")
Back to Journals » The Application of Clinical Genetics » Volume 9
Advances in the management of erythropoietic protoporphyria – role of afamelanotide
Authors Lane AM, McKay JT, Bonkovsky HL
Received 10 September 2016
Accepted for publication 28 October 2016
Published 12 December 2016 Volume 2016:9 Pages 179—189
DOI https://doi.org/10.2147/TACG.S122030
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Martin Maurer
Ashley M Lane,1 Jerome T McKay,2 Herbert L Bonkovsky1
1Department of Internal Medicine, Section on Gastroenterology; 2Department of Microbiology and Immunology, Wake Forest University School of Medicine, Winston-Salem, NC, USA
Abstract: Erythropoietic protoporphyria (EPP) and the phenotypically similar disease X-linked protoporphyria (XLPP) are inherited cutaneous porphyrias characterized clinically by acute non-blistering photosensitivity, intolerance to sunlight, and significantly reduced quality of life. They are due to marked overproduction of protoporphyrin (PP) chiefly by erythroblasts and reticulocytes. In EPP, the underlying genetic defect is in the ferrochelatase gene, which encodes the final enzyme in the heme synthetic pathway. In XLPP, the genetic defect is a gain-of-function mutation, usually a four-base deletion, in the gene that encodes the enzyme 5-aminolevulinic acid synthase-2, the first and rate-controlling enzyme of heme synthesis in developing red blood cells. The excess PP causes acute and painful photosensitivity, being activated by light in the long ultraviolet to blue spectrum (380–420 nm, the Soret band). Although several treatments have been proposed, presently no very effective treatment exists for EPP or XLPP. Afamelanotide (Scenesse®) is a first-in-class synthetic analog of α-melanocyte stimulating hormone. Afamelanotide mimics the naturally occurring hormone to increase skin pigmentation by increasing melanin production in melanocytes, resulting in increased sunlight tolerance in those with EPP/XLPP. Afamelanotide is currently approved for use in the European Union and Switzerland, and it is under review in the United States by the Food and Drug Administration for use in patients with EPP/XLPP. This paper provides a review of the clinical characteristics and current therapies for EPP/XLPP. We discuss the pharmacology, clinical efficacy, safety, and tolerability of afamelanotide and summarize the results of several key Phase II and III clinical trials. These data indicate that afamelanotide is a promising therapy for those with these debilitating diseases.
Keywords: afamelanotide, eumelanin, heme, melanocyte stimulating hormone, photosensitivity, porphyria
Introduction: overview of the epidemiology, etiology, and pathogenesis of erythropoietic protoporphyria
Erythropoietic protoporphyria (EPP, OMIM 177000) is a rare photodermatosis due to an innate defect of heme biosynthesis characterized by excessive production and subsequent accumulation of metal-free protoporphyrin (PP) in erythrocytes, plasma, skin, and liver.1–4 It has been described worldwide with an estimated prevalence ranging from 1 in 75,000 to 1 in 200,000. Males and females are equally affected.5,6 Classical EPP is characterized by decreased activity of ferrochelatase, the final enzyme in the heme synthesis pathway that inserts ferrous iron into PP to generate heme (Figure 1, Table 1). The principal source of excess metal-free PP in the plasma and erythrocytes in EPP is erythroblasts and reticulocytes in the bone marrow.2,4,7
