")
Back to Journals » International Journal of Nanomedicine » Volume 13
Aerosol-synthesized siliceous nanoparticles: impact of morphology and functionalization on biodistribution
Authors Diebolder P , Vazquez-Pufleau M , Bandara N , Mpoy C , Raliya R , Thimsen E, Biswas P, Rogers BE
Received 16 June 2018
Accepted for publication 9 August 2018
Published 12 November 2018 Volume 2018:13 Pages 7375—7393
DOI https://doi.org/10.2147/IJN.S177350
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Philipp Diebolder,1 Miguel Vazquez-Pufleau,2 Nilantha Bandara,1 Cedric Mpoy,1 Ramesh Raliya,2 Elijah Thimsen,2 Pratim Biswas,2 Buck E Rogers1
1Department of Radiation Oncology, Washington University School of Medicine, St Louis, MO, USA; 2Department of Energy, Environmental and Chemical Engineering, Washington University in St Louis, St Louis, MO, USA
Introduction: Siliceous nanoparticles (NPs) have been extensively studied in nanomedicine due to their high biocompatibility and immense biomedical potential. Although numerous technologies have been developed, the synthesis of siliceous NPs for biomedical applications mainly relies on a few core technologies predominantly intended to produce spherical-shaped NPs.
Methods: In this context, the impact of different morphologies of siliceous NPs on biodistribution in vivo is limited. In the present study, we developed a novel technique based on an aerosol silane reactor to produce sintered silicon NPs of similar size but different surface areas due to distinct spherical subunits. Silica-converted particles were functionalized for radiolabeling with copper-64 (64Cu) to systematically analyze their behavior in the passive targeting of A431 tumor xenografts in mice after intravenous injection.
Results: While low nonspecific uptake was observed in most organs, the majority of particles were accumulated in the liver, spleen, and lung. Depending on the morphologies and functionalization, significant differences in the uptake profiles of the particles were observed. In terms of tumor uptake, spherical shapes with lower surface areas showed the highest accumulation and tumor-to-blood ratios of all investigated particles.
Conclusion: This study highlights the importance of shape and fuctionalization of siliceous NPs on organ and tumor accumulation as significant factors for biomedical applications.
Keywords: silicon, silica, human tumor xenograft, PEGylation, 64Cu
Introduction
Nanoparticle (NP) research has emerged as one of the top scientific fields due to the unique properties of nanomaterials with tremendous potential use in medicine and other fields.1,2 In contrast to traditionally used bulk materials, NPs possess unique biochemical, electronic, magnetic, and optical properties that rely on their sub-micrometer size (<1 μm) and their high surface-to-volume ratio.3 Nanoscale materials have now entered clinical trials with more than 100 agents being evaluated or approved by the US Food and Drug Administration.4 The current clinical nanomedicine approaches investigate many different diseases (ClinicalTrials.gov) but mainly focus on the imaging and treatment of cancer.5,6 Compared to most traditional therapeutics, NP-based drug delivery systems (DDSs) facilitate the fine-tuning of many important properties influencing the drug pharmacokinetics including solubility, circulation time, and target-specific distribution.7 Although current commercial nanodrugs are still single functional, NPs can be engineered as multifunctional systems allowing several functions at the same time, eg, specific targeting, controlled drug release, and in vivo imaging.8 However, integrating several nanomaterials into one multifunctional system remains challenging and is the subject of intensive research.9 Nevertheless, much preclinical research is focused on the development of multifunctional agents to merge therapeutic and diagnostic modalities in one single compound and thus bring us closer to the long-conceived dream of “theranostics”.10,11
The immense interest in nanomedicine led to the development of numerous DDSs including micelles, liposomes, proteins, dendrimers, and polymers.12 Last but not least, inorganic materials have been studied for their potential use in nanomedicine since they offer advantages in terms of synthesis and stability.13 Most of the inorganic materials in nanomedicine are either based on noble metals or based on metal oxides, but concerns about the toxicity of some of these compounds arose and are subject of intense discussion.14–17 A special class of inorganic nanomaterials is based on semiconductor silicon (Si) and its oxide silica (Si dioxide [SiO2]).18–21 Siliceous materials have been studied as inorganic alternatives with low toxicity for nanomedicine due to their high biocompatibility and nontoxic degradation products, eg, nontoxic silicic acid that is rapidly excreted by the kidneys.22
Nowadays, silica NPs (SiO2NPs) are the best-investigated class of siliceous nanomaterial in biomedicine due to their high biocompatibility, degradability, tunable morphology, and ease of modification.20–23 Moreover, silica can be integrated with other nanomaterials to achieve multifunctionality, eg, by coating other inorganic materials such as gold and iron oxide to produce core–shell NPs.24 As a most prominent example, Cornell dots (C-dots) possess a fluorescent core and represent the first SiO2NPs under clinical investigation.25,26 Biomedical SiO2NPs are typically produced by various bottom-up approaches where precursor particles grow in size via chemical reactions. Most of the current approaches are based on sol–gel methods evolved from the Stöber process introduced in 1968.27 This approach allows the production of monodisperse silica particles in varying sizes (5–2,000 nm) by the hydrolysis and condensation of silanes in aqueous solution and in the presence of a mineral acid or base as catalyst. By adding surfactants to the process, pores of different shapes and sizes can be integrated into the silica network. This allows the generation of mesoporous SiO2NPs with high surface areas that are promising platforms for robust and controllable drug delivery.28 Recently, a new class of silica hybrid analogs has emerged, using various organosilica precursors during the sol–gel synthesis. These (periodic) mesoporous organosilica NPs contain organic bridges in their silica network that facilitates the functionalization for biomedical applications.21
The other class of siliceous nanomaterial being investigated in biomedicine is based on elemental Si.19 In general, biomedical Si NPs (SiNPs) are characterized by a fast hydrolytic in vivo degradation and renal clearance.22 Porous Si (pSi) NPs are fabricated in various top-down approaches relying on etching and milling. Most frequently electrochemical etching of the surface of a Si wafer is used to produce both spherical-shaped and irregularly shaped pSi.29,30 Recently, pSi NPs have received attention as promising multifunctional platforms for drug delivery and noninvasive imaging due to high drug loading capacity and adjustable pore sizes.19 Although thin layers of native oxide are usually observed, pSi NPs are often oxidized in controlled conditions to produce defined layers of SiO2 of varying thicknesses to archive higher in vivo stability, improved functionalization, and better drug release.19,29 In terms of imaging, oxidized pSi NPs exhibit intrinsic near-infrared photoluminescence due to quantum confinement effects and defects at the Si–SiO2 interface, which can be used for in vivo tracking.31 In contrast, this intrinsic luminescence of pSi NPs is rapidly lost under physiological conditions and other imaging technologies such as MRI, PET, and SPECT have successfully been implemented.24 In contrast to pSi, nonporous SiNPs can be typically produced by numerous technologies including both top-down and bottom-up approaches but also mixtures of both involving decomposition and re-assembly of precursors.32 Currently, these approaches are predominantly used to synthesize smaller (2–8 nm), colloidal quantum dots (QDs), which are highly attractive for in vitro and in vivo imaging applications due to their low toxicity compared to conventional heavy metal QDs.33
In this article, we implemented a novel aerosol reactor-based approach to produce various siliceous NPs for potential biomedical applications. First, nonporous SiNPs were synthesized by thermal decomposition of silane in an aerosol reactor. By adjusting the reactor conditions, we could control the synthesis process to produce three differently shaped SiNPs of similar final size (~170 nm) but consisting of differently sized smaller primary particles. Outer layers of these SiNPs were converted by thermal oxidation into SiO2 to obtain higher biostability in vivo. To analyze their biomedical potential, the functionalized NPs were radiolabelled with copper-64 (64Cu) and injected in tumor-bearing mice. For comparison, in vivo studies were done in parallel with spherical SiO2NP of similar size that was produced by a standard sol–gel approach. Biodistribution of both polyethylene glycol (PEG)ylated and non-PEGylated NPs was analyzed after 4 and 24 hours of circulation and tissue uptake discussed in light of morphology and functionalization. Our data show the value of these aerosol-synthesized Si–SiO2 nanocomposites for potential biomedical applications and give insight into the impact of morphology and functionalization of these NPs on in vivo organ distribution.
Materials and methods
Chemicals and reagents
All chemicals and reagents were purchased from commercial suppliers and used without further purification. Silane in helium (0.9% SiH4), helium, and nitrogen (all ultrahigh purity) were obtained from Airgas (Radnor, PA, USA) and used for synthesizing SiNPs in the gas phase. Ultrapure type I water (resistivity: 18.2 MΩ·cm) was obtained from an in-house PURELAB flex system from Evoqua Water Technologies (Pittsburgh, PA, USA). (3-Aminopropyl)-triethoxysilane (APTES, 99%), ammonium acetate (NH4OAc) (≥99.99% trace metal basis), Chelex 100 sodium form, and dimethyl sulfoxide (DMSO, anhydrous, ≥99.9%) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Absolute ethanol (200 proof) was bought from Decon Labs (King of Prussia, PA, USA), and ITLC-SG paper was bought from Agilent Technologies (Santa Clara, CA, USA). Chelator 2-S-(4-isothiocyanatobenzyl)–1,4,7-triazacyclononane-1,4,7-triacetic acid (p-SCN-Bn-NOTA, ≥94%) was purchased from Macrocyclics (Plano, TX, USA), cross-linker MAL-PEG-SCM (molecular weight: 5 kDa) was purchased from Creative PEGWorks (Chapel Hill, NC, USA), and diethylenetriaminepentaacetic acid (DTPA, 98.5%) was purchased from Strem Chemicals (Newburyport, MA, USA). 64Cu (t1/2=12.7 hours, β+=17%, β−=39%, electron capture =43%, Emax =0.656 MeV) was produced by a (p,n) reaction on enriched 64Ni on a TR-19 biomedical cyclotron (Advanced Cyclotron Systems Inc., Richmond, BC, Canada) at Mallinckrodt Institute of Radiology, Washington University School of Medicine (WUSM) and purified with an automated system using standard procedures.34,35 For radiolabeling, a stock solution of 64CuCl2 was diluted with a 10-fold excess of 0.1 M NH4OAc (pH 5.5). FBS, DMEM, PBS (1×), and trypsin-EDTA were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Sterile, nonpyrogenic saline (0.9% sodium chloride injection) was purchased from Hospira (Lake Forest, IL, USA).
Synthesis of aerosol-synthesized NPs
Nonspherical siliceous NPs (NP1, NP2, and NP3) were synthesized via silane pyrolysis in a silane furnace aerosol reactor. The reactor was made of a stainless steel tube (1″ diameter and 18″ long) inside a Lindberg Furnace (Thermo Fisher Scientific). The reactor was fed by gas from three cylinders with a fixed chemical composition of silane in helium (0.9% SiH4), helium, and nitrogen. Mass flow controllers (MKS Instruments, Andover, MA, USA) were used to independently regulate the flow of each gas stream into the reactor. A pressure transducer (WIKA Instruments, Klingenberg am Main, Germany) was used to monitor pressure. Data acquisition and user interface were achieved via LabVIEW (National Instruments, Austin, TX, USA) and a data acquisition board (National Instruments). NP1 was synthesized by feeding the reactor with 0.004% silane, 19.021% helium, and 80.975% nitrogen using a reactor temperature of 1,000°C. Produced Si particles were collected on quartz filters (Advantec MFS, Dublin, CA, USA) for 10 hours during continuous reactor operation. NP2 and NP3 were produced in a similar way using 0.02% silane and 99.98% helium under constant flow using temperatures for the silane pyrolysis of 600°C (NP2) or 1,000°C (NP3) and 10 hours continuous reactor operation. Outer layers of SiNPs were converted into silica by thermal oxidation using particle-specific conditions. NP1 was isothermally treated for 1 hour at 900°C by increasing the temperature with 1°C/min up to 400°C and 5°C/min to the final temperature. NP2 was oxidized for 5 minutes at 500°C. NP3 was heated for 10 hours at 600°C, followed by 10 hours at 900°C. Right before further experimentation, NPs were diluted in ultrapure water to 0.9 mg/mL (ie, 15 mM) and well-dispersed by ultrasonic treatment.
