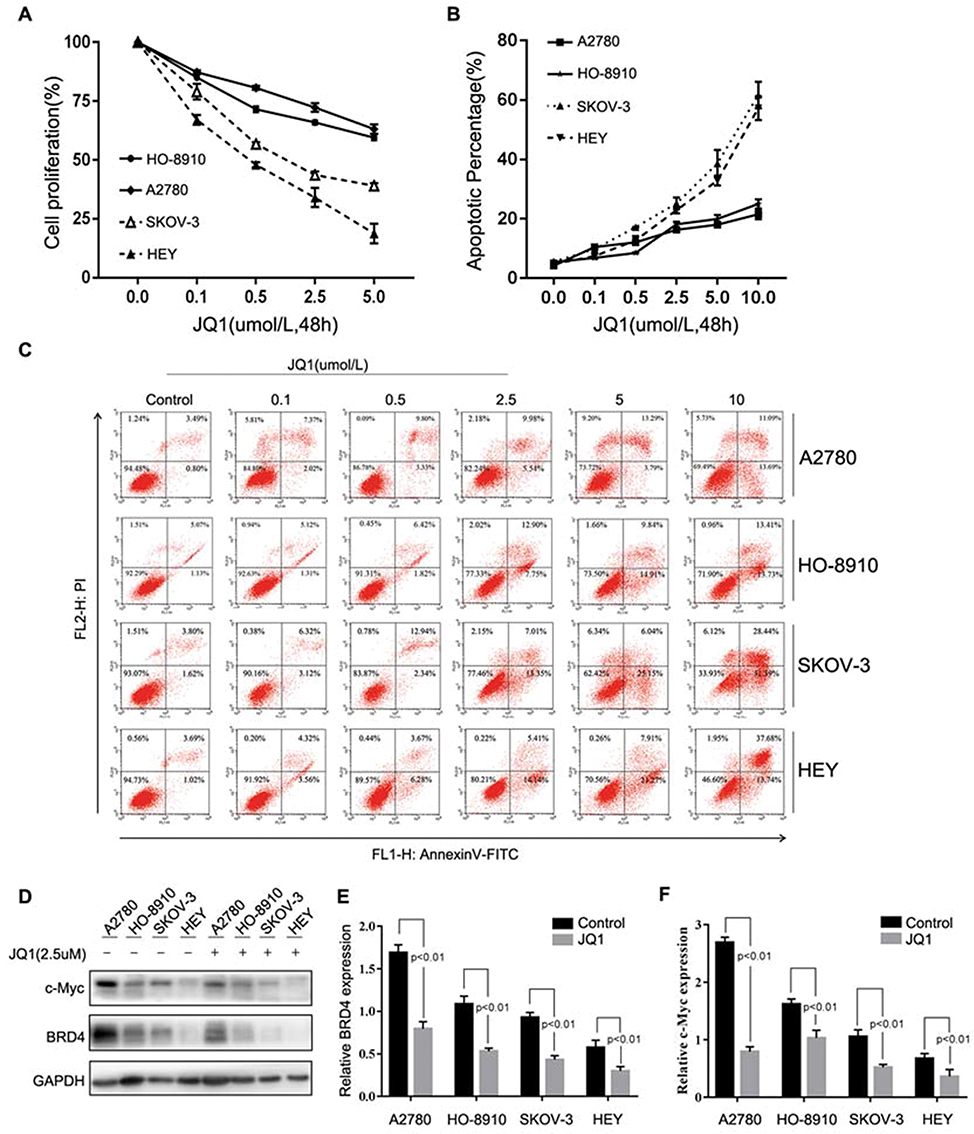 |
Figure 1 Differential sensitivity of human Ovarian Cancer cell lines to JQ1. (A-F) OC cell lines A2780, HO-8910, SKOV-3, and HEY were incubated with different concentrations of JQ1 for 48 h. (A) After treatment, OC cell proliferation was measured by MTT assay. IC50 values were calculated by GraphPad Prism version, 7.0 software. (B and C) Annexin V and PI staining and flow cytometric analysis of the fraction of apoptotic cells in OC cell lines. (D) After treatment of OC cell lines with 2.5 μM JQ1 for 48 h, cell lysates were analysed for c-Myc and BRD4 expression by Western blotting. GAPDH was used as a protein loading control. (E and F) The relative expression of c-Myc and BRD4 were calculated by ImageJ software. P < 0.05 vs control group. |
Because the basal expression of BRD4 and the suppression of MYC were the critical determinants of BET bromodomain inhibitor sensitivity in another malignant tumour,23,24 then we detected the expression of BRD4 and c-Myc and the suppression of BRD4 and c-Myc in the presence of JQ1. As is shown in Figure 1D–F, the different expression of BRD4 and c-Myc had no significance in four cell lines, and the expression of BRD4 and c-MYC were all suppressed in the presence of JQ1. These results indicated other potential mechanisms were mediating the sensitivity of OC cells to JQ1.
