")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 9 » Issue 1
Airway epithelial platelet-activating factor receptor expression is markedly upregulated in chronic obstructive pulmonary disease
Authors Dhar Shukla S , Sohal SS, Mahmood M, Reid D, Muller HK, Walters EH
Received 30 April 2014
Accepted for publication 5 June 2014
Published 13 August 2014 Volume 2014:9(1) Pages 853—861
DOI https://doi.org/10.2147/COPD.S67044
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Shakti Dhar Shukla,1,* Sukhwinder Singh Sohal,1,* Malik Quasir Mahmood,1 David Reid,2 Hans Konrad Muller,1 Eugene Haydn Walters1
1NHMRC Centre for Research Excellence for Chronic Respiratory Disease and Lung Ageing, School of Medicine, University of Tasmania, Hobart, Tasmania, Australia; 2Queensland Institute of Medical Research, Iron Metabolism Laboratory, Brisbane, Queensland, Australia
*Shakti Dhar Shukla and Sukhwinder Singh Sohal are joint first authors
Background: We recently published that platelet-activating factor receptor (PAFr) is upregulated on the epithelium of the proximal airways of current smokers and also in bronchial epithelial cells exposed to cigarette smoke extract. These treated cells also showed upregulation of Streptococcus pneumoniae adhesion. Bacterial wall phosphorylcholine specifically binds to PAFr expressed on airway epithelium, thus facilitating adherence and tissue invasion, which may be relevant to chronic obstructive pulmonary disease (COPD). Moreover, the use of inhaled corticosteroids (ICS) in COPD patients is associated with an increased risk of invasive respiratory pneumococcal infections.
Objective: In this study, we have investigated whether PAFr expression is especially upregulated in airway epithelium in COPD patients and whether this expression may be modulated by ICS therapy.
Methods: We cross-sectionally evaluated PAFr expression in bronchial biopsies from 15 COPD patients who were current smokers (COPD-smokers) and 12 COPD-ex-smokers, and we compared these to biopsies from 16 smokers with normal lung function. We assessed immunostaining with anti-PAFr monoclonal antibody. We also used material from a previous double-blinded randomized placebo-controlled 6-month ICS intervention study in COPD patients to explore the effect of ICS on PAFr expression. We employed computer-aided image analysis to quantify the percentage of epithelium stained for PAFr.
Results: Markedly enhanced expression of PAFr was found in both COPD-smokers (P<0.005) and COPD-ex-smokers (P<0.002) compared to smokers with normal lung function. There was little evidence that PAFr expression was affected by ICS therapy over 6 months.
Conclusion: Epithelial PAFr expression is upregulated in smokers, especially in those with COPD, and is not obviously affected by ICS therapy.
Keywords: adhesion, airway epithelium, inhaled corticosteroids, pneumococcal infections, Streptococcus pneumoniae
Background
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory lung disease characterized by airflow limitation that is not fully reversible. It is a global health crisis with a substantial and increasing burden, which reduces quality of life, increases national economic burden, is a major cause of acute hospital admissions, and has significant mortality, especially during exacerbations.1–5
Emphasis has been laid on the importance of lower respiratory-tract bacterial colonization in the pathogenesis of, subsequent long-term progression of, and exacerbations in COPD, and so it is likely to be one of the important factors driving inflammatory processes in the airways.6–8 However, the mechanisms underlying the initial bacterial epithelial adherence and subsequent infection remain elusive. Haemophilus influenzae and Streptococcus pneumoniae are the most important pulmonary bacteria during both stable phase of disease and exacerbations of COPD.9–11 Moreover, complex interactions between the host, microbes (both bacteria and viruses), and environmental pollution are associated with exacerbations that are marked by substantial increases in inflammatory markers in the airways.12,13
Large intervention trials and follow-up pharmacoepidemiology studies have recommended the use of inhaled corticosteroids (ICS) to reduce frequency of exacerbations and improve quality of life in more severe COPD patients, and ICS has become an established therapy.14 Although ICS therapy undoubtedly has some positive effects, it is accompanied by an increase in the risk of lower respiratory-tract bacterial infections in COPD patients, especially with S. pneumoniae, which increase in incidence over time.15–17
In an international collaborative study, we recently demonstrated increased platelet-activating factor receptor (PAFr) expression and PAFr-dependent S. pneumoniae adhesion to epithelial cells in culture exposed to cigarette smoke extract, and provided preliminary evidence of an increase in airway epithelial expression of PAFr in smokers.18 PAFr is a G-protein-coupled epithelial cell membrane receptor that naturally binds the phosphorylcholine ligand on the eukaryotic proinflammatory chemokine PAF. Of all bacteria, S. pneumoniae (also Haemophilus and Pseudomonas) cell walls specifically express phosphorylcholine (ChoP), which out-competes PAF to bind to the PAFr. This initiates bacterial uptake by the host cell in a β-arrestin-dependent pathway.19–21
In addition, PAFr-deficient mice are less likely to develop invasive pneumococcal infections as compared to their wild-type counterparts.22 PAFr-dependent pneumococcal adhesion is also upregulated by several other factors in addition to cigarette smoke, including acid, respiratory viral infection, interleukin-1α, and exposure to particulate matter.23 This all fits with the pattern of infection and deterioration seen in COPD.