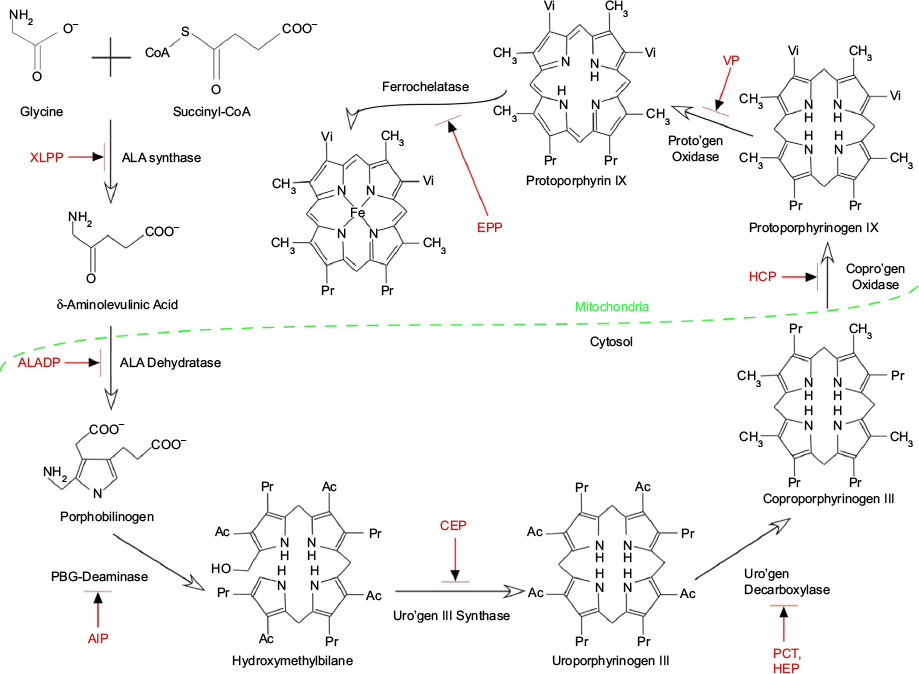 | Figure 1 Summary of the heme synthetic pathway, highlighting enzymatic defects associated with the porphyrias. Notes: The heme synthetic pathway involves eight enzymes, four of which are active in the mitochondria, and four of which are active in the cytoplasm.2,55 The pathway is initiated and completed in the mitochondria. Intermediate steps in the cytoplasm begin with the activity of ALA dehydratase. Open arrows indicate progression through the pathway. Deficiency (indicated by blocked red arrows) in any of the eight enzymes involved in the pathway may contribute to development of acute or chronic hepatic porphyrias or erythropoietic porphyrias, as shown in red. Abbreviations: Ac, acetate; AIP, acute intermittent porphyria; ALA, 5-aminolevulinic acid; ALADP, ALA dehydratase deficiency porphyria; CEP, congenital erythropoietic porphyria; copro’gen, coproporphyrinogen; EPP, erythropoietic protoporphyria; HCP, hereditary coproporphyria; HEP, hepatoreythropoietic porphyria; PBG, porphobilinogen; PCT, porphyria cutanea tarda; Pr, propionate; proto’gen, protoporphyrinogen; Uro’gen, uroporphyrinogen; Vi, vinyl; VP, variegate porphyria; XLPP, X-linked protoporphyria. |
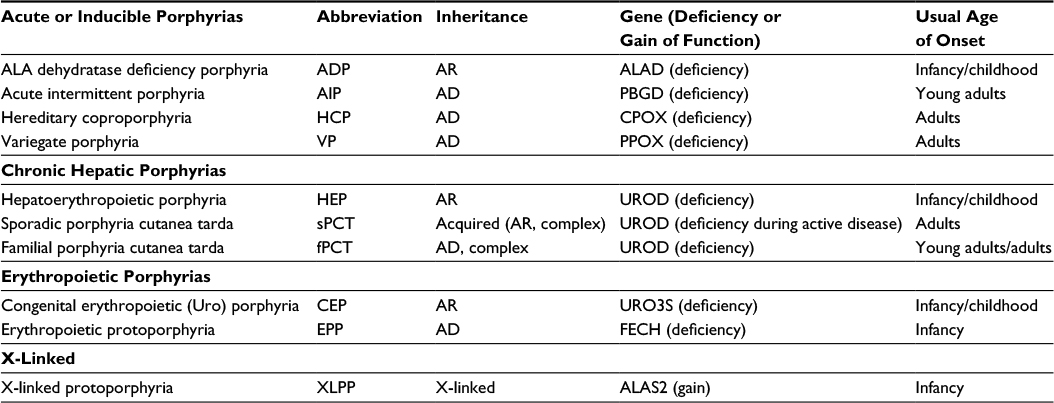 | Table 1 Types of porphyrias Notes: The porphyrias are classified by their major clinical manifestations (acute/chronic/erythropoietic/X-linked). Inheritance patterns and the gene products affected are also listed, as are the usual ages at which the diseases first become manifest. The familial form of PCT (fPCT) requires other risk factors, such as alcohol iron, hepatitis C, or HIV infection for clinical expression of disease. Most subjects with ~50% decrease in activity of UROD do not develop clinical disease, thus the “complex” AD inheritance. Abbreviations: AD, autosomal dominant; ALA, 5-aminolevulinic acid; ALAD, ALA dehydratase; ALAS2, 5-aminolevulinate synthase 2; AR, autosomal recessive; CPOX, coproporphyrinogen oxidase; f, familial; FECH, ferrochelatase; PBGD, porphobilinogen deaminase; PPOX, protoporphyrinogen oxidase; UROD, uroporphyrinogen decarboxylase; URO3S, uroporphyrinogen 3 synthase. |
EPP clinical expression varies, typically presenting in infancy or early childhood as a severe, immediate, painful photosensitivity upon first exposure to sunlight.3,4,8 Cardinal clinical features of EPP and X-linked protoporphyria are summarized in Table 2. Metal-free PP accumulating in the skin and dermal blood vessels undergoes a photodynamic reaction when skin is exposed to sun or visible light. This in turn leads to singlet oxygen (oxygen radicals) and likely other reactive oxygen species that produce tissue and blood vessel damage. Patients are most sensitive to blue light (380–420 nm), corresponding to what is