JQ1-Induced Autophagy In JQ1-Resistant OC Cell Lines
Autophagy plays an important role in the regulation of drug resistance,18 and we investigated whether JQ1 induces autophagy in resistant OC cell lines. Because conversion of cytosolic LC3-I to LC3-II through lipidation by a ubiquitin-like system is a classical marker of autophagosome formation,16 we first evaluated JQ1 effect on the conversion of LC3-I to lipidated LC3-II in four cell lines. As is shown in Figure 2A, LC3 conversion did not occur in SKOV-3 and HEY, OC cell lines. In contrast, increased LC3-II conversion in A2780 and HO-8910 cells was observed after treatment with increasing concentrations of JQ1 (Figure 2A and B). Furthermore, we investigated the expression of SQSTM1/P62, Beclin1, and ATG5 in OC cell lines. Our results showed that treatment with JQ1 led to increased expression of ATG5 and Beclin1, and decreased SQSTM1/P62 in JQ1 resistant OC cell lines. In contrast, the autophagy molecular marker expression changed modestly after JQ1 treatment in JQ1-sensitive cell lines (Figure 2B). Besides, the expression of BRD4 decreased in the four cell lines, showing the effectiveness of JQ1 in this experiment. To further test for JQ1-induced autophagy in the resistant group, we analysed the distribution of endogenous LC3 puncta, another classical marker of autophagosome formation.25 Figure 2C shows a marked increase by JQ1 of the endogenous LC3 puncta in the JQ1-resistant group compared to DMSO-treated control cells. Since the presence of acidic vesicle organelles (AVO) is another characteristic of autophagy, we used acridine orange stain to detect AVO. Figure 2D shows, abundant cytoplasmic AVO in JQ1-resistant group compared to those treated with DMSO.
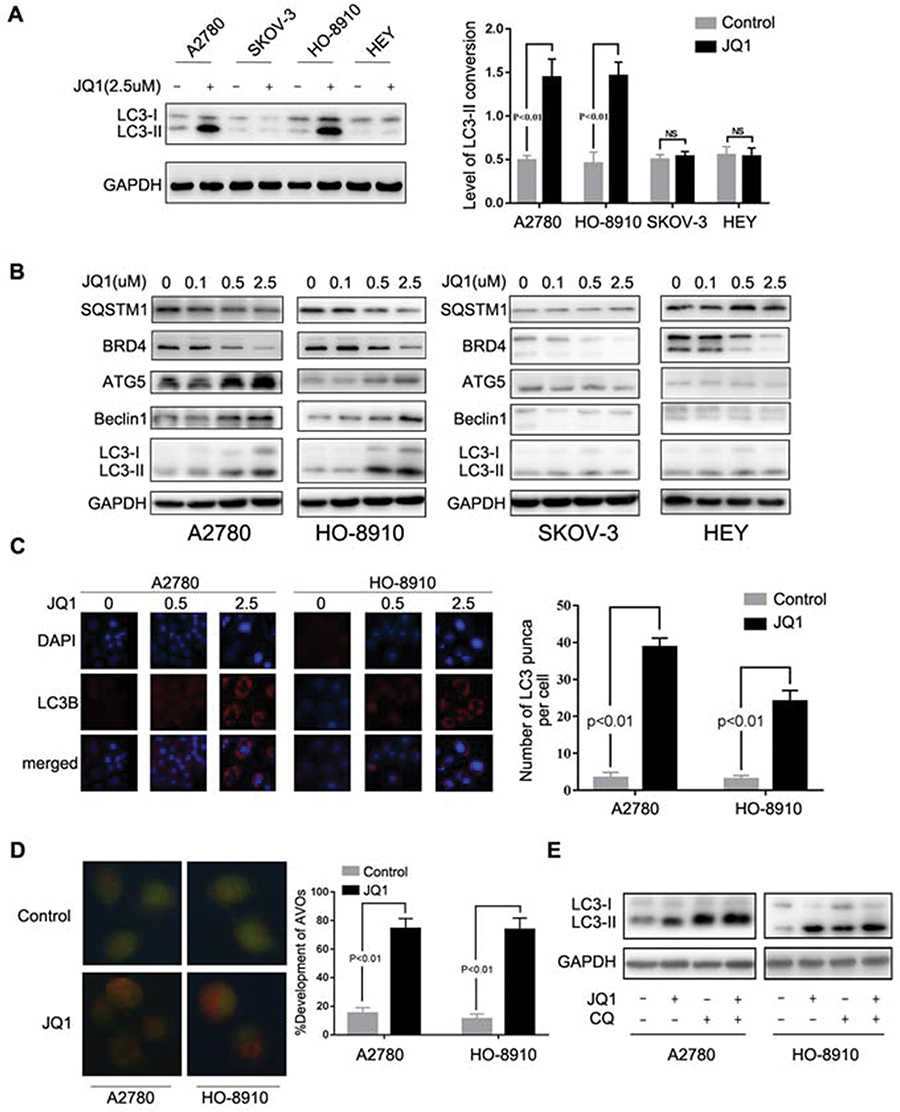 |
Figure 2 JQ1-induced autophagy in the resistant group. (A) After treatment of A2780, HO-8910, SKOV-3, and HEY cells with 2.5 μM JQ1 for 24 h, cell lysates were subjected to Western blotting for LC3-I/II expression. GAPDH was used as a protein loading control. Relative expression of LC3- II was detected by ImageJ software. P < 0.05 vs control group. (B) Ovarian Cancer cell lines were incubated with increased concentrations of JQ1 for 48 h, and cell lysates were subjected to Western blotting using antibodies against the indicated molecules. (C) Immunofluorescence staining of the endogenous expression of LC3B in A2780 and HO-8910 cells after treatment with 2.5 μM JQ1 for 48 h. Confocal images were obtained of the immunofluorescent LC3B (red) and nuclei stained with DAPI (blue). (magnification 60×oil). LC3 dots in each cell were enumerated in at least three distinct visual fields. P < 0.05 vs control group. (D) Cells were treated with DMSO or JQ1 (2.5 μM) for 48 h and stained by acridine orange to observe the formation of acidic vesicle organelles (AVO). P < 0.05 vs control group. (E) After treatment with JQ1 (2.5 μM) and CQ (5 μM), expression of LC3-I/II was detected by Western blotting. |
Reports show LC3-II conversion and the accumulation of LC3 puncta also occur when autophagosome turnover is inhibited in late stages, and suggest that the entire autophagy process was not complete.26 Therefore, we next investigated whether JQ1-induced autophagy was responsible for activation of autophagic flux rather than blocking the degradation of autolysosome. Figure 2E shows that co-treatment with JQ1 and CQ (an autophagy late-stage inhibitor that inhibits lysosome fusion with autophagosome) increased the level of LC3-II conversion compared to CQ treatment alone. SQSTM1/P62 was an indicator of autophagic flux, and the expression of SQSTM1/P62 also decreased after treatment with JQ1 (Figure 2B). Our data demonstrated that JQ1-induced autophagy was responsible for activation of autophagic flux rather than inhibiting degradation of autolysosome.