Synthesis of spherical NPs
Spherical NP4 was synthesized using a sol–gel method following the procedure outlined by Möller and Bein.36 To synthesize, 4 mL of absolute ethanol and 2 mL of ultrapure water were combined in a 50 mL glass vial and sonicated for 3 minutes at room temperature (RT) using a bath sonicator. While maintaining sonication, 200 μL of tetraethyl orthosilicate (TEOS) was pipetted to the solution and sonicated for an additional 3 minutes. After the addition of 0.5 mL of ammonium hydroxide (NH4OH), the solution was sonicated for an additional 7–12 minutes and NP4 was separated by centrifugation. The supernatant was discarded, and the vials were kept at 60°C in an oven for 1 hour to get SiO2NPs in powdered form. NP4 was treated and stored as described earlier for aerosol-synthesized particles.
Surface modification of NPs with amines
The silica surface of the NPs was functionalized with amine groups by treatment with APTES. Typically, 60 μmol of NPs was centrifuged (16,800× g, 10 minutes) and resuspended in 7.6 mL of 5% H2O diluted in absolute ethanol (pH 5.0). For silanization, 400 μL of APTES was added to a final concentration of 5%. The mixture was incubated for 3 days at 60°C in sealed glass vials and vortexed from time to time. Unreacted APTES was removed by washing three times with 70% ethanol. The final product was dispersed in ultrapure water to 0.1 μmol/μL. The presence of primary amines was determined by a Ninhydrin Test Kit (AnaSpec, Fremont, CA, USA) according to the manufacturer’s recommendations.
NOTA and PEG conjugation to amine-functionalized NPs
Stock solutions of the chelator p-SCN-Bn-NOTA (2 mM) and cross-linker MAL-PEG-SCM (20 mM) were prepared by diluting the compounds in DMSO and stored at −80°C until use. Thirty micromoles of the APTES-treated NPs was distributed in acid-washed conical tubes (1.5 mL; Thermo Fisher Scientific), centrifuged (16,800× g, 10 minutes), and washed once with 1 mL of DMSO. NPs were dispersed in 1 mL/tube DMSO, and p-SCN-Bn-NOTA stock solution was added to a molar ratio of 1.2–1 (NP to p-SCN-Bn-NOTA). To prepare PEGylated NPs, MAL-PEG-SCM stock solution was additionally added to a molar ratio of 1.2–1–10 (NP to SCN-Bn-NOTA to MAL-PEG-SCM). After adjusting the pH to 8.5, the tubes were incubated in an Eppendorf ThermoMixer (Hamburg, Germany) at 37°C for 1 hour shaking (1,000 rpm) to prepare the final NOTA or NOTA/PEG-conjugated NPs.
64Cu radiolabeling of NOTA-functionalized NPs
To radiolabel with radioactive 64Cu, NOTA-conjugated NPs (both non-PEGylated and PEGylated) were washed once with 1 mL/tube of radiolabeling buffer (0.1 M NH4OAc, pH 5.5). After centrifugation, NPs were resuspended in 200 μL/tube of radiolabeling buffer containing about 1 mCi of 64Cu (37 MBq). Radiolabeling occurred during an 1 hour incubation at 37°C and shaking at 800 rpm in an Eppendorf ThermoMixer. To complex unreacted 64Cu, pelleted NPs were resuspended in 800 μL/tube PBS and 200 μL of chelex-treated 50 mM DTPA solution (pH 7.0) and incubated for further 5 minutes at 37°C (800 rpm). After washing once with PBS, the final products from tubes containing similarly treated preparations of NPs were combined in one tube. Centrifuged NPs were re-dispersed in sterile saline to obtain 10 μCi (0.37 MBq) per injection (100 μL/mouse). The radiochemical purity of the final products after 64Cu-radiolabeling was analyzed by instant thin layer chromatography (ITLC). Silica gel-impregnated glass fibers were spotted 2 cm from the bottom with 1 μL of final products. The plates were developed in chelex-treated 50 mM DTPA (pH 7.0) solution until the solvent reached 7 cm. The dried plates were analyzed employing a BioScan radio-TLC Imaging Scanner AR-2000 (Eckert & Ziegler, Berlin, Germany) using the WinScan software (Version 3.13).
Characterization of bare and functionalized NPs
The NPs were characterized by different techniques including transmission electron microscopy (TEM), dynamic light scattering (DLS), laser Doppler velocimetry (LDV), and NP tracking analysis (NTA). TEM images were obtained using an FEI Tecnai G2 Spirit instrument (Thermo Fisher Scientific). To prepare TEM samples, 10 μL of each NP sample was pipetted on parafilm. Carbon-coated copper grids (200 mesh; TED Pella, Redding, CA, USA) were placed with the carbon site onto the drops and incubated for 1 minute. Excess samples were carefully removed with the outer tip of Whatman filter papers (GE Healthcare, Chicago, IL, USA) and grids air-dried prior to TEM imaging. A Malvern Zetasizer Nano ZS (ZEN3600; Malvern Instruments, Malvern, UK) was used to measure particle sizes by DLS employing disposable polystyrene cuvettes and ultrapure water as diluent. To analyze the zeta potential (ZP) by LDV, the water-dispersed NPs were filled in disposable polystyrene cuvettes and measured in the Malvern Zetasizer Nano ZS (ZEN3600) equipped with a universal dip cell. Sizes of the bare particle were further determined by NTA in a NanoSight NS300 instrument (Malvern Instruments)after dilution in ultrapure water and illumination by a green (532 nm) laser light.
Cell culture
The epidermoid carcinoma cell line A431 was purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA) and cultivated in complete growth medium (DMEM supplemented with 10% of heat-inactivated FBS) in low passage number in a humidified cell incubator (5% CO2, 37°C). For xenograft mouse models, A431 was expanded in T150 tissue culture flasks and harvested by trypsinization when reaching 70%–90% confluency. Harvested cells were washed three times with PBS and diluted to 5×107/mL in PBS.
Biodistribution
Animal experiments were conducted in compliance with the Guidelines for Care and Use of Research Animals established by Washington University Animal Studies Committee using a protocol approved by the committee. The 5–6-week-old female athymic nude mice (Foxn1nu/Foxn1+) were purchased from Envigo (Huntingdon, UK) and housed under pathogen-free conditions in the WUSM animal facility. Mice with an average weight of 25 g were anesthetized by the inhalation of 1%–2% isoflurane in 100% oxygen and implanted subcutaneously on the right shoulder with 5.0×106 A431 cells suspended in 100 μL of PBS. Tumors were allowed to grow for 2 weeks reaching final tumor weights ~350 mg on average. In vivo biodistribution studies were conducted in the Washington University Small Animal Imaging Facility. Isoflurane-anesthetized, tumor-bearing mice were intravenously injected with about 10 μCi (0.37 MBq) of NP-NOTA-64Cu or NP-NOTA-PEG-64Cu via the tail vein. After each time point (4 and 24 hours), mice were sacrificed by CO2 euthanasia and cervical dislocation. Tumors and selected organs (blood, muscle, lung, liver, spleen, kidney, bladder, heart, bone, pancreas, stomach, and intestine) were collected, weighted, and radioactively counted with a Beckman Gamma-8000 counter (Beckman Coulter, Brea, CA, USA). Tumor and organ uptake were analyzed and calculated as percentage of injected dose per gram (%ID/g).
Statistics
Data are represented as the mean ± SD of at least three independent experiments. Multiple comparisons were conducted by GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA) implementing one-way ANOVA, followed by Tukey’s multiple comparison test (****P<0.0001; ***P<0.001; **P<0.01; *P<0.05; nsP≥0.05).
Results
Synthesis of NPs
A novel aerosol reactor approach was developed for the synthesis of siliceous NPs with varying morphologies.37 The reactor setup (Figure S1A) and synthesis process can be outlined as follows. A stream of 0.9% silane gas in helium is further diluted with a carrier gas stream of either a helium/nitrogen mixture (NP1) or helium (NP2 and NP3). The mixed gas stream then flows continuously through a thermal reactor, made of a stainless steel tube inside a Lindberg furnace. Inside the reactor, as the stream flows into hotter zones, the gas heats up to a critical temperature where silane starts to pyrolise and to undergo nucleation and condensation thus forming monomeric primary particles of several nanometers in diameter. Depending on reactor conditions and silane concentration, primary particles continue to grow by agglomeration to form larger reversible structures (agglomerates). If the residence time and temperature in the reactor are high enough, the agglomerates might coalesce to form solid irreversible NPs (aggregates). In general, the aerosol reactor design enabled the accurate control of several process parameters including temperature (error proportional–integral–derivative controller: <±1°C), gas concentrations (error of mass flow controllers: <±5% of set point), and operation time. By controlling these parameters, specific and highly reproducible morphologies of synthesized SiNPs were obtained. For instance, the change in the reactor temperature from 600°C to 1,100°C resulted in an increase in the primary particle sizes and an increase in sphericity (Figure S1B). The geometric mean of the primary particle size distribution for each temperature could be reproduced even by different operators. For 700°C, the geometric mean of the primary particle size distribution was measured to be 10.3±0.87 nm (± standard error [SE]). At 1,000°C, the geometric mean of the primary particle size distribution was 29.4±0.59 nm (± SE), while final aggregate sizes were almost unchanged typically with an SE of ±0.6% for the geometric mean of the aggregate size distribution. As a common rule, higher silane concentration leads to a reproducible increase in primary particle sizes within the investigated dynamic range of the reactor (ie, 25 ppm to 0.9% SiH4).
In this study, the reactor conditions were adjusted to synthesize three types of NPs with different primary particle sizes as summarized in Table S1. The size distributions of the produced NPs were monitored in real time by a scanning mobility particle sizer (SMPS) to guarantee reproducible size distributions independent of the chosen conditions and run as well as to obtain fairly constant final aggregate sizes between all three types of NPs (Figure S1C). Final products were collected on quartz filters showing black (NP1 and NP2) to gray colors (NP3) (Figure S1D). To convert the Si surface to SiO2, products were treated by thermal oxidation using particle-specific conditions. Successful oxidation of particles was confirmed by changing colors of final products from black/gray (Si) to white (SiO2). The conversion of Si into SiO2 is expected to have occurred in a core–shell process, where the surface oxidizes first and the core of the NP at the end.38 Conversion efficacy of Si into SiO2 was calculated gravimetrically by weighing the filters before and after the oxidation process reaching >90% (NP2) or even >95% (NP1 and NP3). Control SiO2NPs of entirely spherical shape were produced by a standard sol–gel method as outlined in the “Materials and methods” section.
