")
Back to Journals » Cancer Management and Research » Volume 10
Advances in the treatment of soft tissue sarcoma: focus on eribulin
Authors Koliou P, Karavasilis V, Theochari M, Pollack SM, Jones RL , Thway K
Received 1 June 2017
Accepted for publication 18 November 2017
Published 1 February 2018 Volume 2018:10 Pages 207—216
DOI https://doi.org/10.2147/CMAR.S143019
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Harikrishna Nakshatri
Panagiotis Koliou,1,* Vasilios Karavasilis,1,* Maria Theochari,2 Seth M Pollack,3 Robin L Jones,4 Khin Thway4
1The London Sarcoma Service, University College London Hospital, London, UK; 2Hippokration Hospital, University of Athens, Athens, Greece; 3Division of Oncology, University of Washington, Seattle, WA, USA; 4Royal Marsden Hospital, London, UK
*These authors contributed equally to this work
Abstract: Eribulin mesylate is a synthetic derivative of halichondrin B isolated from a marine sponge. Its mechanism of action is through microtubule inhibition, which is different from that of taxanes. Eribulin has been approved for the treatment of metastatic breast cancer and more recently for non-operable or metastatic liposarcoma in patients who have received prior anthracycline chemotherapy. The major side effects of eribulin are bone marrow suppression including neutropenia, leukopenia, anemia, and fatigue/weakness, which can be well managed. In this article, we reviewed evidence from the latest published data on eribulin and its use in the treatment of soft tissue sarcomas. We explored the drug’s mechanism of action, pharmacodynamics, pharmacokinetics, and metabolism. Lastly, we reviewed all preclinical studies as well as clinical trials that investigated eribulin.
Keywords: eribulin, chemotherapy, sarcoma
Introduction
Soft tissue sarcomas (STS) represent a fairly rare and heterogeneous set of connective tissue tumors, including over 50 different histological subtypes with diverse clinical behavior, response to systemic therapy, and overall outcome.1 The backbone of standard treatment for localized disease involves surgery with clear margins with neoadjuvant or adjuvant radiotherapy.2
Nevertheless, even if an optimal local control is achieved, recurrences do happen in about 50% of patients with initially high histological grade cancers. The outcome of patients who either present with or develop metastatic disease is generally poor with the overall survival (OS) ranging between 12 and 18 months.3 The results of a Phase II study with the addition of olaratumab to doxorubicin (a monoclonal antibody that targets platelet-derived growth factor [PDGF] receptor alpha and blocks binding of factors PDGF-AA, -BB)4 have shown a statistically significant improvement in progression-free survival (PFS) of up to 6 months with an increase in OS to 26.9 months.5 First-line chemotherapy with doxorubicin stands as a palliative option, conferring a clinical benefit to almost half of the patients.6
Unfortunately, the durability of response to first-line chemotherapy is brief with a median PFS between 3 and 7 months.7 For those who are still fit to have further chemotherapy, available second-line treatment options are trabectedin, pazopanib, and doublet of gemcitabine/docetaxel, which have expanded the therapeutic portfolio available to treat metastatic STS.8,9
In the past, the clinical impact of novel drugs in the most common sarcoma subtypes has not been well determined. Because many histological subtypes of STS are represented in clinical trials, it can be challenging to determine whether the clinical outcomes are related to a specific sarcoma subtype and thus interpretation of the results has been challenging.10 Currently, more emphasis is given on focused, tailored therapy according to histological subtypes.11 However, there are few data showing an OS benefit for any systemic therapy.12
Figure 1 shows the chemical structures of eribulin (C40H59NO11)13 from halichondrin B (C60H86O19), a compound isolated from a natural marine sponge.14 Its mechanism of action is dual – via tubulin-based antimitotic mechanism, it blocks G2 phase (second subphase of interphase) and M phase (mitosis) and disrupts the mitotic spindle formation in the cell cycle. Moreover, through prolonged mitotic blockage, it causes apoptosis of cancer cells.15
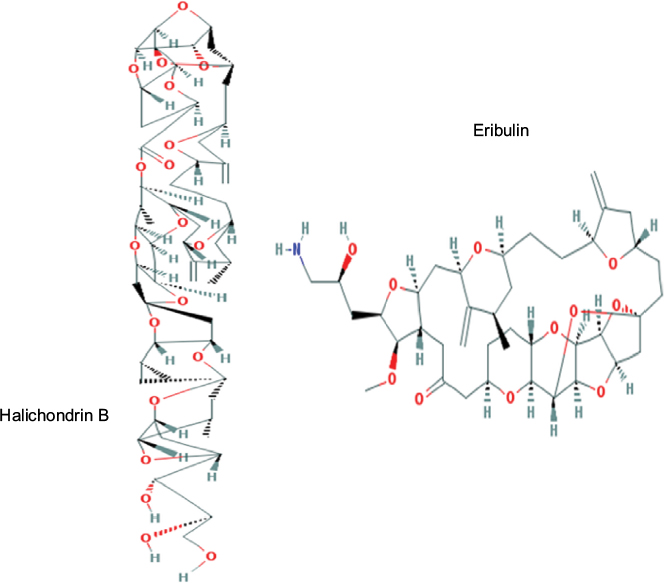 | Figure 1 Molecular structures of halichondrin B and eribulin. Note: Reproduced from National Center for Biotechnology Information. Pubchem Open Chemistry Database –Compound Summary for CID 11354606. https://pubchem.ncbi.nlm.nih.gov/compound/11354606,13 and https://pubchem.ncbi.nlm.nih.gov/compound/5488895.14 |
The approval of the drug for stage IV metastatic breast cancer was based on the results of a Phase III trial that randomized women to receive either eribulin or an investigator’s choice of chemotherapy.16 When compared to the physician’s treatment of choice in previously treated locally recurrent and metastatic breast cancer, eribulin showed an improvement in OS of 2.5 months, which was statistically significant.16 Eribulin was approved for STS as a result of a subsequent Phase III study that recruited only non-operable or metastatic patients with STS subtypes of leiomyosarcoma (LMS) and liposarcoma (LPS).17 A statistically significant improvement of 2 months in median OS for patients who received eribulin was observed when compared to dacarbazine (DTIC).17 There was no significant benefit found with respect to PFS.17
The main aim of this review was to evaluate the potential role of eribulin in certain histological subtypes of STS with particular focus on the results of the abovementioned Phase III trial.