known as the Soret band. Most patients develop stinging, prickling, and burning pain within minutes of exposure to sunlight, followed by erythema, edema, and occasionally petechiae, which can last for hours to several days. Unlike the typical features of other forms of cutaneous porphyrias, such as porphyria cutanea tarda, congenital erythropoietic porphyria (Günther’s disease), and variegate porphyria, vesicles or bullae and hirsutism (especially on the face) rarely, if ever, occur in EPP, and the severity and chronicity of cutaneous changes is less severe. Nevertheless, chronic skin changes such as lichenification, pseudovesicles, and labial grooving can develop after multiple episodes of acute phototoxicity.1–4,8 As PP is a lipophilic molecule excreted by the liver, hepatic manifestations of this disease may include cholelithiasis, cholestatic hepatitis, and pigmentary cirrhosis.4,5
 | Table 2 Cardinal clinical features of EPP/XLPP Note: The diagnostic findings that are hallmarks of EPP/XLPP are delineated. Abbreviations: PP, protoporphyrin; EPP, erythropoietic protoporphyria; XLPP, X-linked protoporphyria; RBC, red blood cell. |
Genetic studies have demonstrated two different patterns of inheritance in EPP caused by mutations in the ferrochelatase gene (FECH).9,10 In the majority of cases, photosensitivity is inherited in an autosomal recessive pattern, dubbed a “pseudo-dominant” pattern, in which a deleterious FECH mutation, which markedly decreases or eliminates FECH activity, is inherited in trans to a highly prevalent noncoding hypomorphic allele (FECH IVS3-48T >C).9,10 Less common is an autosomal recessive inheritance, in which FECH mutations are present in the coding regions of both alleles.8,9 Acquired somatic mutations of FECH secondary to myeloid disorders are unusual causes of EPP.4,9,11,12. Typically, the activity of FECH in clinically affected persons is 10%–30% of normal.2,3
In recent years, a closely related genetic disorder with a similar clinical and biochemical phenotype as EPP, namely, X-linked protoporphyria (XLPP, OMIM 300752), has been identified to occur when overproduction of PP is caused by gain-of-function mutations in the 5-aminolevulinate synthase-2 gene (ALAS2). ALAS2 encodes the initial enzyme of erythroid heme biosynthesis and is located on the X-chromosome, thus the name XLPP.9,10 In this form of the disease, FECH activity is normal, but PP is overproduced in developing red blood cells due to the excessive activity of ALAS2.2–4 The molecular defects vary somewhat, but most commonly are related to a four base pair deletion in the C terminus of the ALAS2 gene (1706-1709 del AGTG). It remains unclear how and why such increases in PP production are not compensated by normal FECH activity with formation of greater amounts of heme or zinc PP. Indeed, there may be other pathogenic factors, such as inadequate supplies of iron (Figure 2).13 In XLPP, levels of PP in red blood cells and plasma are often very high, and the proportion that is the zinc chelate is also high (up to 40%–50% zinc PP). The risk of development of PP hepatopathy in XLPP may be greater than in EPP.