Characterization of bare NPs
The morphology of final NPs was first characterized by TEM. The analysis of TEM pictures revealed nonspherical shapes for all aerosol reactor-produced NPs (NP1, NP2, and NP3). As a common characteristic, the NPs were built from smaller primary particles of fairly spherical shape and different sizes depending on the conditions used during the synthesis. In contrast, the sol–gel-produced control particles (NP4) possessed uniform spherical shape. Figure 1 outlines the typical morphologies of the investigated NPs based on their appearance in TEM using the following color coding throughout this publication: NP1 in green, NP2 in blue, NP3 in red, and NP4 in orange. To determine the size of the final products, DLS analysis of water-dispersed NPs was performed showing comparable final particle sizes 145±33 nm (NP1: 106±17 nm, NP2: 140±11 nm, NP3: 146±6 nm, and NP4: 187±5 nm) (Table 1). A low polydispersity index (PDI) was seen for NP3 (0.10±0.01) and NP4 (0.05±0.02), indicating narrow monodisperse distributions of NPs. In contrast, higher PDIs were observed for the other NPs suggesting moderate polydisperse distributions (NP1: 0.49±0.05; NP2: 0.34±0.04). NPs were additionally visualized by high-resolution NTA to confirm the particle sizes obtained by DLS. The NTA data revealed slightly larger sizes of NPs with on average 174±18 nm. Sizes of NPs with low PDI in DLS measurements were almost identical in NTA (NP3: 148±77 nm; NP4: 188±46 nm). However, NPs with higher PDIs in DLS showed increased particle sizes in NTA, ie, NP1 (184±76 nm) and NP2 (175±65 nm). To gain further information about the particle morphology, TEM pictures were analyzed by ImageJ (National Institute of Health, Bethesda, MD, USA) to determine the average sizes of the single spherical subunits. By measuring the spherical areas (n=100), diameters of primary particles were calculated. NP1 showed the smallest subunit sizes with only 6±2 nm in diameter, followed by NP2 with 14±3 nm and finally NP3 with an average diameter of 41±11 nm (Figure S2). Due to the monodisperse nature of the spherical control particles, the ImageJ analysis was used to additionally confirm the final particles sizes of NP4 with 187±39 nm. Based on the appearance in TEM and the similar final particle sizes, the relative surface areas of final particles were estimated assuming a spherical composition of all final products. The smallest surface area was anticipated for the entirely spherical control particle NP4 and increased for the nonspherical NPs depending on the sizes of their primary subunits from large for NP3 (41 nm), via intermediate for NP2 (14 nm), to small for NP1 (6 nm) (Figure 1).
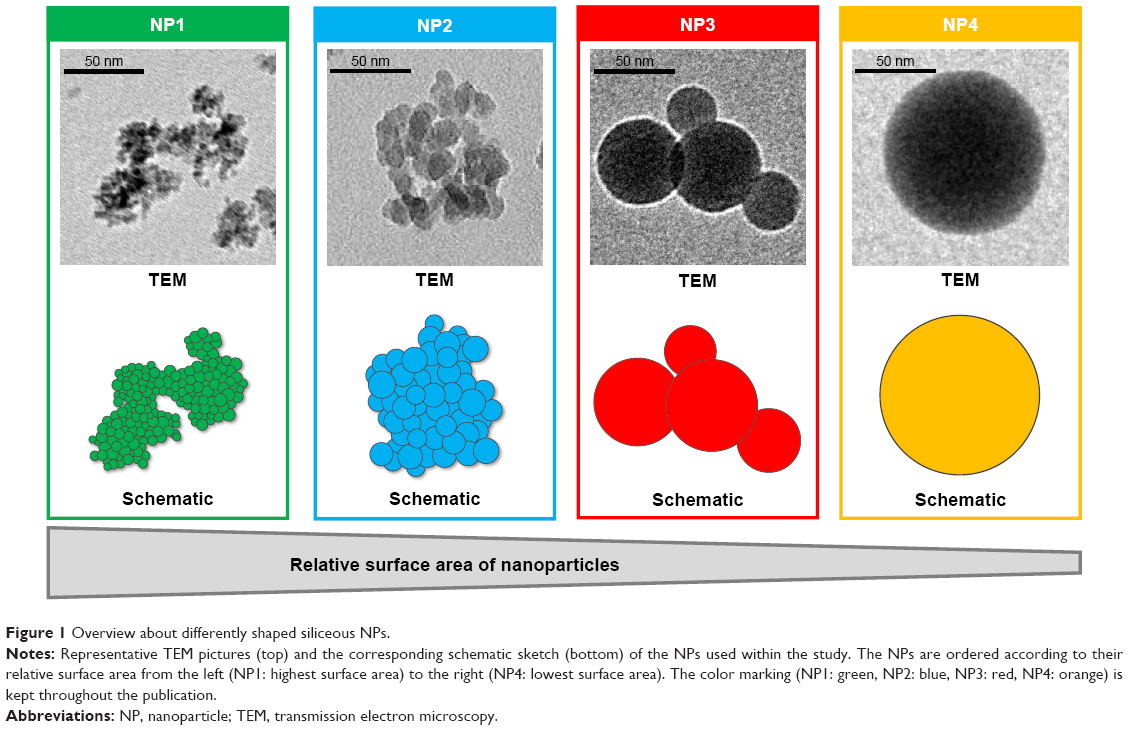 | Figure 1 Overview about differently shaped siliceous NPs. |
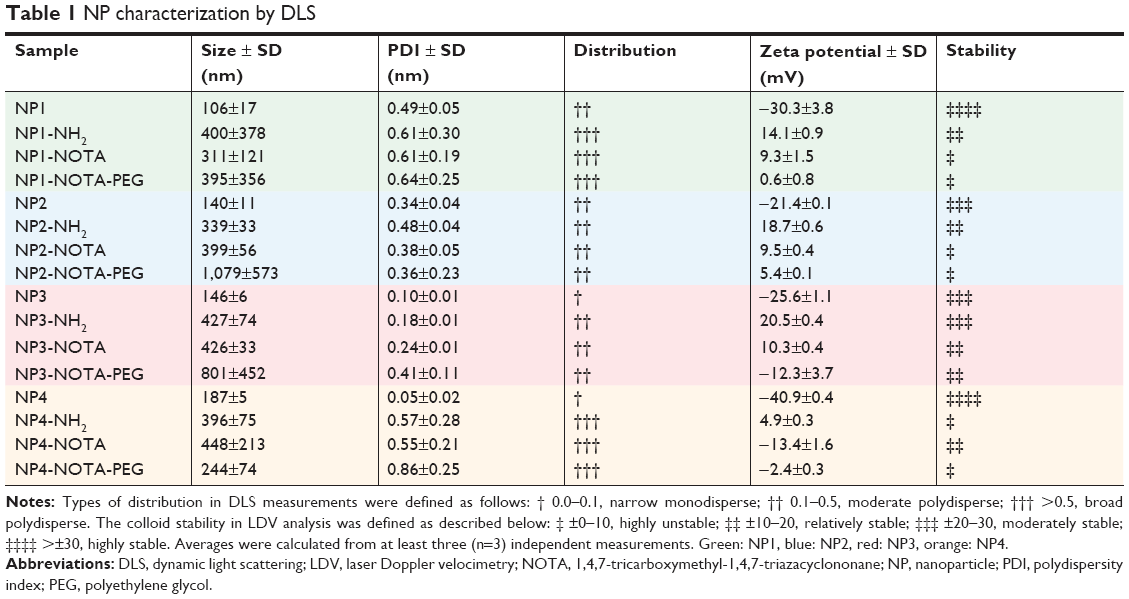 | Table 1 NP characterization by DLS |
Functionalization and characterization of modified NPs
In preparation for 64Cu radiolabeling and in vivo tracking in tumor-bearing mice, NPs were modified by various compounds, including APTES, NOTA, and PEG. First, bare NPs were chemically treated with APTES to cover the surfaces with organofunctional alkoxysilane molecules. Silanization was performed for up to 72 hours at 60°C using 5% APTES dissolved in an ethanol/water mixture. Successful functionalization of the various NPs was confirmed by a semiquantitative ninhydrin test kit (data not shown). In the next step, the silanized NPs were conjugated to the NOTA chelator using the amine-reactive cross-linker p-SCN-Bn-NOTA. Primary amines of silanized NPs were reacted with the isothiocyanate groups of p-SCN-Bn-NOTA to form stable thiourea bonds in DMSO at pH 8.5 for 1 hour at 37°C as described. To further investigate the influence of PEGylation on biodistribution of the NPs, amine-reactive MAL-PEG-SCM was optionally added during the chelator reaction using a molar excess of MAL-PEG-SCM to SCN-Bn-NOTA of 10–1. Primary amines of silanized NPs reacted with the succinimidyl NHS ester group of MAL-PEG-SCM to stable carboxamide bonds, thus adding an additional 5 kDa linear PEG chain to the functionalized NPs.
The NPs were then characterized by DLS in order to determine potential changes in particle sizes. The data for the DLS measurements of water-dispersed NPs are summarized in Table 1. On average, silanization of bare NPs by APTES lead to about a threefold increase in particle size, independent of the particle shape. NOTA conjugation to silanized NPs did not change the size further. An approximate 1.7-fold increase in particle size was observed when PEG was used. The PDI for the DLS measurements was in the moderate polydisperse range (0.1–0.5) for all functionalized samples of NP2 and NP3. In contrast, broad polydispersity (>0.5) was observed for NP1 and NP4 after all functionalization steps.
To investigate the influence of the surface modification on stability, the ZPs of water-dispersed NPs were evaluated (Table 1). As described below, the different functionalization steps changed the ZPs of the NPs in similar ways independent of the particle type. In general, the bare particles gave negative ZPs with an average of −30±8 mV, indicating moderate (NP2 and NP3) to high (NP1 and NP4) stabilities. Compared to these bare particles, surface modification by APTES treatment increased (44±3 mV) the ZPs to 15±7 mV suggesting reduced stabilities (relatively stable: NP1 and NP2; moderately stable: NP3; and highly unstable: NP4). The further addition of chelator NOTA to the silanized NPs decreased (−11±6 mV) the ZPs to an average of 4±12 mV, signaling even lower overall stabilities (relatively stable: NP3 and NP4; highly unstable: NP1 and NP2). Similar to the NOTA conjugation alone, the addition of both NOTA and PEG to the silanized NPs resulted in a comparable decline (−17±11 mV) of the ZPs close to zero (−2±8 mV), indicating again very low colloid stabilities (relatively stable: NP3; highly unstable: NP1, NP2, and NP4).
Radiolabeling and biodistribution of NPs
As a final step, in vivo mouse studies were conducted in order to determine the impact of the NP shape and PEGylation on organ distribution and tumor uptake. Therefore, the various types of NOTA-conjugated NPs were first radiolabeled with 64Cu. To guarantee high radiochemical purity of the final products before injection, ITLC of the 64Cu-radiolabeled NPs was performed. The ITLC profiles confirmed the high efficacy of radiolabeling with an average radiochemical purity of 93%±12% as calculated from the regions of interests (ROIs). For proper comparison, 10 μCi (0.37 MBq) of the final radiolabeled NPs were injected in the tail vein in athymic nude mice bearing A431-tumor xenografts. Mice were sacrificed after 4 or 24 hours to determine the uptake of the radioactive NPs as %ID/g into selected organs and tumors. The complete dataset of this biodistribution study is given in Tables S2–S5, including all collected organs and the tumors. Based on the distinct nature of the NPs, significant differences in the uptake profiles of the particles were observed. In general, the highest uptake of radiolabeled NPs was seen for lung, liver, spleen, kidneys, and intestine. In contrast, tumors and all other investigated organs showed lower overall uptake of radiolabeled NPs. To better emphasize these differences, only the biodistribution data of relevant organs and tumors are discussed separately and, in more detail, below. The biodistribution of non-PEGylated NPs in the major organs is shown in Figure 2A (4 hours) and Figure 2B (24 hours), and that of PEGylated NPs is shown in Figure 3A (4 hours) and Figure 3B (24 hours). Due to the scale difference, the uptake in the tumors after 24 hours is shown separately (Figure 4).