Preclinical development and mechanisms of action of eribulin
Eribulin mesylate is a non-taxane agent, which is a structurally simplified, completely synthetic novel analog of a naturally occurring chemical compound halichondrin B, initially purified from the Japanese marine sponge Halichondria okadai in 1986.18 Unlike other antitubulin agents, it has a completely distinct mechanism of action. It binds to the plus ends of microtubules with high affinity – which are polymers made from α- and β-tubulin proteins – preventing cross-link formations and it inhibits microtubule polymerization without decreasing microtubule length and without affecting depolymerization.
In addition, it also precipitates the sequestration of tubulin into non-functional aggregates. Eribulin inhibits microtubule dynamics via a novel mechanism of action. By inhibiting mitotic spindle formation, it causes irreversible mitotic block that leads to cell cycle arrest in the G2–M phase and apoptosis.19,20 As a result, it interferes with mitotic spindle formation and stops cancer cell growth. Apoptosis is subsequently triggered following a prolonged and irreversible mitotic blockade. Posttranslational modifications of the antiapoptotic BCL-2 family member proteins occur during prolonged mitotic arrest.21 This takes place either via phosphorylation of BCL-2 protein or via ubiquitination and subsequent proteasomal degradation with MCL-1 gene that has previously been reported to result in inhibition of their antiapoptotic function.21 Eribulin causes mitotic arrest prior to BCL-2 and BCL-xL phosphorylation, MCL-1 downregulation, and onset of apoptosis.21 Data support that phosphorylation inactivates anti-apoptotic BCL-2 proteins during mitotic arrest and as such is a key regulatory feature of mitotic death.21 Both the loss of MCL-1 expression and the functional loss of BCL-2 and BCL-xL are controlled by phosphorylation.21 These two events are counterbalanced mechanisms obligatory in the coordination of the inactivation of each other through phosphorylation.21 As a result, it can be postulated that disruption of this balance and unequivocal phosphorylation of these protein members is postulated as a mechanism of drug resistance.21,22 Eribulin has shown potent antiproliferative activity against a broad range of human cancer cell lines and has been associated with tumor regressions and eliminations in a variety of well-established human tumor xenograft models.23
Beyond its antimicrotubule effect, eribulin is also involved in tumor vascular remodeling.24 The hypoxic microenvironment created by abnormal tumor vasculature contributes to tumor progression, metastasis, and drug resistance. The abnormal tumor vasculature, together with tumor and stromal proliferation, results in a high interstitial tumor pressure, further impeding tumor perfusion and contributing to the hypoxic tumor environment.25 The effect of eribulin on tumor vascular remodeling was demonstrated in human breast cancer xenograft models in nude mice.25 The results revealed that eribulin altered the morphology of the tumor vasculature, leading to increased aggregate surface area of the microvasculature, thus explaining the increased tumor perfusion following eribulin treatment.25 An increase in the number of microvessels and subsequent tissue perfusion were observed in eribulin-treated tumors. These changes to the microenvironment alleviate tumor-induced hypoxia and may possibly enhance the efficacy of subsequent drug therapies through the reduction of hypoxia-driven chemoresistance and enhancement of intratumoral delivery of drugs. Eribulin can induce cellular differentiation in LPS and LMS but not in a complete manner.26 Results from both SW 872 LPS and SK-UT-1 LMS cell lines indicate that eribulin treatment induces cellular differentiation down the adipocyte and smooth muscle pathways.26 In addition to the improvement in vascular perfusion, the differentiation-related phenotypic changes observed imply that eribulin may make tumors less aggressive, as degree of differentiation and severity of malignancy are known to be inversely correlated.26
Tumor microenvironments consist of many non-malignant cells and control vascular architecture.27 In xenograft models, the mechanisms underlying the effects of eribulin on non-malignant host (murine) cells were explored by conducting quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)-based gene expression profiling of mouse-derived tumor stroma.25 After analysis of 84 genes associated with angiogenesis regulatory pathways based on known expression in endothelial cells or pericytes, as well as 43 epithelial mesenchymal transition (EMT)-related mesenchymal or epithelial marker genes, it was shown that following eribulin treatment, expression levels of 29 murine genes involved in angiogenesis regulatory pathways were significantly reduced in xenografts in a dose-dependent manner.25
Eribulin acts as a potent antivascular drug by decreasing the expression of signaling pathways involved in pericyte-driven angiogenesis (vascular endothelial growth factor, Wnt, Notch, and ephrin). Although no EMT-related epithelial marker genes showed significantly altered expression after eribulin treatment in xenograft models, 13 EMT-related mesenchymal marker genes showed decreased expression after eribulin treatment in both models.25 The results show that eribulin had an effect on gene expression in signaling pathways related to angiogenesis and EMT in the tumor stroma and reduced the degree of hypoxia. The protein levels of key EMT/mesenchymal epithelial transition (MET) markers behaved similarly to mRNA expression patterns: eribulin increased the levels of E-cadherin protein, while decreasing the levels of N-cadherin and vimentin proteins.28 The results of these morphological observations together with gene and protein expression patterns suggest that eribulin caused reversion of EMT and induction of MET in STS and triple negative breast cancer cells.28,26