Altogether, these results demonstrate that JQ1-induced autophagy in JQ1-resistant OC cell lines, while this effect was absent in JQ1-sensitive group.
Inhibition Of JQ1-Induced Autophagy Enhances Anti-Proliferative Activities And Promotes Cell Apoptosis In JQ1-Resistant Group
Accumulated evidence shows the involvement of autophagy in the context of anticancer therapy and its roles as cytoprotective, non-protective, cytotoxic, and cytostatic.18 Therefore, we determined whether autophagy mediates cellular resistance to JQ1. First, we evaluated the relationship between JQ1-induced autophagy and growth inhibition in the resistant group. As shown in Figure 3A, the induction of autophagy by JQ1 in A2780 and HO-8910 OC cell lines was blocked by autophagic inhibitor 3-MA. In addition, cell growth was evaluated by colony formation assays, to exclude the possibility that the above effect was due to the additive effects of JQ1 and 3-MA or CQ (Figure 3B). Next, we evaluated the effect of the combination of autophagy inhibitors 3-MA (5 µmol/L) and CQ (5 µmol/L) on cell proliferation and growth. MTT assays showed that inhibiting autophagy by 3-MA and CQ significantly suppressed the growth of JQ1-resistant group compared to those treated with JQ1 alone (Figure 3C). Because the JQ1 concentration used in this assay has no apparent effect on cell growth as seen in colony formation assay, the results showed that 3-MA or CQ treatment enhanced the effect of JQ1 and reversed JQ1 resistance in the JQ1-resistant group. Collectively, our data suggest that inhibition of JQ1-autophagy enhances JQ1 promoted cell apoptosis.
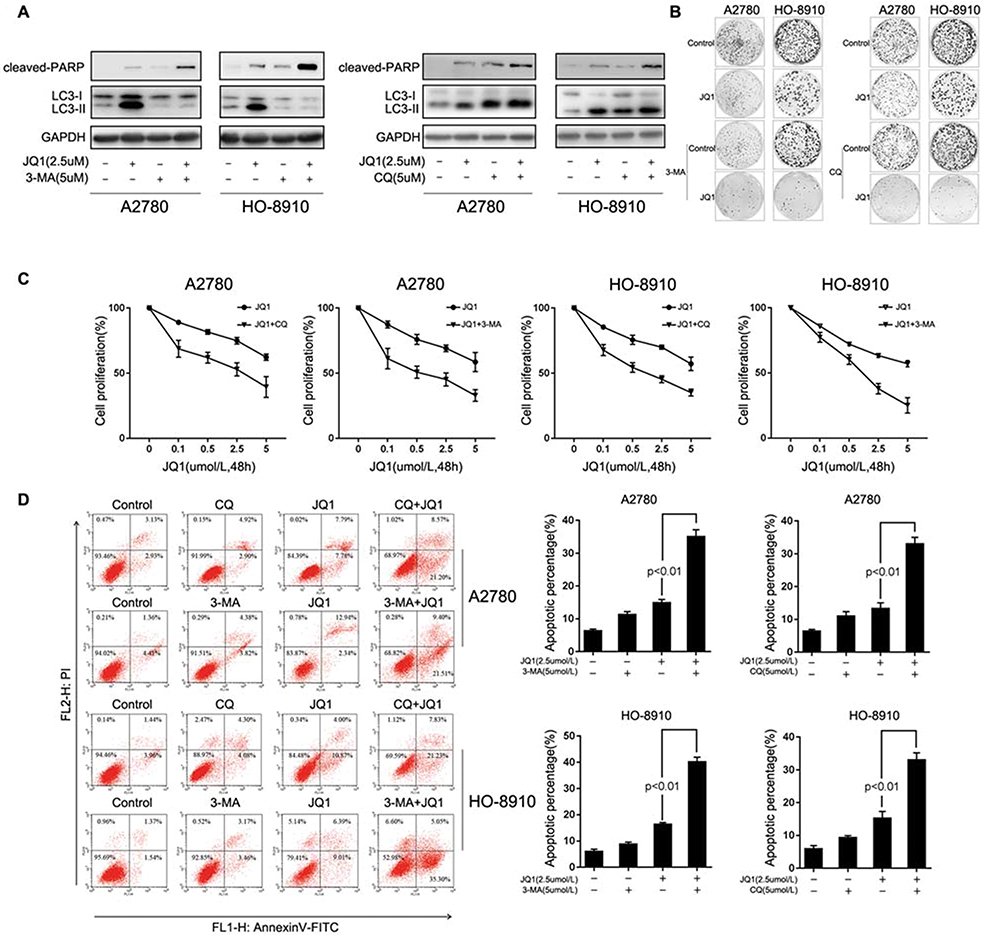 |
Figure 3 Inhibition of JQ1-induced autophagy enhances anti-proliferative activities and promotes cell apoptosis in resistant ovarian cancer cells. (A) A2780 and HO-8910 cells were treated with 2.5 μM JQ1, 5 μM 3-MA, or 5 μM CQ or a combination of JQ1 with 3-MA or CQ for 48 h, and measured the expression of the apoptosis marker cleaved PARP p85 and autophagy marker LC3-I/II. (B) A2780 and HO-8910 cells were plated in 6-well plates and treated with 2.5 μM JQ1, 5 μM 3-MA or 5 μM CQ, or a combination of both JQ1 and autophagy inhibitors for 14 days in a colony formation assay. (C) After co-treatment with increased concentrations of JQ1 and 5 μM 3-MA or 5 μM CQ, MTT assay was used to detect the anti-proliferation effect of JQ1 on resistant OC cells. IC50 was calculated by GraphPad Prism software. (D) A2780 and HO-8910 cells were treated with 2.5 μM JQ1, 5 μM 3-MA, or 5 μM CQ or a combination for 48 h and quantified for apoptosis by fluorescence-activated cell sorting (FACS) based on annexin V staining. P < 0.05 vs JQ1-alone treated group. |