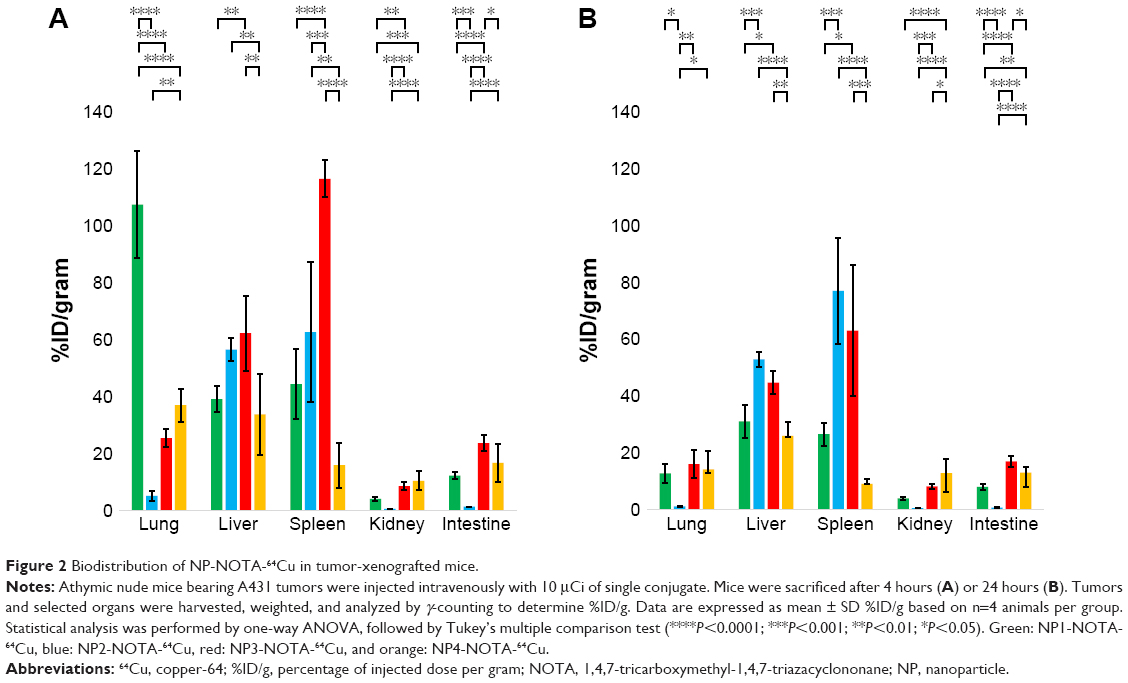 | Figure 2 Biodistribution of NP-NOTA-64Cu in tumor-xenografted mice. |
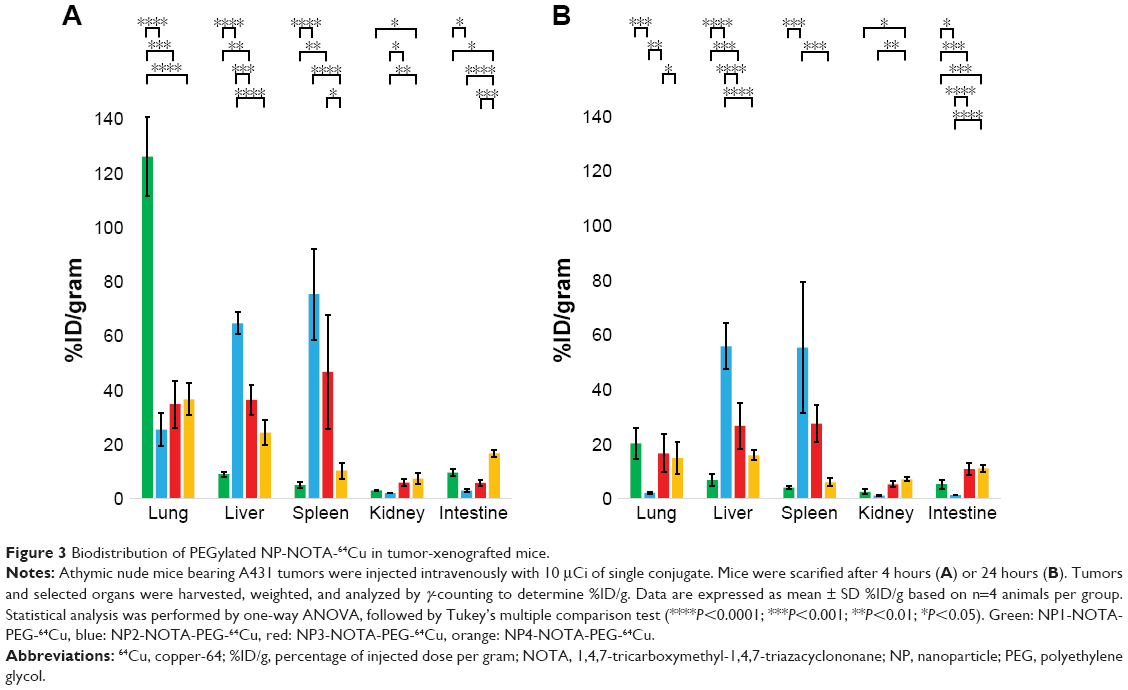 | Figure 3 Biodistribution of PEGylated NP-NOTA-64Cu in tumor-xenografted mice. |
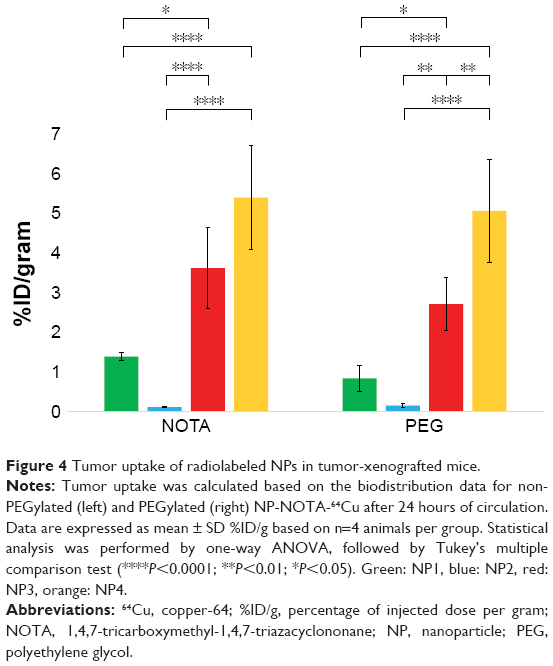 | Figure 4 Tumor uptake of radiolabeled NPs in tumor-xenografted mice. |
Lung uptake
The apparent uptakes of radiolabeled NPs into the lungs showed dramatic differences between various types of NPs. Very high initial lung uptake (non-PEG: 108.84±19.07%ID/g; PEG: 126.06±14.51%ID/g) was observed for aerosol-synthesized NP1 possessing the highest relative surface areas of all particles. In contrast to all other types of NPs (all <37.46%ID/g), the lung uptake of NP1 at the 4 hours time point was significantly increased for both non-PEGylated and PEGylated NPs (all P<0.0001). Interestingly, this high uptake of NP1 into the lungs was mostly dependent on the circulation time but not on the PEGylation status. The uptake in lungs clearly decreased after 24 hours (non-PEG: 12.86±3.37%ID/g; PEG: 20.23±5.72%ID/g), showing less significant differences compared to the other NPs. Moreover, the calculated P-values of the non-PEGylated and PEGylated products confirmed that the lung uptake of NP1 was independent of PEGylation (4 hours: P=0.2484; 24 hours: P=0.4278). According to this, PEGylation had no significant effect on lung uptakes for the other NPs (4 hours: all P>0.1128; 24 hours: all P>0.9999).
In summary, aerosol-synthesized NP1 with the highest surface area of all particles showed superior lung uptake after 4 hours. This effect was almost negated after 24 hours when compared with the other NPs. PEGylation had no significant influence on lung uptake for any particle type.
Liver uptake
By analyzing the liver distributions, very significant differences were calculated for the aerosol-synthesized NP2 after PEGylation compared to all other NPs (all P<0.0007). NP2 showed the highest liver uptake after PEGylation of all NPs that was independent of the analyzed time point (4 hours: 64.54±4.07%ID/g; 24 hours: 55.77±8.33%ID/g). Furthermore, the high liver uptake of NP2 was unaffected by PEGylation (4 hours: P=0.8864; 24 hours: P=0.9943), reaching similar high overall uptakes for the non-PEGylated NP2 (4 hours: 57.28±4.24%ID/g; 24 hours: 52.95±2.68%ID/g). Similarly, the intermediate liver uptake of spherical NP4 (4 hours: 34.17±14.35%ID/g; 24 hours: 26.02±4.96%ID/g) did not significantly change after PEGylation (4 hours: P=0.6345; 24 hours: P=0.1742). The other nonspherical particle NP1 (4 hours: 39.64±4.56%ID/g; 24 hours: 31.11±5.72%ID/g) and NP3 (4 hours: 63.06±13.29%ID/g; 24 hours: 44.74±4.04%ID/g) revealed higher relative accumulation in that organ. However and in contrast to NP2 and NP4, PEGylation led to a significant drop in liver uptake of both NP1 (4 hours: P=0.0003; 24 hours: P<0.0001) and NP3 (4 hours: P=0.0014; 24 hours: P=0.0015).
Overall, the aerosol-synthesized NP2 showed relatively high uptake into the liver that was time and PEGylation independent. Similar to this, the intermediate liver uptake of sol–gel-synthesized NP4 was PEGylation independent. In contrast, the intermediate (NP1) or high (NP3) liver uptake of the remaining aerosol-synthesized NPs was clearly decreased when PEGylated.
Spleen uptake
The analysis of spleen uptakes showed an extremely high accumulation of non-PEGylated NP3 (nonspherical, low surface area) after 4 hours (117.97±6.57%ID/g) that was significantly higher than for all other NPs (all P<0.0003). This effect of increased spleen uptake of NP3 was mostly dependent on time and PEGylation status showing lower uptake after 24 hours (63.01±23.13%ID/g) or PEGylation (4 hours: 46.71±21.08%ID/g; 24 hours: 27.45±6.66%ID/g). Calculating the P-values confirmed that the observed differences between non-PEGylated and PEGylated NP3 were significant (4 hours: P<0.0001; 24 hours: P=0.0237). NP1 revealed intermediate (4 hours: 44.95±12.46%ID/g) or low (24 hours: 26.66±4.04%ID/g) spleen uptake that decreased after PEGylation (4 hours: 4.89±1.02%ID/g, P=0.0106; 24 hours: 4.11±0.58%ID/g, P=0.3267). High apparent uptake (4 hours: 63.45±24.78%ID/g; 24 hours: 77.02±18.71%ID/g) was seen for the NP2 independent of PEGylation (4 hours: P=0.9294; 24 hours: P=0.3702). The spherical control NP4 showed only low spleen uptake (4 hours: 16.15±7.95%ID/g; 24 hours: 9.13±1.60%ID/g) also independent of PEGylation (4 hours: P=0.9987; 24 hours: P>0.9999).
In summary, very high initial spleen uptake was observed for non-PEGylated NP3 that decreased over time. PEGylation reduced the spleen uptake of NP3 dramatically. A similar behavior (ie, higher initial spleen uptake when non-PEGylated) was observed for NP1, although the overall uptake was clearly lower. In contrast, overall high or low spleen uptake was observed for NP2 and NP4, respectively, but independent of time and PEGylation.
Kidney uptake
Uptake of NPs in the kidneys was lower when compared with the lung, liver, and spleen. Interestingly, the kidney uptake seemed to be mainly time independent for all types of NPs. The highest accumulation was seen for the spherical NP4 (4 hours: 10.59±3.37%ID/g; 24 hours: 12.97±4.78%ID/g), followed by the nonspherical NP3 (4 hours: 8.68±1.32%ID/g; 24 hours: 8.28±0.97%ID/g), NP1 (4 hours: 4.13±0.57%ID/g; 24 hours: 4.08±0.47%ID/g), and finally NP2 with very low overall kidney uptake (4 hours: 0.62±0.06%ID/g; 24 hours: 0.60±0.07%ID/g). PEGylation of the nonspherical NPs had no major impact on the kidney uptake (all P>0.1971). However, PEGylation of the spherical NP4 was more relevant especially after 24 hours (4 hours: P=0.0904; 24 hours: P=0.0029), showing an overall reduced kidney accumulation (4 hours: 7.26±2.09%ID/g; 24 hours: 7.11±0.72%ID/g).