It is known that transforming growth factor-β (TGF-β) enhances phosphorylation of receptor-regulated Smad2 and Smad3 proteins, resulting in enhanced complexing with Smad4.29 Translocation of the resulting complex into the nucleus activates the transcription of essential EMT-related genes, including TWIST1, SNAI1, SNAI2, ZEB1, and others.29,30 In xenograft models, eribulin had three significant effects on EMT/MET transition: 1) it reversed the observed phenotype from the previously induced spindle-like EMT morphology to the original cuboidal morphology typical of TGF-β-untreated cells; 2) it upregulated mRNA expression levels of epithelial marker CDH1, while downregulating several mesenchymal markers; and 3) it significantly decreased TGF-β-induced phosphorylation of Smad2 and Smad3.28 This suggests that eribulin at least partially induced MET due to downregulation of the TGF-β/Smad pathway.28
Other drugs used in the treatment of STS, with similar effect but not the same mechanism of action as eribulin, include the taxanes and especially docetaxel alone or in combination with gemcitabine. The mechanism of action of docetaxel involves the promotion of microtubule assembly, inhibition of microtubule polymerization, and attenuation of the effects of BCL-2 and BCL-xL gene expression.31 Taxane-induced microtubule stabilization arrests cells in the G2M phase of the cell cycle and induces BCL-2 phosphorylation, leading to a cascade of events that eventually cause apoptotic cell death.31 Docetaxel has an inhibitory effect on endothelial cell migration, differentiation, or proliferation of cancer cells by promoting apoptosis.31 However, there are other antitumor cytotoxic drugs that inhibit endothelial cell migration, such as cisplatin, cyclophosphamide, doxorubicin, and methotrexate.32 Taxanes are unique as suitable antiangiogenic drugs in that they target the endothelial cells by specifically binding to endothelial cell receptors with higher affinity at a dose that does not affect the other normal cells.33 Docetaxel and paclitaxel have a different effect on different types of tumors (eg, prostate, lung, gastric tumors, or STS), and they inhibit to variable extent the secretion of vascular endothelial growth factor for each of these cancer cell types.33 Although a clear additive effect cannot be excluded, it is suggestive from evidence that docetaxel in combinatorial treatment has a possible synergistic result when clinical response rates have been compared.34 The combination of doxorubicin and ifosfamide, a well-known regimen in the treatment of STS, on the other hand, has an additive effect rather than a synergistic effect.35 Lastly, mechanisms of resistance to taxanes occur through EMT in a similar process as to that described earlier for eribulin. There are two additional important mechanisms of resistance: induction of P-glycoprotein encoded by multidrug resistance protein 1 gene and mutations of beta-tubulin.36
Phase I trials of eribulin
Four Phase I trials, three in Western countries and one in Japan, have been carried out to assess the toxicity-specific profile of eribulin. All Phase I, II, and III eribulin trials are shown in Table 1.37–39 The primary Phase I trial that evaluated eribulin focused on the safety of advanced and refractory solid tumors and was designed with guided dose escalation accompanied by real-time pharmacokinetics (PKs).40 The baseline dose was 0.125 mg/m2/week intravenously (iv). The dose-limiting toxicity (DLT) was observed at 2.0 mg/m2/week subsequent to the development of grades 3 and 4 neutropenia, respectively, in two patients.40 The maximum tolerated dose (MTD) was set at 1.4 mg/m2/week.40 A second Phase I trial determined the dosing of eribulin at 0.25 mg/m2 as 1 h iv infusions on days 1, 8, and 15 of a 4 week cycle.37 The MTD of eribulin was determined at 1.0 mg/m2. At 1.4 mg/m2, neutropenia (grade 4 in two patients and grade 3 in three patients) was the principal DLT.37 Fatigue (53% overall) and nausea (41% overall) were the most frequent adverse effects (AEs).37 A third Phase I trial determined the MTD of eribulin to be 2.0 mg/m2 when administered as an 1 h iv infusion in 3 week cycles.38 Finally, a Japanese Phase I trial demonstrated manageable tolerability of eribulin at 1.4 mg/m2 when treatment was given on days 1 and 8 of a 3 week cycle.39 Neutropenia was again the principal DLT of all the trials. As a result, from these Phase I trials, the recommended dose of eribulin for further clinical evaluation was set at a concentration of 1.4 mg/m2. However, administration of eribulin on days 1, 8, and 15 of a 4 week cycle was found difficult to tolerate, as neutropenia often limited its administration on day 15.41 As a result, the regimen was modified to a 3 week cycle with eribulin given on days 1 and 8, and this schedule was, therefore, established as standard to move forward on Phase II and Phase III trials.42