In this report, we have extended our previous cross-sectional observations to analyze the expression of airway epithelial PAFr in another batch of normal smoker tissue, but also in groups of COPD subjects (smokers and ex-smokers) not on ICS, and in COPD subjects who were involved in a trial of ICS therapy for 6 months.
Materials and methods
Study design
All subjects gave written informed consent prior to participation. The study was approved by the Human Research Ethics Committee (Tasmania) Network.
In the cross-sectional study, 17 current smokers with established COPD (COPD-smokers) and 15 ex-smokers with COPD (COPD-ex-smokers) were recruited by advertisement for bronchoscopy and airway biopsy (Table 1), and their tissue was compared with biopsies collected in a similar manner from 16 normal lung-function smokers (NLFS). Data from this group was presented in the previous study,18 but this assessment was done de novo on new slides by a separate researcher.
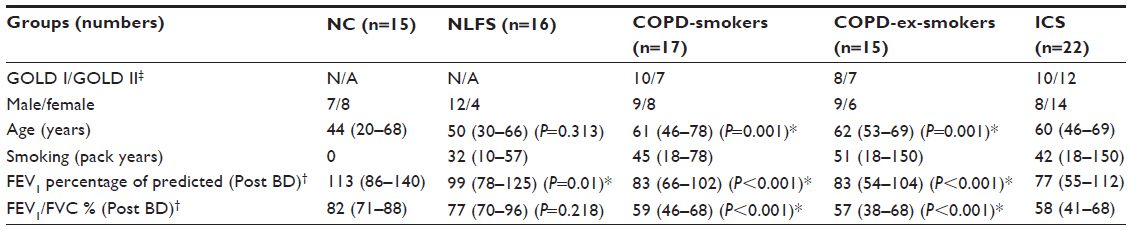 | Table 1 Demographic and lung function data for participants in the cross-sectional study |
Here, we have also included residual airway biopsy tissue from a previous 6-month randomized control trial using inhaled fluticasone24 for a preliminary investigation of the effect of ICS on PAFr expression in COPD subjects. In this longitudinal study, a computer-generated random numbers table was used to assign COPD participants to fluticasone propionate (Accuhaler; GlaxoSmithKline plc, London, UK) 500 μg twice daily or placebo via identical multidose dry powder inhaler devices for 6 months (Figure 1). Before and after treatment, lung function and bronchial biopsies were performed. Half of the participants were current smokers and the other half were ex-smokers. This trial was registered with the Australian New Zealand Clinical Trials Registry (Registry No ACTRN12612001111864). The details for the randomized control trial have been published previously.25 For the current analysis, we had 15 pairs of biopsy tissue in the ICS arm, but only eight in the placebo arm, available for processing.