The kidney uptake was higher for the spherical NP4 than for all nonspherical NPs. Almost no differences between the time points in kidney uptake could be measured. PEGylation had no significant influence on kidney uptake for the nonspherical NPs and moderate effect on the spherical NP4 after 24 hours with a slightly decreased kidney uptake.
Intestinal uptake
Highest intestinal uptake was observed for nonspherical NP3 (4 hours: 23.96±2.90%ID/g; 24 hours: 17.05±1.97%ID/g), followed by spherical NP4 (4 hours: 16.92±6.74%ID/g; 24 hours: 13.12±2.02%ID/g), and nonspherical NP1 (4 hours: 12.43±1.21%ID/g; 24 hours: 8.10±1.01%ID/g). As already seen for the kidneys, the accumulation of NP2 into the intestine was very limited (4 hours: 1.36±0.04%ID/g; 24 hours: 0.82±0.08%ID/g) and significantly lower than for all other NPs. Independent of the particle morphology, the differences of intestinal uptake to the two different time points were mostly not significant (all P>0.4076) except for NP3 with a slight significant decrease over time (P=0.0372). PEGylation did not change intestinal uptake for NP1, NP2, and NP4 (4 hours: P all >0.7973; 24 hours: P all >0.2321). However, a significant decrease in intestinal uptake was observed after PEGylation of NP3 (4 hours: P<0.0001; 24 hours: P=0.0002).
In summary, there was a significantly higher intestinal uptake of NP3 than for all other NPs, especially to the early time point. In contrast to all other particles, there was a significant decrease in the intestinal uptake of NP3 after PEGylation, especially after 4 hours.
Tumor uptake
Good tumor penetration and retention are essential for the successful development of NPs as potential vectors for delivering therapeutic payloads and for imaging cancer. Figure 4 compares the tumor uptakes for the non-PEGylated vs PEGylated NPs after 24 hours. In general, the uptake of NPs into the A431 tumors showed a pattern similar to that observed for the kidneys. The highest uptake in tumors was seen for the spherical NP4 (non-PEG: 5.39±1.31%ID/g; PEG: 5.05±1.30%ID/g), followed by the nonspherical NP3 (non-PEG: 3.61±1.02%ID/g; PEG: 2.70±0.67%ID/g), NP1 (non-PEG: 1.38±0.10%ID/g; PEG: 0.84±0.33%ID/g), and NP2 with very low overall tumor uptake (non-PEG: 0.11±0.01%ID/g; PEG: 0.15±0.05%ID/g). By analyzing the P-values, this pattern was basically independent of time (NOTA: all P>0.2663; PEG: all P>0.5292) or PEGylation (4 hours: all P>0.9765; 24 hours: all P>0.7299).
Overall tumor uptake was higher for spherical NP4 than for the nonspherical NPs (NP3>NP1>NP2). In general, tumor uptakes were basically independent of time or PEGylation.
Tumor-to-organ ratios
Based on the uptakes obtained during the in vivo mice studies, both tumor-to-blood (TTB) and tumor-to-muscle (TTM) ratios were calculated (Table S6), showing nonsignificant differences between both time points (all P>0.0883). Figure 5A highlights the calculated TTB and Figure 5B the TTM ratios after 24 hours. Analogous to the absolute tumor uptakes, the highest ratios were observed for the spherical NP4 (TTB: 3.13±0.62; TTM: 7.76±0.31) that was significantly higher than for all the nonspherical NPs. No significant differences were seen between various nonspherical NPs neither in TTB (NP1: 1.66±0.31, NP2: 1.38±0.19, and NP3: 1.58±0.50) nor in TTM ratios (NP1: 4.12±1.14, NP2: 4.39±1.36, and NP3: 4.34±1.12). By analyzing the P-values, PEGylation of NPs had no significant influence on neither the TTB ratio (all P>0.5984) nor the TTM ratio (all P>0.9891).
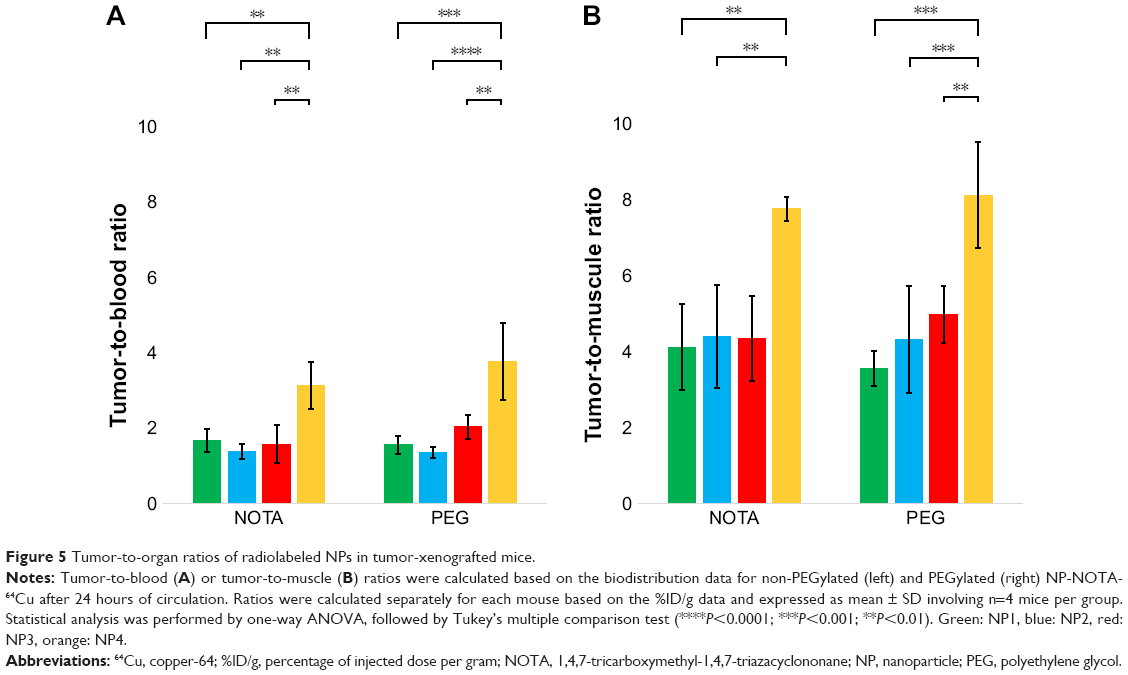 | Figure 5 Tumor-to-organ ratios of radiolabeled NPs in tumor-xenografted mice. |
In summary, sol–gel-produced NP4 showed higher TTB and TTM ratios than the aerosol-synthesized NPs. In fact, no significant difference in ratios of TTB and TTM could be measured between the aerosol-synthesized NPs. Both TTB and TTM ratios were independent of the time point and PEGylation for all investigated NPs.
Discussion
Particle synthesis
In this study, we implemented an aerosol reactor approach to synthesize morphologically different siliceous NPs of similar aggregate sizes (~170 nm) for their evaluation in biomedical applications. Based on the process, these compounds are composed of primary spherical subunits of different sizes (ie, 6, 14, and 41 nm) but form stable aggregates due to thermal-induced sintering during particle synthesis. Most current technologies for the production of biomedical siliceous NPs often rely on liquid phase techniques (ie, sol–gel methods for the synthesis of SiO2NPs)27,39 or top-down approaches (ie, etching and milling for the fabrication of pSi).19,40 In contrast, the de novo growing in the gas phase is mainly used to synthesize smaller (<10 nm) colloidal SiNPs for imaging applications (eg, Si QDs).33 Here, different technologies have been established including supersaturated Si vapor in a microplasma reactor41 or by the pyrolysis of silane gas in a microwave plasma reactor.42 Last but not least, fumed silica may be synthesized by high temperature hydrolysis of SiCl4 in an O2(N2)/H2 flame, where small primary SiO2NPs (5–50 nm) form branched hard-agglomerates but usually grow up to micrometer size (0.1–100 μm) and are mainly used in industrial applications.43 In our approach, we used a furnace aerosol reactor for the pyrolysis (ie, thermal anoxic decomposition) of monosilane (SiH4) gas to grow SiNPs of defined nanometer size and morphologies, which might also be valuable for biomedical applications. Historically, early attempts in the aerosol-assisted production of NPs were often developed empirically since a theoretical understanding of the physico-chemical details was missing. Indeed, aerosol synthesis of SiNPs is composed of several overlapping steps (chemical reaction, nucleation, growth, agglomeration, and under some circumstances, sintering) that are challenging to study due to the high temperatures, short reaction times, and limited access to track the reaction.44 Nevertheless, recent improvements in multiscale modeling of the aerosol synthesis process for SiNPs have allowed better predictions in the synthesis of narrow sized primary particles and controlling the synthesis process.44 In accordance, we have developed a reactor setup and process control that allowed us to produce narrow size distributions of primary SiNPs that differed depending on the reactor temperature. Yet, the final size distributions of sintered aggregates remained practically unaffected during synthesis within a broad temperature range of 600 to 1,100°C. Based on this observation, we developed a stable process to synthesize aggregates of similar size distributions but made of primary particles of different sizes to analyze their potential in biomedical applications.
Particle functionalization
Several approaches are now available to study the biodistribution of NPs, including inductively coupled plasma-optical emission spectroscopy/mass spectrometry, fluorescence, and radiolabeling. Based on our long-standing expertise, we performed radiolabeling with 64Cu to evaluate the potential of our aerosol-synthesized NPs for bio-applications. This technique requires the covalent linkage of copper chelators (eg, NOTA) to the particle surface. In general, surface modification of the SiNPs (eg, colloid Si QDs) can be achieved by various techniques to generate either hydride or halogen-terminated surfaces. In a next ligand exchange step, hydrosilylation of the hydride-terminated or nucleophilic substitution of the halogen-terminated surfaces is performed to establish robust covalent linkages between the Si surface atoms and carbon, nitrogen, or oxygen species.32 These techniques allow the formation of self-assembled monolayers (SAMs) of a variety of distal moieties and are highly useful for subsequent functionalization steps. In contrast to these mentioned approaches, we decided on another widely used strategy of Si surface modification by adding stable silica shells to the Si cores. Here, thermal oxidation is by far the most used process due to its simplicity, adaptability, and relatively low cost of implementation. Thermal oxidation of our aerosol-synthesized Si particles was performed under morphology-dependent conditions and required high temperatures with varying exposure times of up to several hours. In general, successful oxidation of SiNPs can be performed in a wide range of process temperatures (RT to 1,100°C) that requires very little oxygen.45 Thermal oxidation of SiNPs occurs in a two-step shrinking core model46 and might result in stable silica layers of different oxygen contents and sometimes different surface hydride species depending on the condition used for thermal treatment.38 Since all our particles have been processed at a temperature of ≥500°C, it can be anticipated that these particles contain a crystalline Si core, covered by a layer of amorphous Si and an outer Si oxide shell highly valuable for further particle functionalization. Moreover, we preferred the silica conversion strategy for the following two additional reasons: first, SiO2NPs possess prolonged circulation times and in vivo stabilities compared to their Si counterparts (half-life of days compared to hours). Second, there are a variety of well-known silica surface modification technologies on hand including those based on various commercially available alkoxysilanes and halosilanes.47 In the case of alkoxysilanes, stable 1–3 Si−O−Si links with the surface silanol groups (≡Si−OH) are formed in a condensation reaction. Most frequently, APTES or (3-mercaptopropyl)-trimethoxysilane (MPTMS) is used for silica surface modification to add either aminopropyl silane (APS) or mercaptopropyl silane (MPS) films. The covalently linked primary amines (−NH2) or thiol groups (−SH) of such modified silica surfaces facilitate the linker chemistry with other frequently used linking moieties including those based on n-hydroxysuccinimide (NHS), isothiocyanates (both for amine coupling), or maleimides (for thiol coupling). In the current study, the silica surface of our NPs was modified with primary amines by APTES treatment to allow subsequent functionalization with NHS-functionalized NOTA or NHS-functionalized PEG. However, APTES treatment has increased the apparent sizes of our aerosol-synthesized NPs by about threefold. Since this effect was also observed for our spherical control particles, we conclude that the particle aggregation after amine modification was independent of the morphology. The phenomena of aggregation of SiO2NPs after APTES treatment can be explained by an excess of positively charged amino groups on the silica surface at physiological pH.48 Indeed, APTES treatment of our NPs increased the ZPs by an average of 44.1±2.8 mV to the low positive range indicating decreased overall particle stabilities. APTES-modified SiO2NPs are known to back-bond primary amines to surface silanol groups that might induce particle aggregation due to the reduced shear plane and electrostatic repulsion. Subsequent functionalization by NOTA or NOTA/PEG moieties slightly decreased ZPs but still stayed in the range of low overall stability (ie, around ±0 mV). Thus, decreasing the ratio of surface amines to silanol groups should reduce particle aggregation and improve biocompatibility. Although not performed here, quantitative methods to determine the number of functional amine groups were reported49 and could help to optimize the condition for surface modification and to further increase the particle dispersibility. Alternatively, adding other inert functional groups and/or negatively charged moieties (eg, methylphosphonate) can increase the overall solubility by preventing back-bonding48 and might be a valuable strategy to improve the biocompatibility of our aerosol-synthesized NPs.