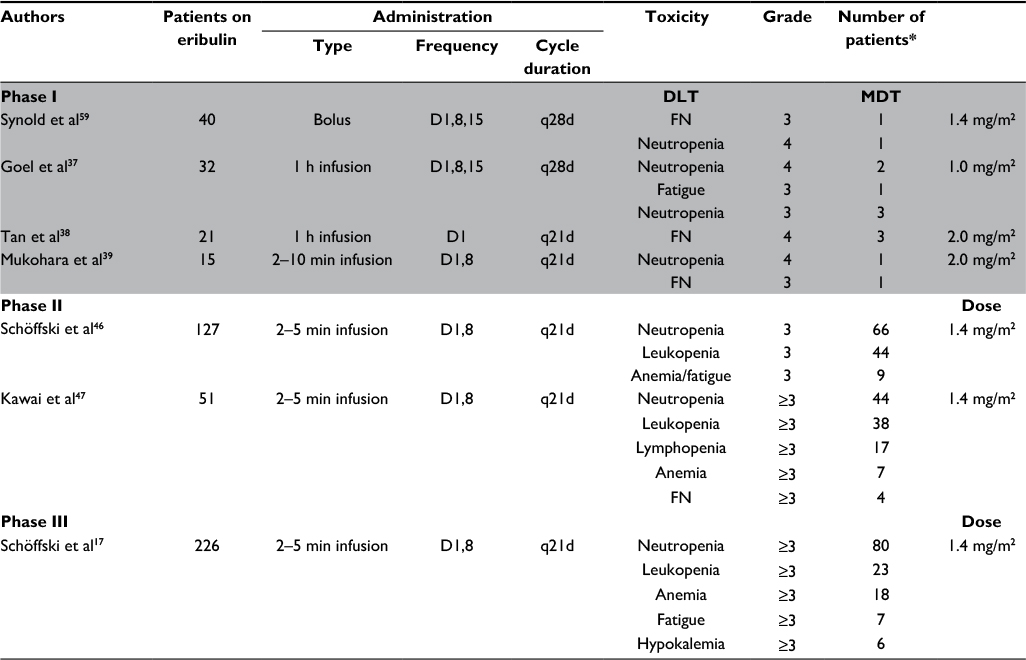 | Table 1 Summary of Phase I, Phase II, and Phase III clinical trials of eribulin in soft tissue sarcoma Notes: Dose-limiting toxicity and maximum tolerated toxicity in Phase I and toxicity in Phase II and III studies; route of administration was intravenous. *The actual number of patients who received eribulin in the trial who had toxicity ≥3. Gray shading indicates that the data apply only to Phase I trial toxicity. Abbreviations: DLT, dose-limiting toxicity; MDT, maximum tolerated toxicity, FN, febrile neutropenia; q21d, cycle of 21 days; q28d, cycle of 28 days; D1, day 1 of the cycle; D1,8, days 1 and 8 of the cycle; D1,8,15, days 1, 8, and 15 of the cycle. |
In humans, eribulin has a rapid distribution phase followed by a prolonged elimination phase, with a mean terminal half-life of about 40 h.37,38 PK studies of eribulin showed that the elimination of the drug mostly follows a three-phase elimination with an initial rapid distribution in the central compartment and later a two-phase elimination.43 In patients with adequate liver and renal functions, eribulin clearance was estimated at 2.98 L/h (central volume of distribution was 3.72 L and volumes of two peripheral compartments were 3.60 L and 126 L) and inter-compartmental clearances at 2.7 L/h and 5.6 L/h.40 However, interpatient drug clearance variability (mean 57%, range 26%–98%) was described across studies, which was attributed to inter-individual variation.40
Any degree of liver impairment decreases drug elimination and clearance, prolonging elimination half-life and thus systemic exposure to eribulin. It is, therefore, recommended to decrease eribulin dose according to the Child–Pugh hepatic impairment classification.43 For mild hepatic dysfunction (class A), the dosage is 1.1 mg/m2, while for moderate hepatic impairment (class B), it is 0.7 mg/m2.43 Eribulin must be avoided in patients with severe hepatic impairment (Child–Pugh class C).43 Renal impairment also decreases drug clearances and prolongs exposure, and it is, therefore, recommended to reduce the dosage of eribulin to 1 mg/m2 in patients with moderate (creatinine clearance [CrCl] 30–50 mL/min) and severe (CrCl 15–30 mL/min) renal impairment.44 After normalizing for body weight, CrCl had no effect on eribulin clearance.44
Population PK analyses showed that eribulin clearance is affected by levels of serum albumin, alkaline phosphatase, and bilirubin.45 The effects of age, sex, race, and concomitant medications (cytochrome P450 inhibitors and inducers) on clearance were not significant.44
Phase II trials of eribulin in STS
A Phase II open-label trial of eribulin in recurrent STS (of intermediate or high grade) was conducted by the European Organisation of Research and Treatment of Cancer (EORTC).46 Patients were eligible for enrollment in the study if they had received previously one line of combination chemotherapy or up to two lines of treatment with monotherapy for advanced disease. The EORTC trial did not include patients with embryonal rhabdomyosarcoma, chondrosarcoma, osteosarcoma, Ewing’s tumors, gastrointestinal stromal tumors, dermatofibrosarcoma protuberans, and inflammatory myofibroblastic tumors.46
Recruited patients were divided into four strata: LPS, LMS, synovial sarcoma (SS), and others. PFS at 12 weeks was set as the primary endpoint of the trial. Eribulin treatment was given iv at a dose of 1.4 mg/m2 for about 2–5 min at days 1 and 8 of a 3 week cycle. Disease response to therapy was assessed through imaging using response evaluation criteria in solid tumors (RECIST) 1.0. In total, 128 patients were recruited and 115 patients were eligible for analysis.46
Patients’ median age was 56.7 years and the median time interval from initial diagnosis was 2 years. In regards to histology, 37 patients had LPS (24 dedifferentiated, four pleomorphic, and six myxoid/round-cell LPSs, two not otherwise specified, and one unknown), 40 patients had LMS (11 with uterine origin), 19 patients had SS, and 32 patients had other types of STS.46 With greater than 600 treatment cycles administered to 127 patients, a median number of four cycles of treatment was administered to each patient. The 12-week progression-free rates (PFRs) were 31.6%, 46.9%, 21.1%, and 19.2%, respectively, for LMS, LPS, SS, and the other subtypes.46 The median PFS was 2.6 months (95% confidence interval [CI]: 1.7–6.2) for patients with LPS, 2.9 months (95% CI: 2.4–4.6) for those with LMS, 2.6 months (95%CI: 2.3–4.3) for SS, and 2.1 months (95% CI: 1.4–2.9) for other subtypes.46
The toxicity of eribulin observed in the aforementioned study was in line with the reported toxicities from the previous Phase I trials with only 6% of recorded grade 3 febrile neutropenia and 6% of grade 3–4 thrombocytopenia.46 There was no intervention necessary for the events of neutropenia and thrombocytopenia reported as they self-resolved. Other common non-hematological treatment-associated adverse events reported were fatigue, alopecia, nausea, and sensory neuropathy.46 Six patients discontinued treatment because of neurotoxic effects, though patients with previous grade 2 sensory neuropathy were allowed to enroll in the trial.46 One-third (33%) of 127 patients involved in the trial received eribulin at full dose with no further dose modifications. For the rest of the patients (67%), toxicity management included dose reductions, dose delays, or both. Specifically, 33 (26%) of the patients had treatment delays, 15 (12%) required dose reductions, and 37 (29%) required both delays and dose reductions.46