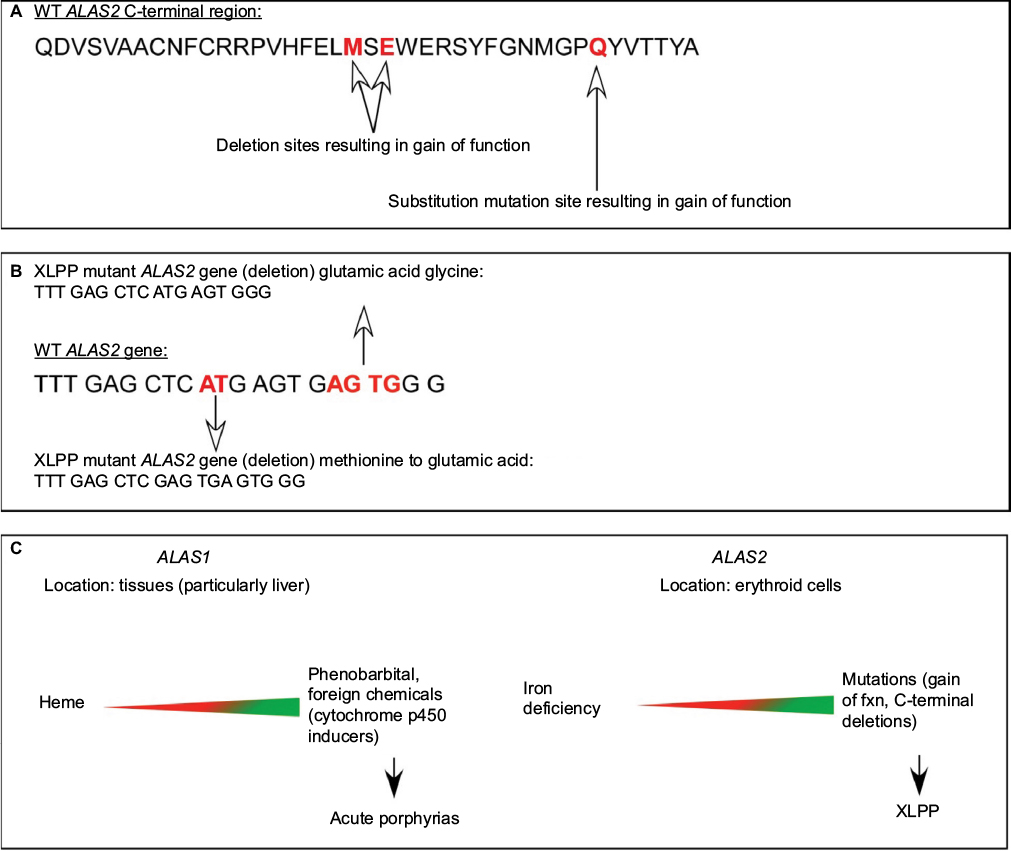 | Figure 2 Different regulation of the housekeeping (ALAS1) and erythroid (ALAS2) forms of 5-aminolevulinate synthase. Notes: The two isozymes for ALAS are encoded by separate genes (ALAS1 on chromosome 3 and ALAS2 on the X chromosome), and provide ALA for eventual heme for hemoproteins and other cellular needs. The genes are uniquely induced and repressed in varying tissues.2,55 (A) The C terminal polypeptide sequence of human ALAS2 with mutation sites (red) known to contribute to XLPP. (B) bp sequence of human ALAS2 spanning known deletion (red) polymorphisms resulting in gain of function. The truncated bp sequences of the XLPP mutants are also displayed with amino acid substitutions. (C) Differential location and regulation of ALAS1 vs ALAS2 are reflected in the potential resulting porphyrias manifested when there is uncontrolled upregulation of the respective genes. Heme markedly downregulates (red) ALAS1, acting at several different levels.2 In contrast, heme has little if any down-regulatory effect on ALAS2, but this gene is upregulated by iron acting through iron regulatory proteins to increase translation and downregulated by lack of iron in developing red blood cells, such that a balance is normally retained between PP synthesis and availability of iron for heme and hemoglobin formation.2,55 Upregulation (green) of either ALAS1 (chemicals) or ALAS2 (gain-of-function mutations) leads to different forms of porphyria (acute porphyrias or XLPP). Abbreviations: XLPP, X-linked protoporphyria; bp, base pair; ALAS, 5-aminolevulinate synthase; PP, protoporphyrin; WT, wild type. |
Natural history and prognosis of EPP and X-linked protoporphyria
Most patients with porphyrias suffer from life-long moderate-to-intense immediate photosensitivity (Figure 3, Table 2). Severity depends upon the nature of the genetic defects, which impacts the levels of PP in red blood cells, plasma, skin, and other tissues, and also upon the complexion and natural skin pigmentation of the patients. For example, deeply pigmented persons of African descent are less severely affected than fair skinned, freckled, redheaded persons of Celtic or Nordic descent. With time, patients with EPP or XLPP learn to avoid sunlight or other strong light source to alleviate the severe sequelae of excess sunlight exposure. Thus, they restrict their outdoor activities during the day (10 am–5 pm). Those with darker complexions often find that by gradually increasing their sun exposure (or even tanning bed exposures) early in the spring seasons, they are able to increase their tolerance. For most patients, though, their lives are marked by chronic limitations.1–4