Biodistribution
Silica offers several advantages for biomedical research compared to other nanomaterials but often suffer from poor biodistribution with high uptake by the cells of the reticuloendothelial system (RES).20 In terms of passive targeting of solid tumors, best hydrodynamic stability in the bloodstream of SiO2NPs is achieved by keeping the particle size between 50 and 300 nm, since particles above 300 nm usually tend to get recognized by the RES whereas smaller particles (<50 nm) might nonspecifically distribute in the body.50 More recent preclinical data suggest that the highest EPR effect in target tissue can be obtained for NPs with rigid core size between 100 and 220 nm while emphasizing the importance of the particle surface charge on biodistribution.51 In this regard, high negative ZPs might improve both stabilities and the EPR effect, whereas high positive ZPs increase either cytotoxicity (smaller NPs) or recognition by the RES (NPs >150 nm core size). Based on this knowledge, we implemented the aerosol reactor technology to generate siliceous NPs with a final rigid core size of on average ~170 nm and high negative ZP (about −30 mV) to increase the EPR effect in our tumor model. However, the DLS data suggest that surface modification (ie, APTES, NOTA, and PEG) increased the hydrodynamic sizes independent of the particle morphology (>300 nm) that might have reduced tumor penetration and facilitated RES recognition. The mouse biodistribution studies revealed that the majority of our radiolabeled SiO2NPs (up to 126%ID/g) accumulated in the organs of the RES (ie, liver, spleen, and lung), followed by intermediate uptake (up to 24%ID/g) in the intestine and kidney (Figures 2 and 3). In contrast, the particle accumulation in the other organs (Tables S2–S5) including blood, muscle, and human A431 xenografts (Figure 4) was rather low (<6%ID/g). Despite this general pattern of organ distribution, the relative organ uptakes of the SiO2NPs varied dependently on the particle morphology. For instance, aerosol-synthesized NP1 composed of small primary particles highly accumulated in lungs at the early time point (109%ID/g) but dramatically decreased after only 24 hours (lung uptake NP1: <13%ID/g), an effect that was much stronger than for any of the other investigated NPs. Initially increased lung concentrations of SiO2NPs that decreased over time have been described previously and were suggested to be caused by the uptake and migration of alveolar phagocytes.52 We conclude that the unique morphology with high surface area of NP1 increased the uptake by alveolar phagocytes, although an initial blockage of small lung capillaries by NP1 might have contributed to this effect as well. Similarly, the unique morphology of aerosol-synthesized NP3 resulted in strong initial spleen uptake (4 hours: 118%ID/g vs 24 hours: 63%ID/g) that was likely caused by the preferred uptake of NP3 through spleen macrophages. Nevertheless, our 24 hour biodistribution data suggest that the majority of injected particles accumulated in the liver independent of the particle morphology. Being the largest organ of the mouse body (around 1 g), calculation of the organ uptake (%ID/organ) indeed revealed high liver accumulation for all particles (NP1: 34%, NP2: 61%, NP3 48%, and NP4: 25%). This is in accordance with other reports indicating that the liver usually sequesters the majority (up to 99%) of administered NPs from the bloodstream and typically less than 5% of the injected NPs are delivered to the diseased tissue.53 As a matter of fact, tumor uptakes were highly dependent on the particle morphology. Here, aerosol-synthesized NPs with large primary subunits and smaller overall surface area (NP3) showed superior tumor penetration and retention compared to those made of smaller primary subunits (ie, NP1 and NP2). Interestingly, PEGylation of our aerosol-synthesized NPs showed minor effects on blood circulation and uptake by tumors. This is in contrast to many studies, although there are some studies that have demonstrated that PEGylation can have a minimal effect on tumor uptake.54,55 Nevertheless, we did observe some differences between PEGylated and non-PEGylated NPs in organs of the RES (eg, reduced liver and spleen uptake by NP1 and NP3). The DLS data suggest that PEGylation might have impaired efficient tumor penetration of the aerosol-synthesized NPs due to an increase in hydrodynamic radius above the critical cutoff size for efficient microvascular permeability (ie, 400–600 nm for most tumors).56 In fact, our aerosol-synthesized NPs, in general, showed a higher susceptibility for aggregation after PEGylation than the spherical NP4 possessing the highest tumor uptake, TTB, and TTM ratios of all particles. In general, nonspecific uptake of SiO2NPs through the RES does not lead to any long-term toxicity since decomposition and clearance of SiO2NPs, though biliary (ie, feces) and renal system (ie, urine) usually occurs within a few weeks.22 We believe that our SiO2NPs would have a similar low toxicity; however, this will need to be confirmed in future studies. Nevertheless, drug-loaded therapeutic NPs might result in systemic toxicity if nonspecifically unloaded in nontarget tissue. In this regard, further fine-tuning of our functionalization procedure is warranted to reduce the particle aggregation and RES uptake.
Conclusion
The goal of this study was to evaluate the general biodistribution of siliceous NPs that were synthesized by a novel aerosol reactor-based approach and to further demonstrate the feasibility of this technology to generate biodegradable NPs with biomedical potential. In vivo biodistribution of the 64Cu-radiolabeled NPs revealed predominant uptake by the phagocytic cells of the RES in liver (Kupffer cells), spleen (red pulp macrophages), and lung (alveolar macrophages). The significant differences of the aerosol-synthesized NPs in organ distribution were mainly caused by the different morphologies of the investigated particles, although the change of other physiochemical characteristics (eg, hydrodynamic size and ZP) might have contributed to this effect as well. In terms of the highest tumor uptake and retention, aerosol-synthesized NPs composed of large primary subunits and smaller overall surface area (NP3) seemed to be superior over those composed of smaller subunits (NP1 and NP2), suggesting the potential of NP3 in the development as a tumor imaging or therapeutic agent. Although similar technologies for the synthesis of siliceous particles by aerosol reactors have been reported previously, this study represents, to our knowledge, the first investigation regarding the potential of such synthesized NPs for biomedical applications. Nevertheless, we observed a high uptake of our NPs by the RES that is in accordance with many other in vivo reports about siliceous NPs for biomedicine. In this regard, further optimization of the synthesis and functionalization process of our aerosol-derived NPs seems to be warranted to improve tumor penetration and retention in upcoming studies. In terms of synthesis, adjusting the reactor conditions to produce larger primary subunits of sintered or even spherical nonsintered particles might be favorable. Last but not least, incorporation of functional groups (eg, organosilica) during the aerosol synthesis process to produce organosilica or silsesquioxane NPs might further expand the potential of this technology in terms of biomedical usage. Overall, the current data suggest that the silane aerosol reactor approach is valuable in generating sintered siliceous aggregates on a nanoscale that are safe and promising for use in future imaging and/or therapeutic applications.
Acknowledgments
This work was supported by the Department of Radiation Oncology at WUSM. The authors would like to thank the small animal imaging facility at WUSM for excellent technical assistance in conducting biodistribution studies, in particular, Amanda Klaas, Margaret Morris, Lori Strong, and Nikki Fettig. We would also like to acknowledge the Isotope Production Group at Washington University for the production of 64Cu. The authors acknowledge Marisa Ilag and Jalen Scott for providing technical assistance with NP functionalization. Financial support to Ramesh Raliya was provided by the CMMN grant NIH-NCI U54CA199092.
Disclosure
The authors report no conflicts of interest in this work.