In a similar fashion, another open-label, multicenter, Phase II trial conducted in Japan that evaluated the safety and efficacy of eribulin in pretreated STS patients has been recently published. Once again, the primary endpoint of this trial was progression free rate (PFR) at 12 weeks. Secondary endpoints included OS, PFS, and safety. Efficacy analysis took place according to histology.47 In this trial design, there were two groups of patients, one consisting of LPS and LMS patients and the second including all other STS subtypes.47 Patients received eribulin mesilate 1.4 mg/m2 iv over 2–5 min on days 1 and 8 of a 3 week cycle. Overall, 52 patients were enrolled and 51 patients were treated. The study recruited 35 patients with LPS/LMS who had characteristics similar to the 16 patients with other histological subtypes. The only exception was the higher percentage of women than men (63% vs 38%) and patients with a performance status (PS) of 0 according to Eastern Cooperative Oncology Group compared to all others (57% vs 44%).47 PFS rate at 12 weeks was 60% in patients with LPS/LMS, 31% in other subtypes, and 51% overall.47 Median PFS survival was 5.5 months in LPS/LMS patients, 2.0 months in other subtypes, and 4.1 months overall.47 Median OS was 17.0 months in patients with LPS/LMS, 7.6 months in other subtypes, and 13.2 months overall. The most common grade 3–4 AEs were neutropenia (86%), leukopenia (75%), lymphopenia (33%), anemia (14%), and febrile neutropenia (8%).47 It was shown that eribulin had clinical activity with manageable side effects in Japanese patients who were previously treated for advanced/metastatic STS.47
Phase III trials of eribulin in STS
The data from the Phase II trial in STS met the primary endpoint with promising results in LMS and LPS. This led to the initiation of the Phase III trial randomizing eribulin and DTIC in LPS and LMS of intermediate or high grade. Patients previously treated with at least two lines of chemotherapy were randomized to receive either eribulin 1.4 mg/m2 iv on days 1 and 8 every 3 weeks or DTIC 800–1200 mg/m2 on day 1 every 3 weeks (subject to investigator’s choice).17 The response was assessed using RECIST 1.1. The primary endpoint was OS with selected secondary endpoints, including PFS and PFRs at 12 weeks.17
From a total of 594 patients who were screened, 452 patients were randomized. Both randomized groups of patients were equivocal with respect to clinical characteristics. Most of the patients (78.8%) were under the age of 65 years with a good PS. LPS and LMS groups accounted for 67.7% and 33.8% of cases, respectively.17 In the LMS subgroup, 43.1% of cases were uterine in origin.17 The study cohort was heavily pretreated, with 45.8% of patients having received more than two prior lines of therapy.17 Three hundred and fifty-one patients (77.7%) received anthracycline before enrollment.
Between the two study arms, there was a statistically significant difference in survival (median OS: eribulin, 13.5 months; DTIC, 11.5 months; hazard ratio [HR]: 0.768; 95% CI: 0.618–0.954, P=0.0169).17 However, there was no significant difference in median PFS (2.6 months for both eribulin and DTIC; HR: 0.877; 95% CI: 0.710–1.085, P=0.2287).17 In addition, the response rates when the two agents were compared were found to be nonsignificant (overall response rates 3.9% vs 4.9%; stable disease 52.2% vs 47.8%).17 In the preplanned OS subgroup analysis, LPS patients benefited from eribulin treatment (15.6 vs 8.4 months; HR: 0.511; 95% CI: 0.346–0.753) compared to LMS patients (12.7 vs 13.0 months; HR: 0.927; 95% CI: 0.714–1.203).17 The benefit from eribulin chemotherapy was observed across all LPS subtypes, favoring dedifferentiated and pleomorphic LPS histologies. Furthermore, PS was also found to be a prognostic factor in this study. Patients with a PS of 0 had a better survival outcome with eribulin (19.9 vs 13.1 months; HR: 0.579; 95% CI: 0.407–0.823) when compared to those with a PS of 1–2 (9.2 vs 9.9 months; HR: 1.09; 95% CI: 0.82–1.44).17
No unexpected or previously unreported toxicity was documented for either drug in this randomized trial. Grades 3 and 4 AEs were observed in 38.9% and 23.9% of patients receiving eribulin and in 35.7% and 19.2% of patients receiving DTIC.17 In both groups, neutropenia, fatigue, and nausea were the most commonly observed AEs.17 Only four (1.8%) patients developed ≥grade 3 sensory neuropathy with eribulin. Death of two patients was ascribed to eribulin (one neutropenic sepsis and one septic shock).17
Discussion
Eribulin is an inhibitor of microtubules, demonstrating controllable AEs across several clinical studies, and the United States Food and Drug Administration (US FDA) approved its use in women with stage IV breast cancer previously treated with anthracyclines or taxanes. Recently eribulin has been licensed as well in patients with metastatic or non-operable LPS who had received prior chemotherapy with anthracycline-containing medication.48,49 The Phase III trial in LPS and LMS published by Schöffski et al showed that eribulin treatment resulted in a statistically significant survival benefit in patients who had received anthracyclines and one or more lines of treatment.17 This study, despite the OS benefit, did not show any significance in PFS. Furthermore, when within group analysis took place for histology, it was found that the LPS patients contributed to the benefit in OS seen and not those with the LMS subtype. Interestingly, in a similar fashion, the breast cancer Phase III trial demonstrated OS benefits, but no improvement in PFS was observed.50
The explanation of this phenomenon is not clear and some suggestions are discussed as follows. Although the starting dose of DTIC varied according to the investigator’s choice, post hoc analysis showed no significant impact on survival. Both eribulin and DTIC demonstrated similar response rates and, therefore, support the findings of similar PFS.