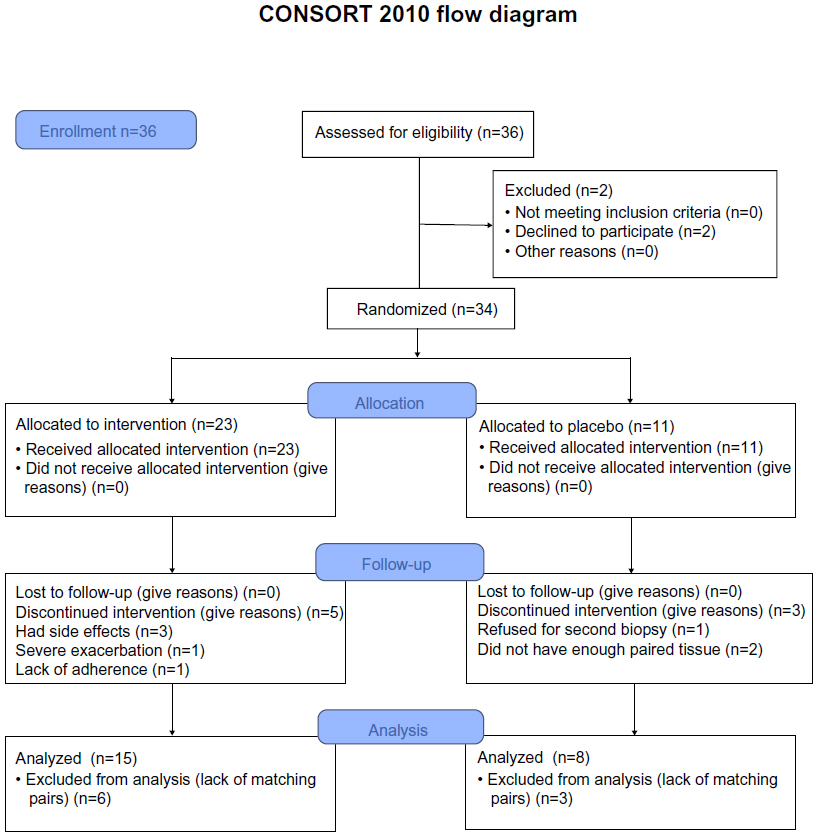 | Figure 1 Study design. |
Subjects
The diagnosis of COPD was made according to the Global Initiative for Chronic Obstructive Lung Disease guidelines1 based on forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio, and was categorized into two groups on the basis of current versus ex-smoking history. For normal lung function current smokers, the inclusion criteria were: a minimum 10 pack-year history of cigarette smoking with spirometry within normal limits, ie, FEV1 >80% of predicted and FEV1/FVC >70%; and no scalloping out of the expiratory descending limb of the flow-volume curve to suggest small airway dysfunction. Normal healthy volunteers had no history of respiratory illness or smoking and had normal lung function.
Exclusion criteria
The exclusion criteria were:
- subjects with a history suggestive of asthma, which includes symptoms in childhood, related atopic disorders, eczema or hay fever, substantial day-to-day variability, or prominent nocturnal symptoms;
- or a history of wheeze rather than progressive breathlessness;
- and any who had previously used ICS (oral or inhaled).
In our study population, there were no major comorbidities such as uncontrolled diabetes, angina, or cardiac failure, nor other coexisting respiratory disorders including pulmonary fibrosis, lung cancer, or bronchiectasis, and subjects were not on medication relating to previously mentioned conditions.
Subjects with an inability to give written informed consent were also excluded.
Inclusion criteria
The inclusion criteria were:
- COPD-smokers: aged 40–70 years with a smoking history ≥15 pack-years; subsequently obtained bronchoalveolar lavage fluid had to be free of culturable bacteria; FEV1 40%–80% of predicted, with forced expiratory ratio (ratio of FEV1 to FVC) ≤70% post-bronchodilator and with definite scalloping out of the descending limb of flow-volume loop on spirometry;
- COPD-ex-smokers: up to 6 months of smoking cessation.
Normal healthy never-smoking controls also underwent bronchoscopic examination and physiological evaluation. Subjects were at least 18 years old with FEV1/FVC ratio ≥70% and FEV1 ≥80% of predicted; they had no history of respiratory illness.
Bronchoscopy
Bronchoscopy was performed using standard techniques.26 Briefly, subjects were premedicated with nebulized salbutamol (5 mg) 15–30 minutes before the procedure. Sedation was achieved with intravenous midazolam (3–10 mg) and fentanyl (25–100 μg). Lignocaine (4%) was used for topical anesthesia above the vocal cords and lignocaine (2%) was used to anesthetize the airways below the cords, in 2 mL aliquots as required, up to a maximum of 6 mL. Subjects were monitored by pulse oximetry throughout the procedure and oxygen was administered routinely. Eight biopsies from secondary carinae of segmental and subsegmental bronchi in the right lower lobe were obtained. There were no complications from the procedures. Bronchial biopsies were fixed in 4% neutral buffered formalin for 2 hours and subsequently processed into paraffin through graded alcohol and xylene using a Leica ASP 200S tissue processor (Leica Biosystems Nussloch GmbH, Nussloch, Germany).