References
Petros RA, Desimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. | ||
Heiligtag FJ, Niederberger M. The fascinating world of nanoparticle research. Mater Today. 2013;16(7–8):262–271. | ||
Stark WJ, Stoessel PR, Wohlleben W, Hafner A. Industrial applications of nanoparticles. Chem Soc Rev. 2015;44(16):5793–5805. | ||
Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm Res. 2016;33(10):2373–2387. | ||
Thakor AS, Jokerst JV, Ghanouni P, Campbell JL, Mittra E, Gambhir SS. Clinically Approved Nanoparticle Imaging Agents. J Nucl Med. 2016;57(12):1833–1837. | ||
Tran S, Degiovanni PJ, Piel B, Rai P. Cancer nanomedicine: a review of recent success in drug delivery. Clin Transl Med. 2017;6(1):44. | ||
Singh R, Lillard JW. Nanoparticle-based targeted drug delivery. Exp Mol Pathol. 2009;86(3):215–223. | ||
Bao G, Mitragotri S, Tong S. Multifunctional nanoparticles for drug delivery and molecular imaging. Annu Rev Biomed Eng. 2013;15:253–282. | ||
Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. | ||
Lammers T, Aime S, Hennink WE, Storm G, Kiessling F. Theranostic nanomedicine. Acc Chem Res. 2011;44(10):1029–1038. | ||
Arranja AG, Pathak V, Lammers T, Shi Y. Tumor-targeted nanomedicines for cancer theranostics. Pharmacol Res. 2017;115:87–95. | ||
Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. | ||
Liang R, Wei M, Evans DG, Duan X. Inorganic nanomaterials for bioimaging, targeted drug delivery and therapeutics. Chem Commun. 2014;50(91):14071–14081. | ||
Wang D, Lin Z, Wang T, et al. Where does the toxicity of metal oxide nanoparticles come from: The nanoparticles, the ions, or a combination of both? J Hazard Mater. 2016;308:328–334. | ||
Golbamaki N, Rasulev B, Cassano A, et al. Genotoxicity of metal oxide nanomaterials: review of recent data and discussion of possible mechanisms. Nanoscale. 2015;7(6):2154–2198. | ||
Fratoddi I, Venditti I, Cametti C, Russo MV. How toxic are gold nanoparticles? The state-of-the-art. Nano Research. 2015;8(6):1771–1799. | ||
Kim S, Ryu DY. Silver nanoparticle-induced oxidative stress, genotoxicity and apoptosis in cultured cells and animal tissues. J Appl Toxicol. 2013;33(2):78–89. | ||
Li W, Liu Z, Fontana F, et al. Tailoring Porous Silicon for Biomedical Applications: From Drug Delivery to Cancer Immunotherapy. Adv Mater. 2018;30(24):e1703740. | ||
Santos HA, Mäkilä E, Airaksinen AJ, Bimbo LM, Hirvonen J. Porous silicon nanoparticles for nanomedicine: preparation and biomedical applications. Nanomedicine. 2014;9(4):535–554. | ||
Chen F, Hableel G, Zhao ER, Jokerst JV. Multifunctional nanomedicine with silica: Role of silica in nanoparticles for theranostic, imaging, and drug monitoring. J Colloid Interface Sci. 2018;521:261–279. | ||
Croissant JG, Fatieiev Y, Almalik A, Khashab NM. Mesoporous Silica and Organosilica Nanoparticles: Physical Chemistry, Biosafety, Delivery Strategies, and Biomedical Applications. Adv Healthc Mater. 2018;7(4):1700831. | ||
Croissant JG, Fatieiev Y, Khashab NM. Degradability and Clearance of Silicon, Organosilica, Silsesquioxane, Silica Mixed Oxide, and Mesoporous Silica Nanoparticles. Adv Mater. 2017;29(9):1604634. | ||
Asefa T, Tao Z. Biocompatibility of mesoporous silica nanoparticles. Chem Res Toxicol. 2012;25(11):2265–2284. | ||
Karaman DŞ, Sarparanta MP, Rosenholm JM, Airaksinen AJ. Multimodality Imaging of Silica and Silicon Materials In Vivo. Adv Mater. 2018;30(24):e1703651. | ||
Ow H, Larson DR, Srivastava M, Baird BA, Webb WW, Wiesner U. Bright and stable core-shell fluorescent silica nanoparticles. Nano Lett. 2005;5(1):113–117. | ||
Phillips E, Penate-Medina O, Zanzonico PB, et al. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Sci Transl Med. 2014;6(260):ra149. | ||
Stöber W, Fink A, Bohn E. Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci. 1968;26(1):62–69. | ||
Li Z, Barnes JC, Bosoy A, Stoddart JF, Zink JI. Mesoporous silica nanoparticles in biomedical applications. Chem Soc Rev. 2012;41(7):2590–2605. | ||
Hon NK, Shaposhnik Z, Diebold ED, Tamanoi F, Jalali B. Tailoring the biodegradability of porous silicon nanoparticles. J Biomed Mater Res Part A. 2012;100(12):3416–3421. | ||
Bimbo LM, Sarparanta M, Santos HA, et al. Biocompatibility of thermally hydrocarbonized porous silicon nanoparticles and their biodistribution in rats. ACS Nano. 2010;4(6):3023–3032. | ||
Park JH, Gu L, von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ. Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat Mater. 2009;8(4):331–336. | ||
Cheng X, Lowe SB, Reece PJ, Gooding JJ. Colloidal silicon quantum dots: from preparation to the modification of self-assembled monolayers (SAMs) for bio-applications. Chem Soc Rev. 2014;43(8):2680–2700. | ||