Next, we examined whether JQ1-induced autophagy contributed to survival in JQ1 resistant group by inhibiting autophagy with 3-MA and CQ. The addition of 3-MA or CQ to JQ1 resulted in a significant increase in the level of apoptosis in JQ1-resistant group (Figure 3D) compared with JQ1 treatment alone. As expected, no significant differences in JQ1-induced apoptosis following co-treatment with 3-MA or CQ was observed in JQ1-sensitive group. Because cleavage of PARP by cleaved caspase-3 is an indicator of apoptosis, we evaluated changes in the levels of these markers of apoptosis using Western blot analysis after treatment with JQ1 in the presence or absence of 3-MA and CQ. Figure 3A shows, after treatment with JQ1 or autophagy inhibitor alone for 48 hrs in JQ1-resistant group, cleaved PARP was marginally detectable. However, the combination of JQ1 and 3-MA or CQ increased the expression level of cleaved PARP in JQ1-resistant group, indicating that the observed synergy is due to induction of apoptosis by the combination treatment. Activation of JQ1-induced autophagy mediated resistance to apoptotic cell death. We also detected the expression of cleaved-PARP in JQ1-sensitive group under similar condition. Figure S1A shows that after treatment with JQ1, the expression of cleaved-PARP increased compared to the control group or autophagy inhibitor alone group. Besides, the combination of JQ1 with autophagy inhibitor did not increase the expression of cleaved-PARP, suggesting that JQ1-induced autophagy in the resistant group could partly explain the different response to JQ1. Thus, we conclude that JQ1 and autophagy inhibitors are synergistic in suppressing the growth of JQ1 resistant cell lines by inducing apoptotic cell death.
Inactivation Of The Akt/mTOR Pathway Is Associated With JQ1-Induced Autophagy In The Resistant Group
The previous study showed a negative regulatory role of the Akt/mTOR pathway in autophagy.19 Therefore we investigated whether Akt/mTOR pathway is involved in JQ1-induced autophagy in the resistant group. First, Western blot was used to examine the phosphorylation status of Akt, mTOR, and p70S6K (a characterised target of the mTOR1 complex). Figure 4A shows inhibition of the Akt/mTOR pathway in JQ1 resistant OC cell lines after treatment with JQ1 and confirmed by decreased phosphorylation levels of Akt, mTOR, and p70S6K. We also tested whether JQ1 shows a similar effect on Akt/mTOR pathway in JQ1 sensitive group. As shown in Figure S1B, JQ1 did not show the same effect on Akt/mTOR pathway in JQ1 sensitive group. Next, we rescued JQ1-induced Akt/mTOR inhibition by overexpressing Akt1 cDNA and observed decreased LC3-II conversion and endogenous LC3 puncta accumulation in JQ1 treated resistant cells (Figure 4B and C). Figure S1C showed that after transfection with AKT1 plasmid, an increase in the level of p-Akt and activation of Akt/mTOR pathway. In contrast, co-treatment of JQ1-resistant cells with LY294002 (an inhibitor of PI3K/Akt pathway) and JQ1 significantly increased the level of LC3-II conversion in JQ1 treated resistant cells (Figure 4D). Overexpression of Akt1 also rescued the anti-proliferative effect of JQ1 in A2780, and HO-8910 cells since the level of apoptosis increased significantly in Akt1 overexpressed cells after treatment with JQ1 (Figure 4E). While transfected with Akt1 plasmid had no such effect in sensitive group (Figure S1D and E). These results showed an association of JQ1-induced autophagy in resistant cells with inactivation of the Akt/mTOR pathway in resistant OC cell lines.