Immunostaining
Sections were cut at 3 mm intervals from individual paraffin blocks, stained with hematoxylin and eosin, and morphologically assessed for quality and lack of damage.
Following removal of paraffin and rehydration, immunostaining for PAFr was done using an anti-PAFr monoclonal antibody (11A4, clone 21, Cayman Chemicals, Michigan, USA, Catalogue No 160600, 1/80 dilution, for 1 hour at 20°C with no heat retrieval). In each case, a negative control with the primary antibody replaced by a species-appropriate isotype-matched immunoglobulin G2 (X0931 clone DAK-GO1; Dako Denmark A/S, Glostrup, Denmark) was included. Tissue from previously resected lung was used as positive control. Endogenous peroxidase blocking was done with 3% hydrogen peroxide in Milli-RO water (EMD Millipore, Billerica, MA, USA) for 15 minutes at 20°C to eliminate nonspecific staining, but no serum block was necessary. Bound antibodies were elaborated using peroxidase-labeled Envision+ (catalogue number K4001; Dako Denmark A/S) and liquid DAB+ (catalogue number K3468; Dako Denmark A/S).
Biopsy section quantification
Computer-assisted image analysis was performed with a Leica DM 2500 microscope (Leica Microsystems, Wetzlar, Germany), Spot Insight-12 digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI, USA), and Image Pro Plus 7.0 (Media Cybernetics, Inc., Rockville, MD, USA) software. Expression of PAFr was assessed on randomized and coded slides by an operator blinded to smoking and clinical status, and was expressed as the percentage of epithelium with PAFr expression. The cell distribution of staining was also noted qualitatively.
Statistical analysis
For cross-sectional data, the distributions were generally skewed, so results are presented as medians and ranges; non-parametric analyses of variance were performed (Kruskal–Wallis Test comparing medians across all the groups of interest) and specific group differences were then explored as appropriate according to prior hypotheses using the Mann–Whitney U test. To avoid multiple comparisons as much as possible, cross-sectional COPD groups were compared independently with the NLFS group and the results are presented as scatter plots. In the COPD groups, we performed regression analysis for PAFr expression against age, FEV1, and smoking history. For multiple comparisons (two COPD groups versus a single NLFS group), a Bonferroni correction was made (P<0.001).
For the longitudinal study, data were log-transformed because of skewness and the Student’s t-test was used for within group comparisons. The post-ICS group data has also been included in Figure 3 for the cross-sectional analysis, to give an overall perspective.
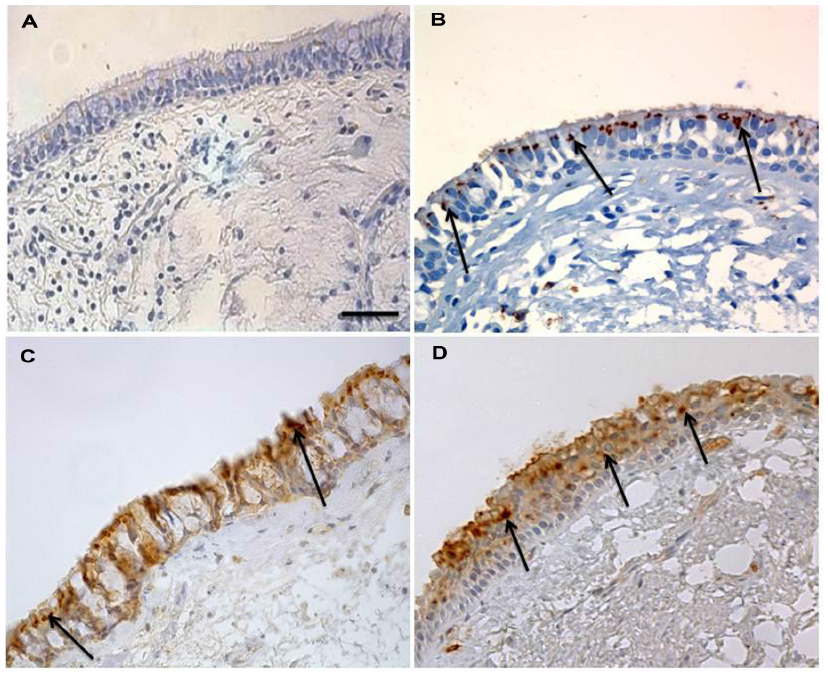 | Figure 2 PAFr expression in epithelium of bronchial biopsy. |
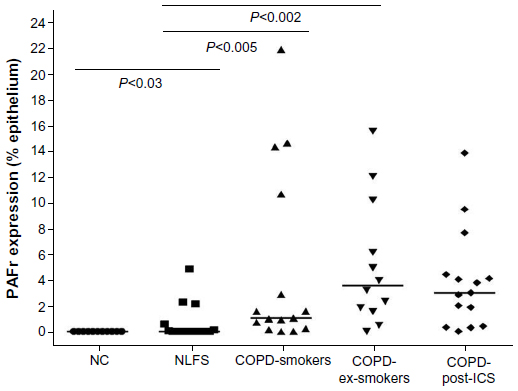 | Figure 3 Percentage of epithelium showing PAFr expression in cross-sectional study. |