It can be argued that post-trial treatments may contribute to differences in OS. However, the initiation of any post-trial therapy was comparable between the two arms of the study, apart from a higher number of patients of the eribulin arm who crossed over to receive DTIC (34.2% vs 7.6%, respectively).
A difference was found in OS without any difference in disease progression according to RECIST in both breast and sarcoma cancers. This might suggest that the effect of eribulin on OS could be related to modifications that the drug causes in the tumor microenvironment. It is well established that anti-microtubule drugs might be involved indirectly in targeting tumor angiogenesis via two postulated mechanisms: either by preventing neovascularization or by disrupting the already neoplastic tumor vessels.25 Preclinical data in breast cancer and STS cell lines have suggested that the mechanism of action of eribulin on angiogenesis is different from the other anti-microtubule drugs. It does not disrupt the preexisting vasculature and does not inhibit neoplastic microvessels, but supports their normalization into vessels with architecture that stops leakage and increases tumor perfusion. This alteration in perfusion on the tumor bed results in higher drug actions in previously not reached microenviroment with prevention of cell metastasis.25 Lastly, eribulin has been shown to disrupt metastasis in vitro and in vivo by altering EMT.25–28,51,52 Taken all together, it can be postulated that patients who were previously treated with eribulin were sensitized to post-trial therapies, thereby providing an explanation for the increased survival, despite no significant difference in PFS.53
Another interesting finding of the Phase III trial in STS is the absence of survival benefit in the LMS patients. This may be due to a different response of LMS to DTIC. A prior study that used DTIC as second- and third-line treatments in STC patients did not reveal any activity in the seven patients with LPS. Out of the 14 patients with LMS, two patients showed partial response and two had stable disease. Actually, of the total three patients with partial response, two had LMS histology of different STS types. As explained previously, in the Phase III STS study of eribulin, patients who progressed crossed over to receive DTIC. Exposure to DTIC may have, therefore, achieved an OS benefit from the LMS cohort of patients.53
There are two other new agents, trabectedin and pazopanib, that have been studied in Phase III trials and have been approved by the US FDA and European Medical Association (EMA) for STS in the US and Europe.54,55 Trabectedin was compared to DTIC in patients with LPS or LMS who had received doxorubicin and at least one other prior line of treatment. In contrast to the eribulin Phase III trial, there was a PFS benefit observed, but no OS advantage was documented. The PALETTE study,55 which tested pazopanib – an oral multi tyrosine kinase inhibitor – versus placebo in refractory sarcoma demonstrated PFS but no OS advantage. The PALETTE study, 55 however, excluded STS patients with adipocytic sarcomas, because in the Phase II trial, this group did not show any response, and as a result the PFS benefit with pazopanib remains for all other types of STS.
Similarly, the Phase III data of eribulin followed the cohorts of the Phase II study and was not extended to other STS subtypes as the primary end point was not met.17,46 The efficacy of eribulin in STS prolonged OS with no effect on PFS. So, eribulin, trabectidin, and pazopanib offer different benefits in patients with certain histologic STS subtypes and in whom specific treatment goals have to be reached.9
Although historically the prolongation of survival is of the highest importance for the primary end points in drug trials, the control of disease has also been acknowledged as a reasonable outcome for a treatment, in virtue of providing management of symptoms related to disease, such as shortness of breath or pain. It must be noted at this point that trabectedin, pazopanib, and eribulin did not show significant response rate and decrease in tumor size, with only a small percentage of patients in the respective studies achieving a partial response: 9.9% with trabectedin, 6% with pazopanib, and 4% with eribulin.17,54,55 However, there are considerable percentages of patients in the aforementioned trials who obtained clinical benefit from treatment. These findings are in contrast to well-documented trial results of studies that have used doxorubicin as first- and second-line treatments for STS, which have demonstrated partial responses and some complete responses in patients who were involved.6,8
Regarding the use of eribulin in a combination regimen, in vitro data have suggested its synergy in combination with other agents. It has been postulated that eribulin may promote vascular perfusion of the tumor microenviroment. Its combination with other chemotherapeutic agents could have synergistic effects. By normalizing neovessels into vasculature that permits nutrient and oxygen perfusion to a higher extent, there is a greater chance for the drug to hit its target and reverse tumor growth, while preventing the hypoxic environment from adding further “mitogenic stress” on tumor cells. A Phase I study has combined gemcitabine with eribulin, and data for other novel combinations are awaited.56,57
Conclusion
The eribulin trial is the first Phase III trial in the past years that has shown a clinically significant, albeit moderate, survival benefit in the treatment of advanced STS of LPS histologies, and these results are considered encouraging. However, discrepancy between the PFS and OS results remains difficult to explain. Eribulin has recently been granted US FDA and EMA approval for the treatment of advanced LPS in patients pretreated with anthracycline chemotherapy.58 It can be argued that the cost-effectiveness of the drug could be a potential obstacle to widespread use of eribulin, and this needs to be addressed in subsequent trials, including quality of life measurements. However, a Phase III trial has shown a significant survival benefit and an obvious question is the possible role of eribulin in the neoadjuvant/adjuvant setting and its role in the first-line setting compared to anthracycline therapy in LPS patients.17 Further studies are required to evaluate putative biomarkers of response to eribulin and the role of this agent in early stage disease.