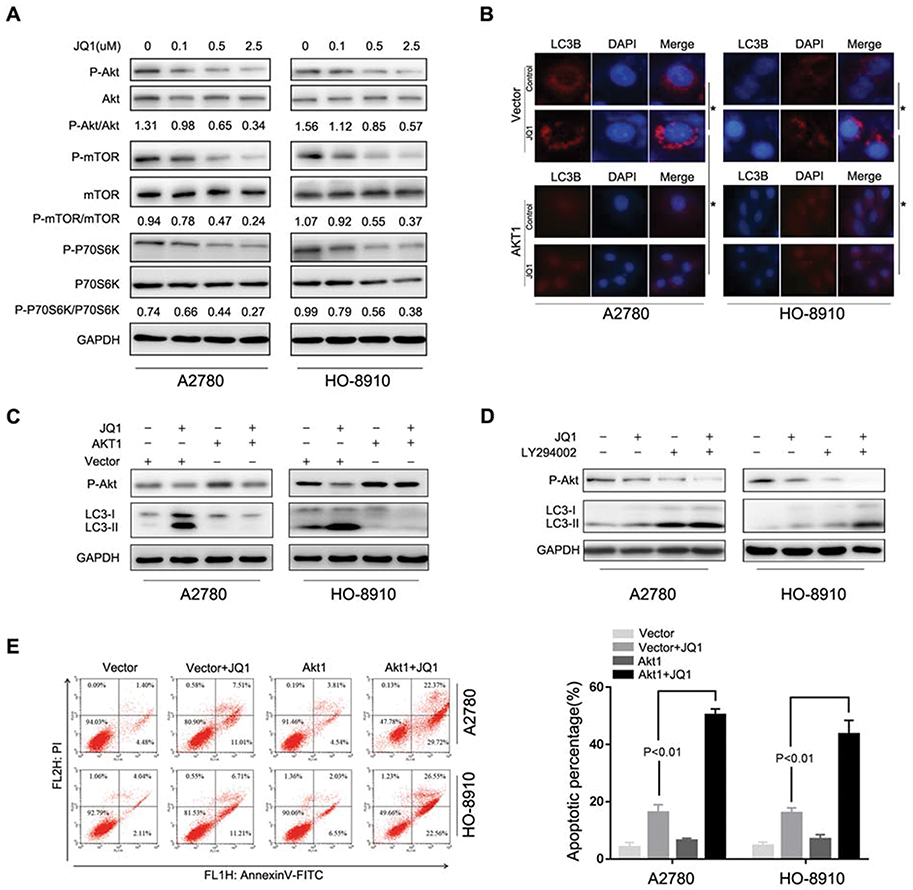 |
Figure 4 JQ1 induces autophagy through the inhibition of the Akt/mTOR pathway. (A) A2780 and HO-8910 cells were treated with the indicated concentration of JQ1 for 48 h. Cell lysates were used to detect Akt (S473), mTOR (S2448), P70S6K (S424/T421), and their phosphorylated counterparts by Western blot. (B and C) After treatment with 2.5 μM JQ1 in combination with transfection by Akt1 plasmid or control vector for 48 h. Immunofluorescence staining was used to detect the endogenous expression of LC3B, and Western blotting was used to identify the phosphorylated Akt (P-Akt), LC3- I, and LC3-II in A2780 and HO-8910 cells. *P < 0.05. (D) Cells were treated with or without JQ1 (2.5 μM) in combination with LY294002 (a PI3K/Akt inhibitor) for 48 h. Phosphorylated Akt (P-Akt), LC3- I, and LC3-II were detected by Western blot. P < 0.05 vs JQ1-alone treatment group. (E) Annexin V-FITC/PI staining were used to detect the level of apoptosis after treatment with JQ1 (2. μM) in combination with transfection by Akt1 plasmid or control vector for 48 h. P < 0.05 vs JQ1-alone treatment group. |
Combination Therapy With JQ1 And Chloroquine Enhances Antitumor Activities In Vivo
A2780 cells were implanted subcutaneously into immunodeficient (BALB/c Nu/Nu) mice, and the xenografted tumours were established over 15 days. The mice were randomised into four treatment groups: DMSO control, JQ1 (20 mg/kg), CQ (80 mg/kg), and combination of JQ1 (20 mg/kg) and CQ (80 mg/kg) treated groups respectively. The mice were treated at 3-day intervals and received a total of ten treatments and observed until 45 days. Figure 5 A shows the slowest growth in the group treated with JQ1 and CQ. The JQ1 (20 mg/kg) or CQ (80 mg/kg) treated groups showed a modest effect on ovarian tumour growth. Consistently, the tumour burden decreased to a significant extent, as measured by the weight of the dissected tumours (Figure 5B and C). However, there was no significant difference in body weight of nude mice among the different treatment groups, which proved that there was no overt toxicity of the combination treatment (Figure 5D). Our data demonstrate that blocking JQ1-induced autophagy by CQ in JQ1-resistant ovarian cancer xenograft animal model enhances the anti-tumour activity of JQ1. Autophagy inhibitor sensitises EOC tumours to the BET inhibitor JQ1 in vivo.