Statistical analyses were performed using SPSS 15.0 for Windows, 2003 (IBM Corporation, Armonk, NY, USA), with a two-tailed P-value ≤0.05 being considered statistically significant.
Results
Immunostaining was prominent only in the apical part of the bronchial epithelium of NLFS, but staining was darker and more generalized, apparently around the perimeter of cells, in the COPD groups. There was negligible PAFr staining in normal controls. These staining patterns are illustrated in Figure 2.
Image analysis confirmed that there was no difference in PAFr expression between the COPD-smokers and COPD-ex-smokers study groups (nonsignificant P-value), but the difference between the NLFS group and the COPD groups (median 0.0%, range 0%–4.9%), was significant (ie, COPD-smokers [P<0.005; median 1.03%, range 0%–21.8%], COPD-ex-smokers [P<0.002; median 3.6%, range 0%–15.5%]). The NLFS group also showed significantly higher PAFr when compared to normal controls (median 0.0%, range 0%–0%). PAFr expression in the ICS-treated group was very similar to the other COPD groups, so no formal statistical testing was done (Figure 3). In the NLFS group, only seven of the 16 epithelia stained detectably, while, in the two COPD groups combined, 24 had detectable staining out of 27 (χ2; P<0.001); five samples from the COPD groups (two out of 17 COPD-smokers and three out of 15 COPD-ex-smokers) did not have enough tissue for analysis.
PAFr expression in the placebo-controlled ICS intervention study
There was considerable variation in PAFr expression within the COPD groups both before and after the treatment phase. The median percent epithelial expression rose in both the groups, by approximately threefold in the ICS limb (P<0.15) but also by approximately twofold in the placebo group over time (Figure 4). Although the final median level in the ICS groups was greater than in the placebo group, this was neither significant nor were the changes in expression over this time between the groups (P<0.4). It is possible, because of the small number of paired tissues that were available to us, that we saw a type-2 statistical error; thus, a power calculation using nontransformed data was undertaken for a potential real increase in epithelial PAFr expression with ICS, which indicated that, for these estimates of effect size, a bronchoscopic study of over 200 subjects per group would be needed to show a significant positive effect of ICS on PAFr expression.
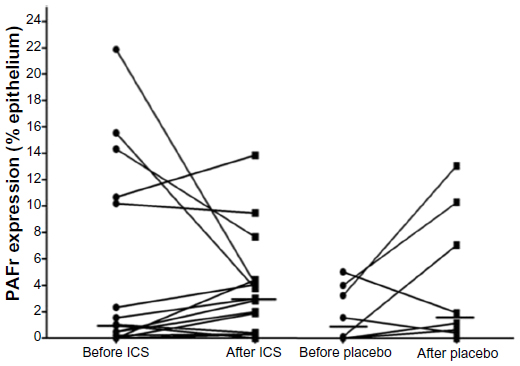 | Figure 4 Relative PAFr expression in the intervention study. |
The regression analyses showed that age and FEV1 had no significant effects on PAFr expression (r=0.23 and 0.11, respectively). However, for pack-years of smoking in the COPD-ex-smokers group, there was quite a strong relationship (r=0.56; P<0.05), but this was not seen in the COPD-smokers subjects (r=0.09; P<0.81).