Disclosure
The authors report no conflicts of interest in this work.
References
Jo VY, Fletcher CD. WHO classification of tumours of soft tissue an update based on the 2013 (4th) edition. Pathology. 2014;46(2):95–104. | ||
Colombo C, Randall RL, Andtbacka RHI, Gronchi A. Surgery in soft tissue sarcoma: more conservative in extremities, more extended in the retroperitoneum. Expert Rev Anticancer Ther. 2012;12(8):1079–1087. | ||
Crago AM, Brennan MF. Principles in management of soft tissue sarcoma. Adv Surg. 2015;49:107–122. | ||
Loizos N, Xu Y, Huber J, et al. Targeting the platelet-derived growth factor receptor alpha with a neutralizing human monoclonal antibody inhibits the growth of tumor xenografts: implications as a potential therapeutic target. Mol Cancer Ther. 2005;4(3):369–379. | ||
Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488–497. | ||
Karavasilis V, Seddon BM, Ashley S, Al-Muderis O, Fisher C, Judson I. Significant clinical benefit of first-line palliative chemotherapy in advanced soft-tissue sarcoma: retrospective analysis and identification of prognostic factors in 488 patients. Cancer. 2008;112(7):1585–1591. | ||
Leahy M, Garcia Del Muro X, Reichardt P, et al. Chemotherapy treatment patterns and clinical outcomes in patients with metastatic soft tissue sarcoma. The Sarcoma treatment and Burden of Illness in North America and Europe (SABINE) study. Ann Oncol Off J Eur Soc Med Oncol. 2012;23(10):2763–2770. | ||
Minchom A, Jones R, Fisher C, et al. Clinical benefit of second-line palliative chemotherapy in advanced soft-tissue sarcoma. Sarcoma. 2010;2010:264360. | ||
Ratan R, Patel SR. Chemotherapy for soft tissue sarcoma. Cancer. 2016;122(19):2952–2960. | ||
Vlenterie M, Litiere S, Rizzo E et al. Outcome of chemotherapy in advanced synovial sarcoma patients: review of 15 clinical trials from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group; setting a new landmark for studies in this entity. Eur J Cancer. 2016;58:62–72. | ||
Yasui H, Naka N, Imura Y, et al. Tailored therapeutic strategies for synovial sarcoma: receptor tyrosine kinase pathway analyses predict sensitivity to the mTOR inhibitor RAD001. Cancer Lett. 2014;347(1):114–122. | ||
Casali PG. Adjuvant chemotherapy for soft tissue sarcoma. Am Soc Clin Oncol Educ B. 2015:e629–e633. | ||
National Center for Biotechnology Information. Pubchem Open Chemistry Database –Compound Summary for CID 11354606. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/11354606. Accessed: August 30, 2017. | ||
National Center for Biotechnology Information. Pubchem Open Chemistry Database –Compound Summary for CID 5488895. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/5488895. Accessed: August 30, 2017. | ||
Mcbride A, Butler SK. Eribulin mesylate: a novel halichondrin B analogue for the treatment of metastatic breast cancer. Am J Heal Pharm. 2012;69(9):745–755. | ||
Twelves C, Cortes J, Vahdat LT, Wanders J, Akerele C, Kaufman PA. Phase III trials of eribulin mesylate (E7389) in extensively pretreated patients with locally recurrent or metastatic brcast Cancer. Clin Breast Cancer. 2010;10(2):160–163. | ||
Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial.Lancet. 2016;387:1629–1637. | ||
Hirata Y, Uemura D. Halichondrins – antitumor polyether macrolides from a marine sponge. Pure Appl Chem. 1986;58(5):701–710. | ||
Jordan MA, Kamath K, Manna T, et al. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol Cancer Ther. 2005;4(7):1086–1095. | ||
Dabydeen DA, Burnett JC, Bai R, et al. Comparison of the activities of the truncated halichondrin B analog NSC 707389 (E7389) with those of the parent compound and a proposed binding site on tubulin. Mol Pharmacol. 2006;70(6):1866–1875. | ||
Wertz IE, Kusam S, Lam C, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471(7336):110–114. | ||
Eichhorn JM, Sakurikar N, Alford SE, Chu R, Chambers TC. Critical role of anti-apoptotic Bcl-2 protein phosphorylation in mitotic death. Cell Death Dis. 2013;4(10):e834. | ||
Towle MJ, Salvato KA, Budrow J, et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001;61(3):1013–1021. | ||
Dybdal-Hargreaves NF, Risinger AL, Mooberry SL. Eribulin mesylate: mechanism of action of a unique microtubule-targeting agent. Clin Cancer Res. 2015;21(11):2445–2452. | ||
Funahashi Y, Okamoto K, Adachi Y, et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014;105(10):1334–1342. | ||
Kawano S, Asano M, Adachi Y, Matsui J. Antimitotic and non-mitotic effects of eribulin mesilate in soft tissue sarcoma. Anticancer Res. 2016;36(4):1553–1561. | ||
Casazza A, Di Conza G, Wenes M, Finisguerra V, Deschoemaeker S, Mazzone M. Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene. 2014;33(14):1743–1754. | ||
Yoshida T, Ozawa Y, Kimura T, et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial–mesenchymal transition (EMT) to mesenchymal–epithelial transition (MET) states. Br J Cancer. 2014;110(6):1497–1505. | ||
Takano S, Kanai F, Jazag A, et al. Smad4 is essential for down-regulation of E-cadherin induced by TGF-β in pancreatic cancer cell line PANC-1. J Biochem. 2007;141(3):345–351. | ||
Gregory PA, Bracken CP, Smith E, et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22(10):1686–1698. | ||