Chinnathambi S, Chen S, Ganesan S, Hanagata N. Silicon quantum dots for biological applications. Adv Healthc Mater. 2014;3(1):10–29. | ||
Kume M, Carey PC, Gaehle G, et al. A semi-automated system for the routine production of copper-64. Appl Radiat Isot. 2012;70(8):1803–1806. | ||
McCarthy DW, Shefer RE, Klinkowstein RE, et al. Efficient production of high specific activity 64Cu using a biomedical cyclotron. Nucl Med Biol. 1997;24(1):35–43. | ||
Möller K, Bein T. Talented Mesoporous Silica Nanoparticles. Chem Mater. 2017;29(1):371–388. | ||
Vazquez-Pufleau M. Applications of Aerosol Technologies in the Silicon Industry [Dissertation]. Energy, Environmental & Chemical Engineering, Washington University in St. Louis; 2016. doi.org/10.7936/K79K48N7 | ||
Winters BJ, Holm J, Roberts JT. Thermal processing and native oxidation of silicon nanoparticles. J Nanopart Res. 2011;13(10):5473–5484. | ||
Rahman IA, Padavettan V. Synthesis of Silica Nanoparticles by Sol-Gel: Size-Dependent Properties, Surface Modification, and Applications in Silica-Polymer Nanocomposites – A Review. J Nanomater. 2012;2012(30):15. | ||
Alhmoud H, Delalat B, Elnathan R, et al. Porous Silicon Nanodiscs for Targeted Drug Delivery. Adv Funct Mater. 2015;25(7):1137–1145. | ||
Nozaki T, Sasaki K, Ogino T, Asahi D, Okazaki K. Microplasma synthesis of tunable photoluminescent silicon nanocrystals. Nanotechnology. 2007;18(23):235603. | ||
Gupta A, Swihart MT, Wiggers H. Luminescent Colloidal Dispersion of Silicon Quantum Dots from Microwave Plasma Synthesis: Exploring the Photoluminescence Behavior Across the Visible Spectrum. Adv Funct Mater. 2009;19(5):696–703. | ||
Strobel R, Pratsinis SE. Flame aerosol synthesis of smart nanostructured materials. J Mater Chem. 2007;17(45):4743–4756. | ||
Körmer R, Schmid HJ, Peukert W. Aerosol synthesis of silicon nanoparticles with narrow size distribution – Part 2: Theoretical analysis of the formation mechanism. J Aero Sci. 2010;41(11):1008–1019. | ||
Holm J, Roberts JT. Thermal oxidation of 6 nm aerosolized silicon nanoparticles: size and surface chemistry changes. Langmuir. 2007;23(22):11217–11224. | ||
Liao YC, Nienow AM, Roberts JT. Surface chemistry of aerosolized nanoparticles: thermal oxidation of silicon. J Phys Chem B. 2006;110(12):6190–6197. | ||
Liberman A, Mendez N, Trogler WC, Kummel AC. Synthesis and surface functionalization of silica nanoparticles for nanomedicine. Surf Sci Rep. 2014;69(2–3):132–158. | ||
Bagwe RP, Hilliard LR, Tan W. Surface modification of silica nanoparticles to reduce aggregation and nonspecific binding. Langmuir. 2006;22(9):4357–4362. | ||
Jung H-S, Moon D-S, Lee J-K. Quantitative analysis and efficient surface modification of silica nanoparticles. J Nanomaterials. 2012;2012:48–48. | ||
Barbé C, Bartlett J, Kong L, et al. Silica particles: a novel drug-delivery system. Adv Mater. 2004;16(21):1959–1966. | ||
McNeil SE. Nanoparticle therapeutics: a personal perspective. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1(3):264–271. | ||
Borchardt G, Brandriss S, Kreuter J, Margel S. Body distribution of 75Se-radiolabeled silica nanoparticles covalently coated with omega-functionalized surfactants after intravenous injection in rats. J Drug Target. 1994;2(1):61–77. | ||
Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–348. | ||
Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl. 2010;49(36):6288–6308. | ||
Verhoef JJ, Anchordoquy TJ. Questioning the Use of PEGylation for Drug Delivery. Drug Deliv Transl Res. 2013;3(6):499–503. | ||
Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–3756. |
Supplementary materials
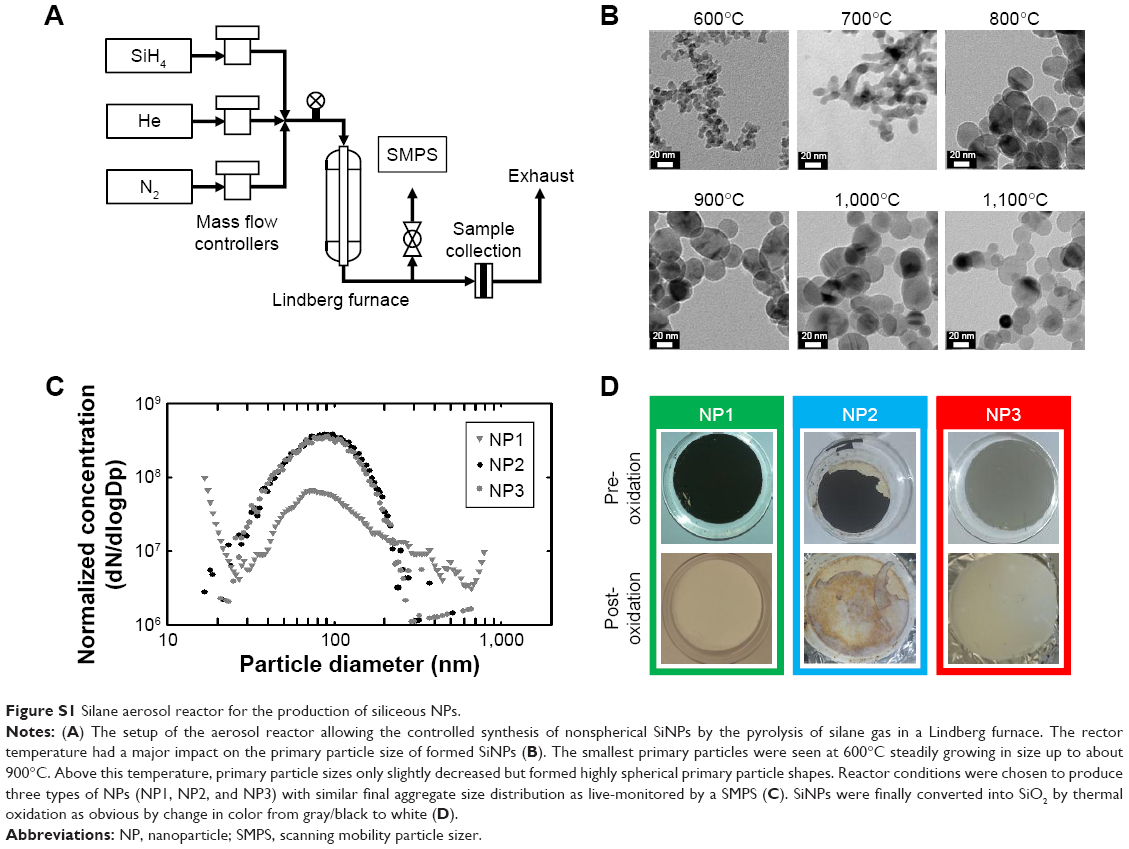 | Figure S1 Silane aerosol reactor for the production of siliceous NPs. |
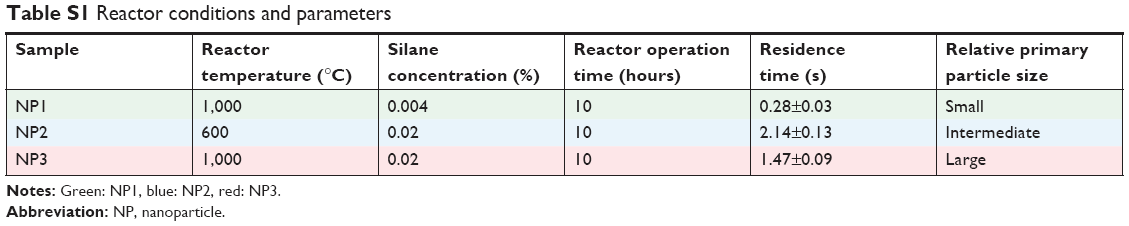 | Table S1 Reactor conditions and parameters |
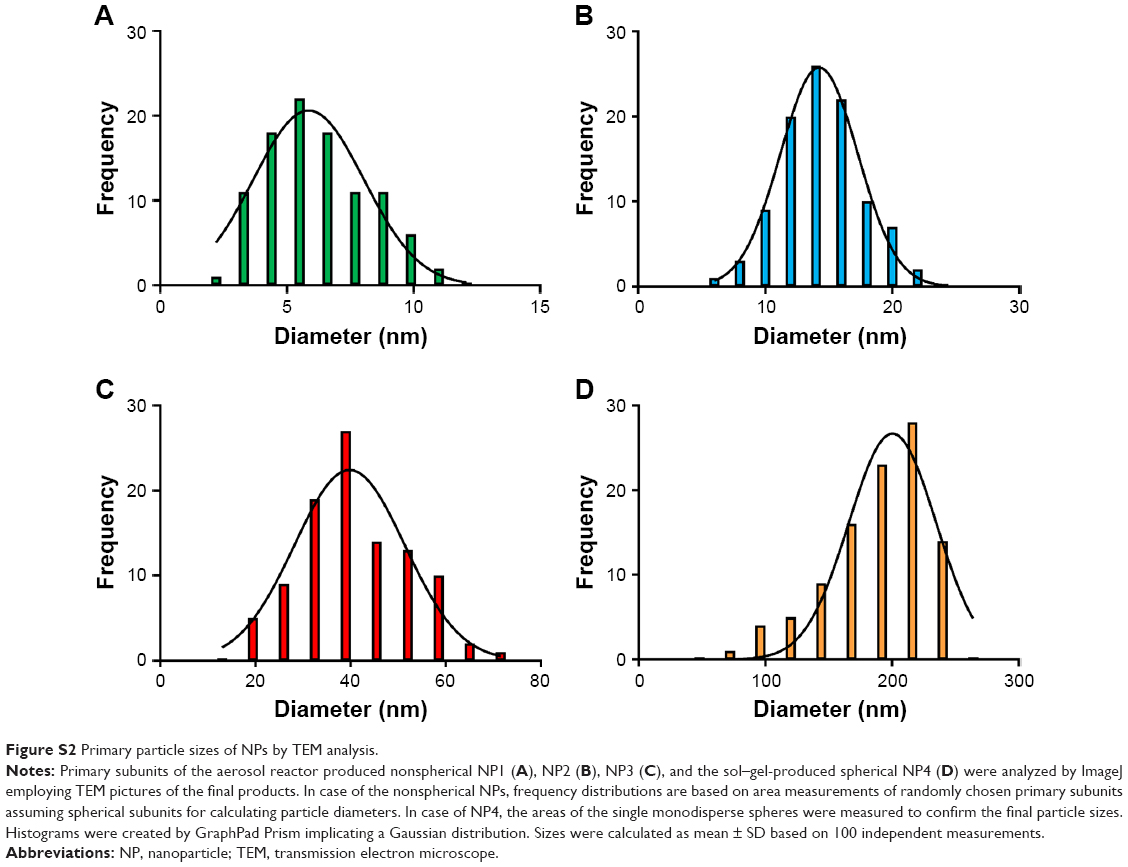 | Figure S2 Primary particle sizes of NPs by TEM analysis. |
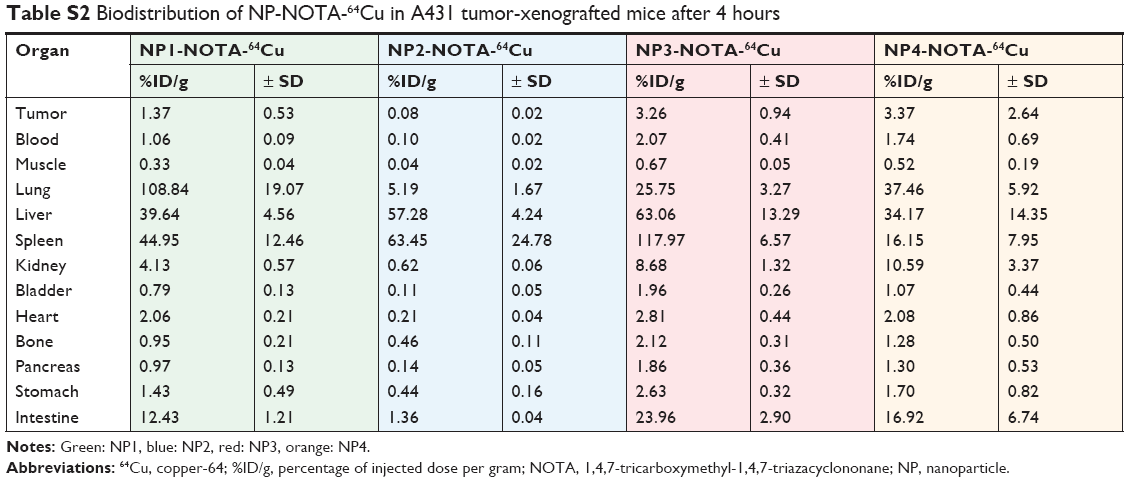 | Table S2 Biodistribution of NP-NOTA-64Cu in A431 tumor-xenografted mice after 4 hours |
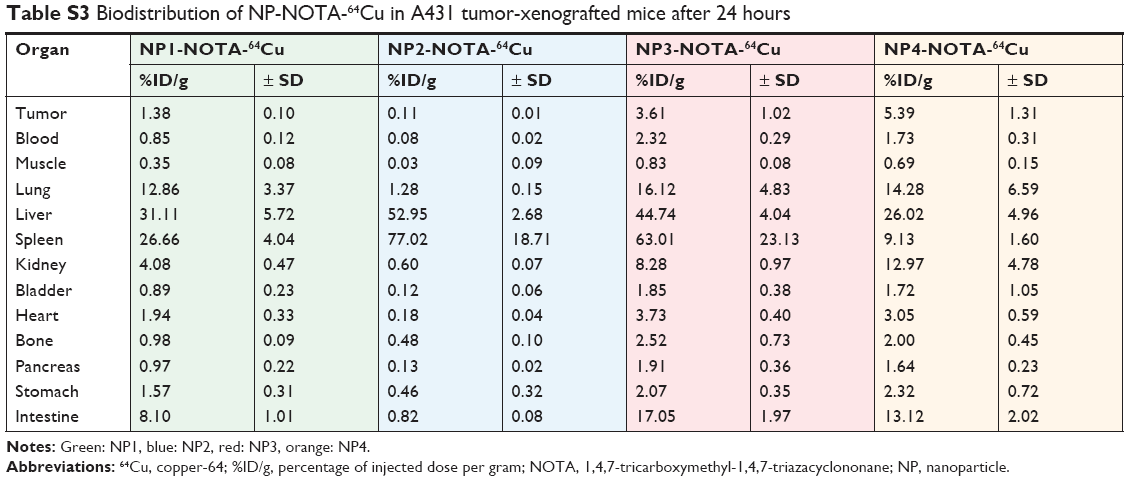 | Table S3 Biodistribution of NP-NOTA-64Cu in A431 tumor-xenografted mice after 24 hours |
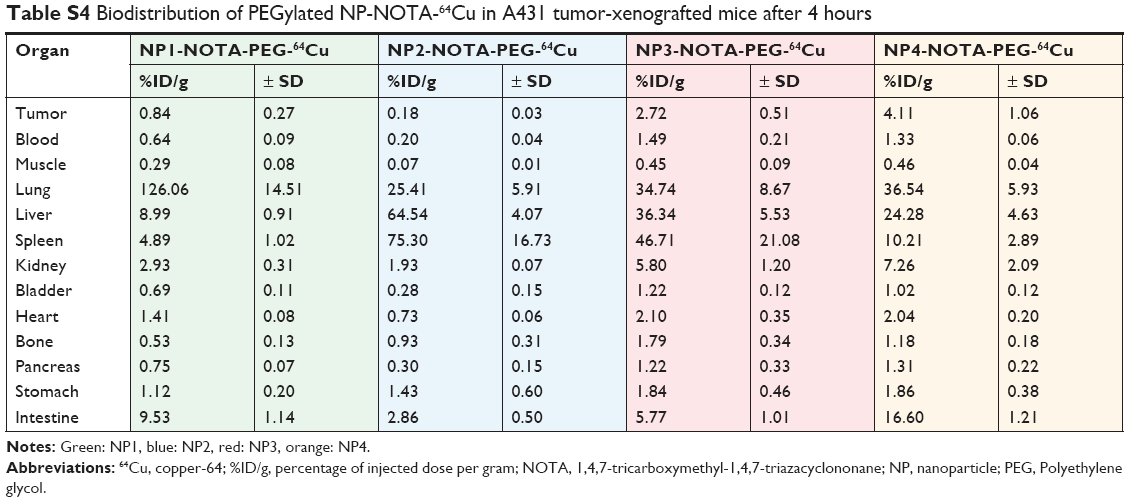 | Table S4 Biodistribution of PEGylated NP-NOTA-64Cu in A431 tumor-xenografted mice after 4 hours |
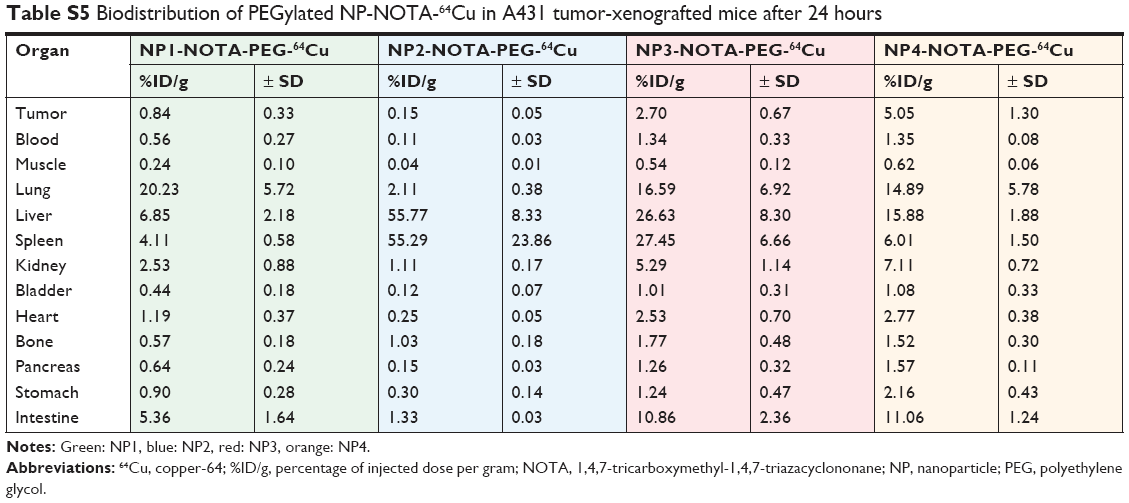 | Table S5 Biodistribution of PEGylated NP-NOTA-64Cu in A431 tumor-xenografted mice after 24 hours |
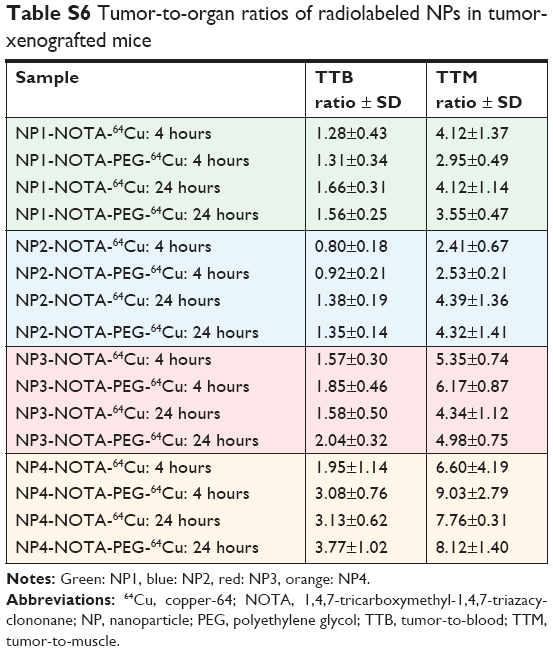 | Table S6 Tumor-to-organ ratios of radiolabeled NPs in tumor-xenografted mice |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.