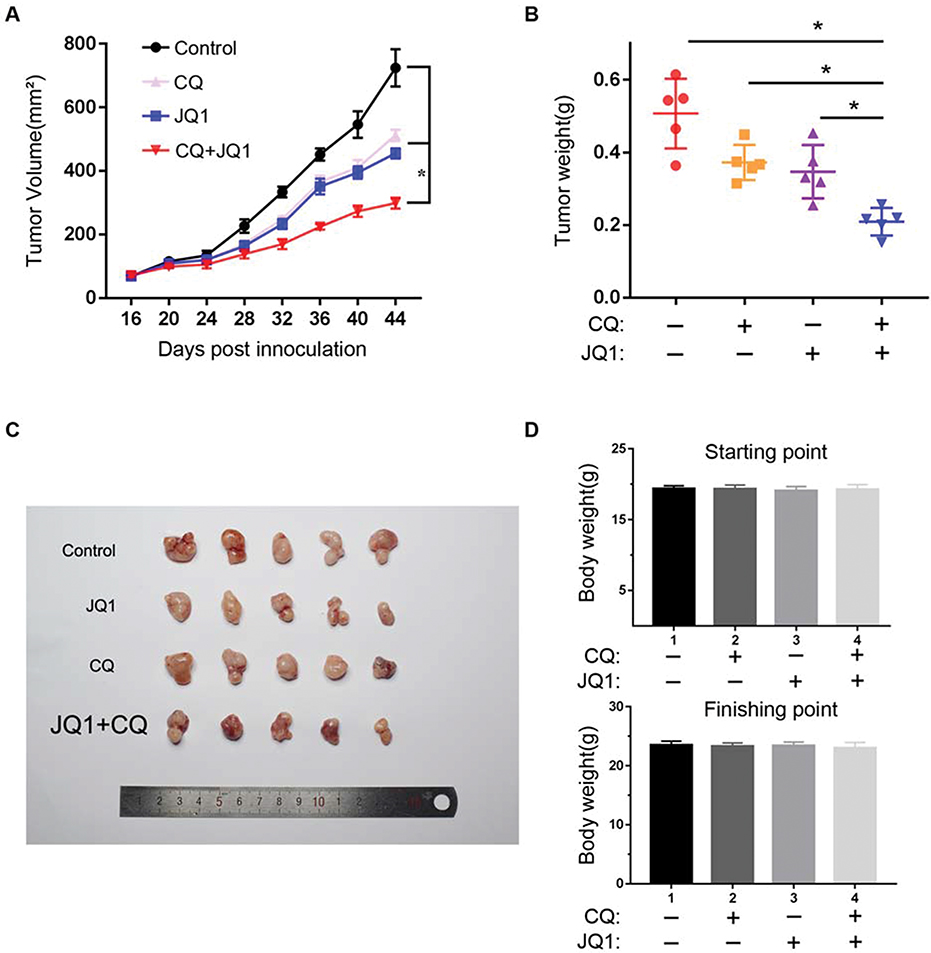 |
Figure 5 Autophagy inhibitor CQ synergises with BET inhibitor JQ1 in suppressing the growth of JQ1-resistant xenograft ovarian tumours in vivo. (A) A2780 cells were injected subcutaneously into the flank of NSG mice. The mice were randomised into four indicated treatment groups (n=5) after 2nd week and treated daily with vehicle control, 20 mg/kg JQ1, 80 mg/kg CQ, or in combination for 30 days. The tumour sizes were measured at the indicated time points. *P < 0.05. (B) Tumour weight was measured as a surrogate for tumour burden at the end of treatment. *P < 0.05. (C) Tumour mass images of A2780 model mice at day 45. P < 0.05 vs control group or JQ1-alone or CQ-alone treatment group. (D) Bodyweight of the mice from the indicated treatment groups at the starting and finishing points of the treatments. There is no statistical difference between the different treatment groups. Data are represented as mean with SEM (n = 5 mice/group). |
Discussion
Although members of the BET family, like BRD4, have been identified as potential therapeutic targets in ovarian carcinoma, and BET inhibitor JQ1 exhibits anti-ovarian cancer activity both in vitro and in vivo,6 emergence of drug resistance limits their clinical value.27 However, the underlying mechanisms of resistance to BET inhibitors remain poorly understood, and the means to optimise BET inhibitors clinical efficacy remains a considerable challenge. Our study showed, for the first time, the involvement of inactivated Akt/mTOR pathway in induction of autophagy, and conferring JQ1 resistance in ovarian cancer cells.
Recent studies have shown that resistance to BET inhibitors is mediated by kinome reprogramming in ovarian cancer.9 Specifically, the activation of receptor tyrosine kinases (RTKs) and downstream signalling by phosphatidylinositol 3-kinase (PI3K), Akt, and ERK mediate resistance to BET inhibitors. Santin AD pointed out that gain of c-Myc was a potential target for BET inhibitors and the inability of BET inhibitors in suppressing c-Myc might also be a mechanism of drug resistance.28 Huang H showed that stable expression of BRD4, an important member of BET family targeted by JQ1, mediates the resistance to BET inhibitors.23 These data suggested that compensatory activation of transcriptional pathways or inability to suppress c-Myc result in stable expression of c-Myc or dysregulation of BRD4, thus mediating drug resistance. Our results confirm the differential ovarian cells response to JQ1. A2780 and HO-8910 were defined as JQ1-resistant group and SKOV-3 and HEY as JQ1-sensitive group. Although we also observed considerable c-Myc and BRD4 downregulation in both resistant and sensitive groups, and the expression of altered basal BRD4 and c-Myc levels could not explain the different response to JQ1. Furthermore, JQ1 could not induce apoptosis in JQ1-resistant group, even at a 2-fold higher concentration than the IC50 (10 μM), whereas expression of BRD4 and c-Myc decreased simultaneously. These findings supported that in JQ1-resistant group, inducing BRD4 downregulation and c-Myc related synthetic lethality was ineffective for BET inhibitors.