Discussion
In the present study, we found that in vivo expression of PAFr is significantly upregulated in COPD patients as compared to NLFS. Expression in COPD-smokers and -ex-smokers was similar but quite variable in both groups. This might suggest that smoking cessation may be ineffective in downregulation of PAFr once COPD has developed. Alternatively, it is possible that especially high expression of PAFr in some smokers is both a risk factor for COPD development in the first place, and its progression, even after smoking has ceased.
Overall, our data on the effects of ICS are negative, with at the very most only modest evidence that ICS upregulate PAFr expression – though they certainly do not decrease it – over 6 months, as suggested in earlier in vitro and animal models.27 The present study may also have been relatively short to demonstrate an effect of ICS on PAFr expression, which could potentially elevate risk of pneumococcal disease in COPD patients. The incidence of pneumonia in patients on ICS escalates over years rather than weeks or months.17 It is also possible that the effects of ICS could be more marked in peripheral airways or lung parenchymal epithelium where the clinical problem occurs, while our biopsies were taken only from relatively central airways. Pneumonia occurs at a rate of approximately 1% per year in severe COPD patients on ICS,28 and it is not known whether there is some aberrant change in this small group of individuals or if some generic change occurs on ICS that manifests clinically only in a small percentage. This issue needs further work.
The relationship between cigarette smoking and infection/colonization with S. pneumoniae and H. influenzae has been well documented, and exposure to tobacco smoke is considered as a major risk factor for invasive pneumococcal disease.29,30 Persistence of chronic inflammation and continuous deterioration of lung function following smoking cessation may be attributed to chronic colonization and invasion of lung tissue by respiratory bacterial pathogens, again most commonly S. pneumoniae and H. influenzae.30 Bacterial adherence to lung epithelium and subsequent tissue invasion is thought to be a crucial step in the pathogenesis of respiratory infections. The major respiratory bacterial pathogens are equipped with the surface ligand known as ChoP, which is capable of binding to epithelial PAFr. Especially virulent strains of S. pneumoniae have been shown to adhere to its bacterial cell surface ChoP more firmly with epithelial surface PAFr.19,21,23 We suggest that our data on PAFr expression in the airway epithelium in COPD may be relevant to this as, out of an estimated 109 different bacterial species,31 it is S. pneumoniae and H. influenzae especially (and also Pseudomonas aeruginosa, which is more relevant in cystic fibrosis) that express the ChoP ligand.
In a collaborative study with Grigg et al,18 we recently described significant increases in PAFr-dependent pneumococcal adhesion, PAFr transcript level, and PAFr protein expression in epithelial cell cultures exposed to cigarette smoke extract. Moreover, a specific PAFr antagonist attenuated pneumococcal adhesion. In the same study, we showed enhanced airway epithelial PAFr expression in smokers’ airways for the first time. The present study takes this observation further and confirms much greater upregulation of PAFr expression in the COPD groups than in NLFS. In contrast, Suri et al32 reported no difference in PAFr messenger RNA transcript levels between NLFS and COPD-smokers. However, they did not quantify the PAFr immunoreactivity on bronchial biopsies, and, ultimately, it is protein expression on the epithelial surface that will matter functionally.32 Moreover, the micrograph provided by Suri et al32 seemed to have more PAFr expression in their normal controls than we have found, but the most staining did appear to be in COPD, consistent with our finding. We now need ex vivo epithelial cultures from these patients to see if, functionally, there is greater bacterial adhesion in COPD airways in the group with the greatest PAFr expression.
The regression analysis in the two cross-sectional COPD groups produced interesting outcomes. There was no relationship between either age or severity of airflow obstruction and PAFr-expression, but pack-years of smoking was strongly related, at least in COPD-ex-smokers. The lack of this relationship in current smokers suggests that this long-term effect was confounded by the effect of current smoking. Thus, both current smoking and history of smoking seem important, but, since only a minority of smokers develop COPD, there must also be an additional individual susceptibility effect.