Herbst RS, Khuri FR. Mode of action of docetaxel – a basis for combination with novel anticancer agents. Cancer Treat Rev. 2003;29(5):407–415. | ||
Miller KD, Sweeney CJ, Sledge J. Redefining the target: chemotherapeutics as antiangiogenics. J Clin Oncol. 2001;19(4):1195–1206. | ||
Grant DS, Williams TL, Zahaczewsky M, Dicker AP. Comparison of antiangiogenic activities using paclitaxel (taxol) and docetaxel (taxotere). Int J Cancer. 2003;104(1):121–129. | ||
Maki RG. Gemcitabine and docetaxel in metastatic sarcoma: past, present, and future. Oncologist. 2007;12(8):999–1006. | ||
Bramwell VHC, Anderson D, Charette ML. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev. 2003;(3):CD003293. | ||
Hwang C. Overcoming docetaxel resistance in prostate cancer: a perspective review. Ther Adv Med Oncol. 2012;4(6):329–340. | ||
Goel S, Mita AC, Mita M, et al. A phase I study of eribulin mesylate (E7389), a mechanistically novel inhibitor of microtubule dynamics, in patients with advanced solid malignancies. Clin Cancer Res. 2009;15(12):4207–4212. | ||
Tan AR, Rubin EH, Walton DC, et al. Phase I study of eribulin mesylate administered once every 21 days in patients with advanced solid tumors. Clin Cancer Res. 2009;15(12):4213–4219. | ||
Mukohara T, Nagai S, Mukai H, Namiki M, Minami H. Eribulin mesylate in patients with refractory cancers: a Phase I study. Invest New Drugs. 2012;30(5):1926–1933. | ||
Morgan RJ, Synold TW, Longmate JA, et al. Pharmacodynamics (PD) and pharmacokinetics (PK) of E7389 (eribulin, halichondrin B analog) during a phase Ι trial in patients with advanced solid tumors: a California Cancer Consortium Trial. Cancer Chemother Pharmacol. 2015;76(5):897–907. | ||
Vahdat LT, Pruitt B, Fabian CJ, et al. Phase II study of eribulin mesylate, a halichondrin B analog, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2009;27(18):2954–2961. | ||
Cigler T, Vahdat LT. Eribulin mesylate for the treatment of breast cancer. Expert Opin Pharmacother. 2010;11(9):1587–1593. | ||
Devriese LA, Witteveen PO, Marchetti S, et al. Pharmacokinetics of eribulin mesylate in patients with solid tumors and hepatic impairment. Cancer Chemother Pharmacol. 2012;70(6):823–832. | ||
Tan A, Sarantopoulos J, Lee L, et al. Pharmacokinetics of eribulin mesylate in cancer patients with normal and impaired renal function. Cancer Chemother Pharmacol. 2015;76(5):1051. | ||
Majid O, Gupta A, Reyderman L, Olivo M, Hussein Z. Population pharmacometric analyses of eribulin in patients with locally advanced or metastatic breast cancer previously treated with anthracyclines and taxanes. J Clin Pharmacol. 2014;54(10):1134–1143. | ||
Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histological subtypes. Lancet Oncol. 2011;12(11):1045–1052. | ||
Kawai A, Araki N, Naito Y, et al. Phase 2 study of eribulin in patients with previously treated advanced or metastatic soft tissue sarcoma. Jpn J Clin Oncol. 2016;47(2):137–144. | ||
Young RJ, Woll PJ. Eribulin in soft-tissue sarcoma. Lancet. 2016;387:1594–1596. | ||
Swami U, Shah U, Goel S. Eribulin in cancer treatment. Mar Drugs. 2015;13(8):5016–5058. | ||
Kaufman PA, Awada A, Twelves C, et al. Phase III open-label randomized study of eribulin mesylate versus capecitabine in patients with locally advanced or metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2015;33(6):594–601. | ||
Kim J Il, Choi KU, Lee IS, et al. Expression of hypoxic markers and their prognostic significance in soft tissue sarcoma. Oncol Lett. 2015;9(4):1699–1706. | ||
Kitahara H, Hirai M, Kato K, Bou-Gharios G, Nakamura H, Kawashiri S. Eribulin sensitizes oral squamous cell carcinoma cells to cetuximab via induction of mesenchymal-to-epithelial transition. Oncol Rep. 2016;6:3139–3144. | ||
Zucali PA, Bertuzzi A, Parra HJS, Campagnoli E, Quagliuolo V, Santoro A. The “old drug” dacarbazine as a second/third line chemotherapy in advanced soft tissue sarcomas. Invest New Drugs. 2008;26(2):175–181. | ||
Demetri GD, Von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786–793. | ||
van der Graaf WTA, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–1886. | ||
Lheureux S, Oza AM, Laurie SA, et al. A phase I combination dose-escalation study of eribulin mesylate and gemcitabine in patients with advanced solid tumours: a study of the Princess Margaret Consortium. Br J Cancer. 2015;113(11):1534–1540. | ||
Roviello G, Milani M, Gobbi A, et al. A phase Ib open-label study to assess the safety and tolerability of everolimus in combination with eribulin in triple-negative breast cancers. Clin Breast Cancer. 2016;16(3):e57–e59. | ||
Osgood CL, Chuk MK, Theoret MR, et al. FDA approval summary: eribulin for patients with unresectable or metastatic liposarcoma who have received a prior anthracycline-containing regimen. Clin Cancer Res. 2017;23(21):6384–6389. | ||
Synold TW, Morgan RJ, Newman EM, et al. A phase I pharmacokinetic and target validation study of the novel anti-tubulin agent E7389: a California Cancer Consortium Trial. In: Proceedings of the ASCO Annual Meeting; May 13–17, 2005; Orlando, FL, USA. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.