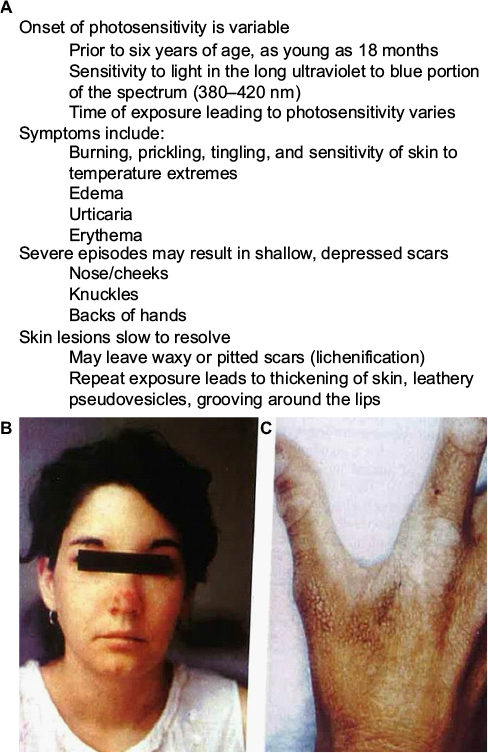 | Figure 3 Cardinal features and characteristics of skin lesions of EPP/XLPP. Notes: (A) The common signs and symptoms found in both erythropoietic protoporphyria and X-linked protoporphyria. (B, C) Common cutaneous lesions in protoporphyria. (B) The image shows an acute photosensitivity reaction resulting in edema of the face and erythema on the bridge of the nose following sun exposure. (C) The image demonstrates the chronic skin changes on the hand of a patient with protoporphyric liver disease. There are thickening and lichenification of the dorsum of the hand in areas that had experienced repeated sun exposure. (Photos kindly provided by JR Bloomer, MD). Abbreviations: EPP, erythropoietic protoporphyria; XLPP, X-linked protoporphyria. |
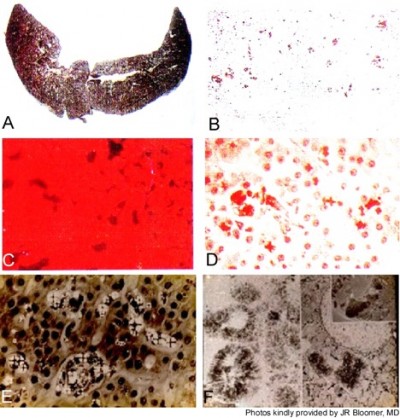
Roughly 1%–3% of subjects develop PP hepatopathy with clinical and laboratory evidence of progressive liver injury (Figure 4). As might be anticipated, any intercurrent conditions that cause cholestasis, such as biliary obstruction, acute viral or cholestatic hepatitis or drug-induced liver injury, and alcoholic hepatitis, may exacerbate their underlying PP disease. Development of PP gallstones at early age is especially frequent, and many require laparoscopic cholecystectomy before the age of 40 years.1–4
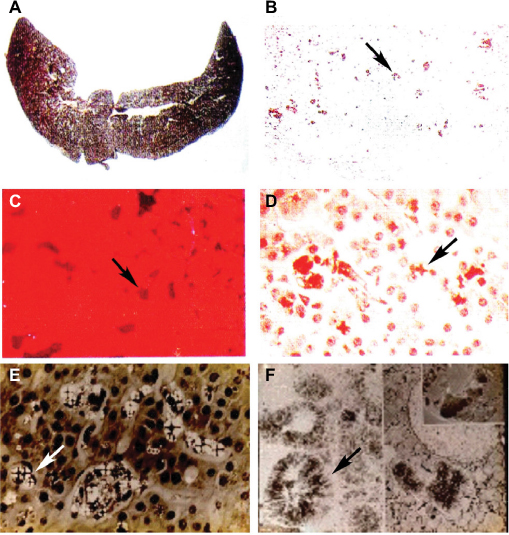 | Figure 4 Liver findings in protoporphyric hepatopathy. Notes: (A) Gross appearance of the liver showing pigmentary cirrhosis. (B–D) Representative microscopic images of the liver, including (B) the pigmented deposits of PP crystals (arrow) under light microscopy, (C) characteristic pink fluorescence indicative of differential excretion of PP in the liver under fluorescence microscopy (arrow), and (D) polarization microscopy. (E) Birefringence on polarization microscopy reveals visible Maltese cross patterns (arrow; D and E) within hepatocytes due to crystals of PP. (F) Electron microscopy demonstrating crystals of PP within hepatocytes (arrow). (Photos kindly provided by JR Bloomer, MD). Abbreviation: PP, protoporphyrin. |