The role that ICS can play in COPD has been extensively studied in the recent past, and ICS are especially effective at reducing exacerbation rates.14 Other described benefits are more tentative, such as reduction in rate of decline in lung function, decreased cancer rates,33,34 and decreased mortality.14 However, a paradoxical effect is that ICS therapy also increases the risk of lower respiratory bacterial infections, especially with S. pneumonia,14,16 an effect that worsens over time.17 An association between ICS and increased risk of hospitalizations due to pneumonia in elderly COPD patients with a high mortality rate, has also been reported.15 Animal models also demonstrate a similar effect of elevated risk of pneumonia with administration of ICS.35
On the basis of our current data, we cannot say that this increased infective risk of ICS is related to enhanced expression of respiratory epithelial PAFr, but it remains a possibility. A much larger clinical study would be needed to determine this, but such a large bronchoscopic investigation is rather unlikely to occur. A better approach may be to see if corticosteroids have any effect on human primary epithelial cell cultures in this regard. It is notable that PAFr expression seemed to increase in both ICS- and placebo-treated COPD subjects over 6 months. Patients are recruited into such studies when they are especially well and free from infection, and this apparently spontaneous change may be a regression back to their more usual status of increased vulnerability.
There were several limiting issues in the study, especially the small numbers of samples available in the ICS intervention study. Furthermore, only large airways were studied, but we are in the process of extending this work to small airway samples and alveolar epithelium obtained at lung resection. Strengths of the present investigation include fairly robust numbers in our cross-sectional study that followed on from the smoking effect we published previously with this tissue bank. The data for the NLFS were obtained de novo and by a separate operator, but are very similar to that published earlier.18
Conclusion
In summary, airway epithelial PAFr expression is significantly upregulated in smokers, but is especially marked in COPD subjects, whether current smokers or not. The significance of this in the pathogenesis and evolution of COPD needs further study, but could be very important in the etiology of chronic and invasive infection with S. pneumoniae and H. influenzae. Whether the increased expression of PAFr is a contributing cause of COPD, or a result of it, needs further investigation.
Author contributions
Mr Shukla performed the literature search, histological and statistical analysis, wrote and revised the manuscript. Dr Sohal designed the study, performed the histology and assisted in writing the manuscript. Dr Reid performed bronchoscopies and clinical assessments and contributed to the writing of the paper. Dr Mahmood assisted in histological analyses and writing of manuscript. Professor Muller advised on histology strategy, quality control and assisted in writing of manuscript. Professor Walters devised the overall study and clinical assessments, and supervised all analyses and writing of the manuscript.
Acknowledgments
Funding source: National Health and Medical Research Council (NHMRC), Australia (Grant No 490023). The sponsors had no role in the study design, in the collection, analysis, and interpretation of the data, or in the decision to submit the article for publication. Trial registration: Australian New Zealand Clinical Trials Registry (ACTRN12612001111864).
Disclosure
The authors report no conflicts of interest in this work.
References
http://www.goldcopd.org [homepage on the Internet]. Global initiative for Chronic Obstructive Pulmonary Disease. 2011. Available from: http://www.goldcopd.org. Accessed January 22, 2013. | |
Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. | |
Pasquale MK, Sun SX, Song F, Hartnett HJ, Stemkowski SA. Impact of exacerbations on health care cost and resource utilization in chronic obstructive pulmonary disease patients with chronic bronchitis from a predominantly Medicare population. Int J Chron Obstruct Pulmon Dis. 2012;7:757–764. | |
Suissa S, Dell’Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax. 2012;67:957–963. | |
Soler-Cataluña JJ1, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax. 2005;60:925–931. | |
Cabello H, Torres A, Celis R, et al. Bacterial colonization of distal airways in healthy subjects and chronic lung disease: a bronchoscopic study. Eur Respir J. 1997;10:1137–1144. | |