Autophagy plays a complex and controversial role in tumorigenesis and treatment of malignant tumour.16 On the one hand, excessive autophagy can commit cancer cells to “autophagic cell death (ACD)” or “type II programmed cell death.” On the other hand, autophagy can play a protective role by eliminating damaged organelles and recycling degradation products in normal cells.18 Autophagy can be affected by various pathways. For example, AMPK pathway and AKT pathway can regulate the initiation of autophagy. During drug treatment, autophagy can protect cancer cells and attenuate drug-induced apoptosis and promote survival.29 Autophagy has been a mechanism of drug resistance in cancer therapy.22 Our findings indicated that JQ1 in ovarian cancer cell lines downregulated c-Myc. However, previous studies reported the induction of cytoprotective autophagy by c-Myc. Hence c-Myc inhibition should decrease autophagy in cancer cells.30 Consistent with this finding, we found that in the JQ1-sensitive group, c-Myc was downregulated and autophagy slightly attenuated after JQ1 treatment, and the ratio of apoptosis increased significantly as the concentration of JQ1 increases. Whether inhibiting c-Myc or BET inhibitors like JQ1 could induce autophagy in ovarian cancer has not been investigated. Interestingly, the JQ1-resistant group showed reduced expression of c-Myc and increased autophagy since the markers of autophagy, for example, conversion of LC3-I to lipidated LC3-II and ATG5, and Beclin1 increased. Furthermore, degradation of P62/SQSTM1 was observed in the JQ1-resistant group after JQ1 treatment, another indication of increased autophagy. Co-treatment of autophagy inhibitors 3-MA and CQ with JQ1 indicated that JQ1-induced autophagy played a cytoprotective role during drug treatment. Autophagy inhibition reverses the resistance of JQ1-resistant A2780 and HO-8910 ovarian cancer cell lines to BET inhibition to increase apoptosis. In vivo studies confirmed the effects of the combination of JQ1 and autophagy inhibitors. Detection of tumour-weight in different treatment groups confirmed that the combination of JQ1 and CQ was safe. Altogether, our results indicated that JQ1-induced autophagy is a critical mechanism involved in JQ1 resistance in ovarian cancer cells.
Recent studies have shown the association of resistance to BET inhibitor with Akt1/mTOR pathway in ovarian cancer.9 It was shown that the proliferation and apoptosis of cancer cells were regulated by Akt/mTOR pathway.31 In addition to the Akt pathway, activated AMPK pathway also participates in resistance to BET inhibitor in AML.32 However, activation of the LKB1/AMPK pathway-induced autophagy showed anti-tumour effect in bladder cancer.33 Since Akt1/mTOR pathway can regulate autophagy and has been tested in ovarian cancer,9 we investigated whether Akt1/mTOR pathway is involved in JQ1-induced cytoprotective autophagy in JQ1-resistant ovarian cancer cell lines. Our data showed detection of decreased phosphorylated levels of Akt, mTOR, and p70S6K in JQ1-resistant group after treatment with JQ1. Consistent, overexpression of Akt1 also led to a decrease in accumulated levels of LC3 puncta and LC3-II conversion induced by JQ1. In contrast, co-treatment of A2780 and HO-8910 cells with LY294002 (a PI3K/Akt pathway inhibitor) and JQ1 significantly increased the levels of LC3-II conversion in JQ1 treated cells. Besides, overexpressing Akt1 also increased the sensitivity of JQ1 resistant cells to JQ1 with improvement in the ratio of apoptotic cells. These data strongly suggested the involvement of Akt/mTOR signalling pathway in JQ1-induced autophagy in ovarian cancer.
Altogether, our data showed that ovarian cancer cell lines have different sensitivity to BET inhibitor JQ1. Different levels of JQ1-induced autophagy were partly responsible for this differential response. Besides, autophagy inhibitor 3-MA or CQ markedly enhanced JQ1-induced apoptosis and attenuated the resistance to BET inhibitor in JQ1-resistant ovarian cancer cells, and in vivo studies also confirmed the combination effect. Combination of autophagy inhibitor and JQ1 treatment may be a useful approach for the treatment of ovarian cancer. However, further studies are needed to determine how autophagy is regulated differentially in resistant and sensitive groups. Additionally, reliable biomarkers are lacking for explicit confirmation of clinical patients who might benefit from this combination treatment. Furthermore, more in vivo studies are necessary to confirm the safety of the combination treatment further. JQ1 displays antitumor activities in a variety of human cancers with different modes of action, and a significant challenge for further development and clinical testing of JQ1 is the lack of knowledge on intrinsic resistance JQ1 itself and lack of reliable biomarkers to predict its sensitivity. Our findings are valuable for indicating the potential molecular mechanisms involved in resistance to BET inhibitor JQ1 in the ovarian tumour and suggest modified treatment strategies for overcoming this resistance.
Conclusion
JQ1-induced autophagy in ovarian cancer cell lines A2780 and HO-8910 attenuated the anti-tumour effect of JQ1. Akt/mTOR pathway was responsible for this. Inhibition of autophagy by 3-MA or CQ or overexpression of Akt1 significantly improved the A2780 and HO-8910 cell response to JQ1. The combined effect was also confirmed in vivo and may provide a potential strategy to sensitise OC to BET targeting therapy.
Ethics Approval And Informed Consent
The Experimental Ethics Committee of Qilu Hospital of Shandong University approved this research (approval number: DWLL-2015-004).
Acknowledgments
This work was supported by China Postdoctoral Science Fund (21510077311145 and 21300076311047) and Natural Science Foundation of Shandong Province (ZR2016HM27) and Science Foundation of Qilu Hospital of Shandong Province.