Those who develop PP hepatopathy may sometimes be “rescued” with plasma or red blood cell exchange transfusions and/or with infusions of intravenous heme.14–16 Some require liver transplantation, resulting in improved hepatic and clinical status. However, because the PP overproduction originates in the bone marrow, liver transplantation is not curative in EPP or XLPP, unlike the acute porphyrias. Cure of the underlying condition requires successful bone marrow or hematopoietic stem cell transplantation. This may reasonably be considered in patients with very severe phenotypes or in those who have undergone or are being considered for liver transplantation, the former especially if they show evidence of recurring and progressive liver disease.2–4
Overview of management options
Keys to the management of EPP/XLPP are summarized in Table 3. In addition to the points already described regarding prevention and treatment of cholestatic conditions, oral β-carotene therapy (Lumitene®, Tishcon Corporation, Westbury, NY, USA) has been used due to the pioneering work of Mathews-Roth et al.17,18 For β-carotene to have an effect, a high daily dose is needed (~180 mg or more per day). This leads to a noticeable yellow hue to the skin (carotenemia), which many persons find objectionable. In addition, many patients do not find that β-carotene offers much benefit. In our experience, patients who have taken both β-carotene and afamelanotide have judged the latter to be much more effective in decreasing phototoxicity. Additionally, a proof-of-concept study has shown potential for colestipol as a therapeutic to improve sun sensitivity, presumably through the binding of intestinal PP.19
 | Table 3 Principles of management of EPP and XLPP Note: Clinical management strategies for effective management of both EPP and XLPP are laid out. Abbreviations: PP, protoporphyrin; EPP, erythropoietic protoporphyria; XLPP, X-linked protoporphyria; HBV, hepatitis B virus; HAV, hepatitis A virus. |
Recently, Tu et al20 suggested that cimetidine in high daily doses (30–40 mg/kg body weight per day) was of some benefit in children with EPP. Its use seems to have been predicated upon earlier reports that cimetidine inhibits certain isoforms of cytochrome P-450 and ameliorated acute porphyria in rodent models and in scattered case reports in humans with acute intermittent porphyria.21–24 On the basis of the different sources of excess heme precursors in acute hepatic porphyrias (liver) vs in EPP/XLPP (bone marrow), and on the lack of demonstrable effects of cimetidine on FECH or ALAS2 activities, it seems doubtful that cimetidine would benefit patients with EPP or XLPP. Perhaps there may be some effects on inflammatory effector pathways, related to histamine, which are blocked by high doses of cimetidine. Questions and concerns about the preliminary report of Tu et al25 have already been raised.