Patel IS, Seemungal TA, Wilks M, Lloyd-Owen SJ, Donaldson GC, Wedzicha JA. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax. 2002;57:759–764. | |
Erkan L, Uzun O, Findik S, Katar D, Sanic A, Atici AG. Role of bacteria in acute exacerbations of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2008;3:463–467. | |
Banerjee D, Khair OA, Honeybourne D. Impact of sputum bacteria on airway inflammation and health status in clinical stable COPD. Eur Respir J. 2004;23:685–691. | |
Soler N, Ewig S, Torres A, Filella X, Gonzalez J, Zaubet A. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur Respir J. 1999;14:1015–1022. | |
Murphy TF, Brauer AL, Schiffmacher AT, Sethi S. Persistent colonization by Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170:266–272. | |
Sapey E, Stockley RA. COPD exacerbations. 2: aetiology. Thorax. 2006;61:250–258. | |
Hurst JR, Perera WR, Wilkinson TM, Donaldson GC, Wedzicha JA. Systemic and upper and lower airway inflammation at exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:71–78. | |
Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356:775–789. | |
Ernst P, Gonzalez AV, Brassard P, Suissa S. Inhaled corticosteroid use in chronic obstructive pulmonary disease and the risk of hospitalization for pneumonia. Am J Respir Crit Care Med. 2007;176:162–166. | |
Singh S, Amin AV, Loke YK. Long-term use of inhaled corticosteroids and the risk of pneumonia in chronic obstructive pulmonary disease: a meta-analysis. Arch Intern Med. 2009;169:219–229. | |
Cates C. Inhaled corticosteroids in COPD: quantifying risks and benefits. Thorax. 2013;68:499–500. | |
Grigg J, Walters H, Sohal SS, et al. Cigarette smoke and platelet-activating factor receptor dependent adhesion of Streptococcus pneumoniae to lower airway cells. Thorax. 2012;67:908–913. | |
Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. 1995;377:435–438. | |
Radin JN, Orihuela CJ, Murti G, Guglielmo C, Murray PJ, Tuomanen EI. Beta-Arrestin 1 participates in platelet-activating factor receptor-mediated endocytosis of Streptococcus pneumoniae. Infect Immun. 2005;73:7827–7835. | |
Swords WE, Buscher BA, Ver Steeg Ii K, et al. Non-typeable Haemophilus influenzae adhere to and invade human bronchial epithelial cells via an interaction of lipooligosaccharide with the PAF receptor. Mol Microbiol. 2000;37:13–27. | |
Rijneveld AW, Weijer S, Florquin S, et al. Improved host defense against pneumococcal pneumonia in platelet-activating factor receptor-deficient mice. J Infect Dis. 2004;189:711–716. | |
Grigg J. The platelet activating factor receptor: a new anti-infective target in respiratory disease? Thorax. 2012;67:840–841. | |
Reid DW, Wen Y, Johns DP, Williams TJ, Ward C, Walters EH. Bronchodilator reversibility, airway eosinophilia and anti-inflammatory effects of inhaled fluticasone in COPD are not related. Respirology. 2008;13:799–809. | |
Sohal SS, Reid D, Soltani A, et al. Changes in airway histone deacetylase2 in smokers and COPD with inhaled corticosteroids: a randomized controlled trial. PLoS One. 2013;8:e64833. | |
Feltis BN, Wignarajah D, Zheng L, et al. Increased vascular endothelial growth factor and receptors: relationship to angiogenesis in asthma. Am J Respir Crit Care Med. 2006;173:1201–1207. | |
Barbier M, Agusti A, Alberti S. Fluticasone propionate reduces bacterial airway epithelial invasion. Eur Respir J. 2008;32:1283–1288. | |
Kew KM, Seniukovich A. Inhaled steroids and risk of pneumonia for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2014;3:CD010115. | |
Nuorti JP, Butler JC, Farley MM, et al. Cigarette smoking and invasive pneumococcal disease. Active Bacterial Core Surveillance Team. N Engl J Med. 2000;342:681–689. | |
Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med. 2004;164:2206–2216. | |
Curtis TP, Sloan WT, Scannell JW. Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci U S A. 2002;99:10494–10499. | |
Suri R, Mallia P, Martin JE, et al. Bronchial platelet-activating factor receptor in chronic obstructive pulmonary disease. Respir Med. 2014;108:898–904. | |
Celli BR, Thomas NE, Anderson JA, et al. Effect of pharmacotherapy on rate of decline of lung function in chronic obstructive pulmonary disease: results from the TORCH study. Am J Respir Crit Care Med. 2008;178:332–338. | |
Parimon T, Chien JW, Bryson CL, McDonell MB, Udris EM, Au DH. Inhaled corticosteroids and risk of lung cancer among patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:712–719. | |
Patterson CM, Morrison RL, D’Souza A, Teng XS, Happel KI. Inhaled fluticasone propionate impairs pulmonary clearance of Klebsiella pneumoniae in mice. Respir Res. 2012;13:40. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.