Author Contributions
Peishu Liu, Hongluan Mao, and Wenqing Luan conceived and designed the experiments. Wenqing Luan, Yingxin Pang, Rui Li, Xuan Wei, Xiaoxiao Jiao, Juanjuan Shi, and Jiangtao Yu performed the experiments and analysed the data. Wenqing Luan and Yingxin Pang wrote the paper. Yingxin Pang revised the paper. All authors have read and approved the final manuscript. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer. 2010;10(11):803–808. doi:10.1038/nrc2946
2. Ledermann JA. Front-line therapy of advanced ovarian cancer: new approaches. Ann Oncol. 2017;28(suppl_8):viii46–viii50. doi:10.1093/annonc/mdx452
3. Bowtell DD, Bohm S, Ahmed AA, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015;15(11):668–679. doi:10.1038/nrc4019
4. Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12(7):465–477. doi:10.1038/nrc3256
5. Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. doi:10.1016/j.cell.2013.03.036
6. Baratta MG, Schinzel AC, Zwang Y, et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci U S A. 2015;112(1):232–237. doi:10.1073/pnas.1422165112
7. Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54(5):728–736. doi:10.1016/j.molcel.2014.05.016
8. Shu S, Lin CY, He HH, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529(7586):413–417. doi:10.1038/nature16508
9. Kurimchak AM, Shelton C, Duncan KE, et al. Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer. Cell Rep. 2016;16(5):1273–1286. doi:10.1016/j.celrep.2016.06.091
10. Zhang Z, Ma P, Jing Y, et al. BET bromodomain inhibition as a therapeutic strategy in ovarian cancer by downregulating FoxM1. Theranostics. 2016;6(2):219–230. doi:10.7150/thno.13178
11. Qiu H, Jackson AL, Kilgore JE, et al. JQ1 suppresses tumor growth through downregulating LDHA in ovarian cancer. Oncotarget. 2015;6(9):6915–6930. doi:10.18632/oncotarget.3126
12. Zhu H, Bengsch F, Svoronos N, et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16(11):2829–2837. doi:10.1016/j.celrep.2016.08.032
13. Karakashev S, Zhu H, Yokoyama Y, et al. BET bromodomain inhibition synergizes with PARP inhibitor in epithelial ovarian cancer. Cell Rep. 2017;21(12):3398–3405. doi:10.1016/j.celrep.2017.11.095
14. Dai X, Gan W, Li X, et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med. 2017;23(9):1063–1071. doi:10.1038/nm.4378
15. Kumar K, Raza SS, Knab LM, et al. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci Rep. 2015;5:9489. doi:10.1038/srep09489
16. Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(7):713–720. doi:10.1038/ncb2788
17. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. doi:10.1038/nrm3735
18. Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res. 2014;74(3):647–651. doi:10.1158/0008-5472.CAN-13-2966
19. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–1295. doi:10.1016/j.febslet.2010.01.017
20. Sun Y, Huang YH, Huang FY, et al. 3ʹ-epi-12beta-hydroxyfroside, a new cardenolide, induces cytoprotective autophagy via blocking the Hsp90/Akt/mTOR axis in lung cancer cells. Theranostics. 2018;8(7):2044–2060. doi:10.7150/thno.23304
21. Suman M, Mahesh S, Amrita C, et al. Reciprocal regulation of AMP-activated protein kinase and phospholipase D. J Biol Chem. 2015;290(11):6986–6993. doi:10.1074/jbc.M114.622571
22. Li YJ, Lei YH, Yao N, et al. Autophagy and multidrug resistance in cancer. Chin J Cancer. 2017;36(1):52. doi:10.1186/s40880-017-0219-2
23. Jin X, Yan Y, Wang D, et al. DUB3 promotes BET inhibitor resistance and cancer progression by deubiquitinating BRD4. Mol Cell. 2018;71(4):592–605 e594. doi:10.1016/j.molcel.2018.06.036
24. Wang B, Fan P, Zhao J, Wu H, Jin X, Wu H. FBP1 loss contributes to BET inhibitors resistance by undermining c-Myc expression in pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res. 2018;37(1):224. doi:10.1186/s13046-018-0888-y
25. Mauthe M, Jacob A, Freiberger S, et al. Resveratrol-mediated autophagy requires WIPI-1-regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy. 2011;7(12):1448–1461. doi:10.4161/auto.7.12.17802
26. Sidjanin DJ, Park AK, Ronchetti A, Martins J, Jackson WT. TBC1D20 mediates autophagy as a key regulator of autophagosome maturation. Autophagy. 2016;12(10):1759–1775. doi:10.1080/15548627.2016.1199300
27. Fong CY, Gilan O, Lam EY, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525(7570):538–542. doi:10.1038/nature14888
28. Li C, Bonazzoli E, Bellone S, et al. Mutational landscape of primary, metastatic, and recurrent ovarian cancer reveals c-MYC gains as potential target for BET inhibitors. Proc Natl Acad Sci U S A. 2019;116(2):619–624. doi:10.1073/pnas.1814027116
29. Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. 2014;289(24):17163–17173. doi:10.1074/jbc.M114.558288
30. Toh PP, Luo S, Menzies FM, Rasko T, Wanker EE, Rubinsztein DC. Myc inhibition impairs autophagosome formation. Hum Mol Genet. 2013;22(25):5237–5248. doi:10.1093/hmg/ddt381
31. Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy. 2007;3(6):635–637. doi:10.4161/auto.4916
32. Jang JE, Eom JI, Jeung HK, et al. AMPK-ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin Cancer Res. 2017;23(11):2781–2794. doi:10.1158/1078-0432.CCR-16-1903
33. Feng L, Chao Y, Hai B, et al. BET inhibitor JQ1 suppresses cell proliferation via inducing autophagy and activating LKB1/AMPK in bladder cancer cells. Cancer Med. 2019;8(10):4792–4805. doi:10.1002/cam4.2385
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.