Mild anemia, often with iron deficiency, is common in EPP/XLPP.2,6 One might anticipate that such deficiency would exacerbate clinical disease due to increased accumulation of PP, which is not converted to heme by FECH, owing to a lack of the necessary iron substrate. Indeed, several case reports have been published showing improvement in symptoms and decreases in PP levels in patients treated with iron supplements.26–28 However, there is controversy in the field regarding the efficacy of iron therapies. Limited evidence shows that some EPP patients had lower red blood cell (RBC) PP levels when they were iron deficient,28,29 while others noted improvements with iron-replacement therapy.13,26,27,30,31 It was recently shown that subjects with EPP or XLPP are able to absorb iron well from the gastrointestinal tract and lack inappropriately high levels of hepcidin.32 The role of iron therapy in EPP/XLPP thus remains less than certain and is in need of further study. Perhaps, only a subset of patients will show a favorable response. Currently, it is not clear what factors may influence response to iron therapy.
Review of the pharmacology and mechanism of action of afamelanotide
Afamelanotide (Scenesse®, Clinuvel Pharmaceuticals, Melbourne, Australia) [Nleu4, D-Phe7 α-MSH] is an analog of human α-melanocyte-stimulating hormone (α-MSH), the naturally occurring hormone that stimulates the synthesis of melanin.33,34 First described in 1980, afamelanotide is a synthetic tridecapeptide that differs from human α-MSH at the fourth and seventh amino acids, where norleucine and D-phenylalanine replace methionine and L-phenylalanine, respectively (Figure 5).35,36 These changes confer unique biological properties including resistance to enzymatic degradation, prolonged plasma half-life, and increased duration of action allowing afamelanotide to emerge as a potent melanocortin ligand.35,37
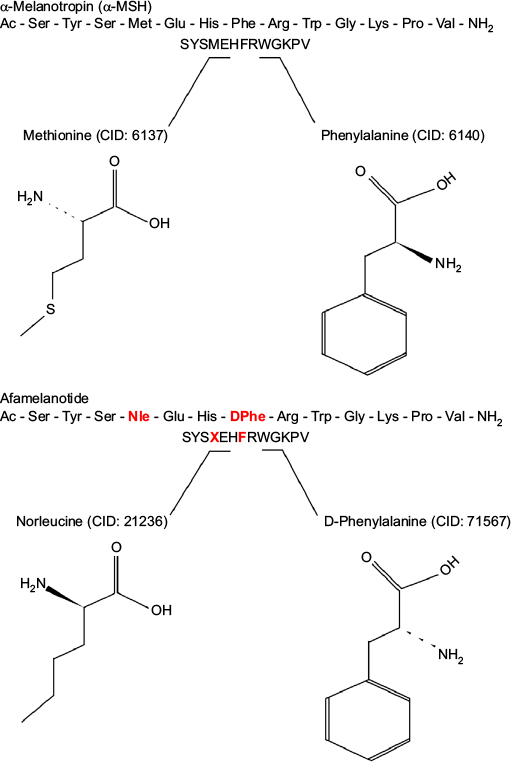 | Figure 5 Structural differences between afamelanotide and α-melanocyte stimulating hormone. Notes: Afamelanotide differs from human α-MSH at the fourth and seventh amino acids, where norleucine replaces methionine (red) and D-phenylalanine replaces L-phenylalanine (red), respectively. These substitutions in the polypeptide confer unique biological properties, including resistance to enzymatic degradation, prolonged plasma half-life, and increased duration of action.35,36 Abbreviations: CID, PubChem compound identifier number; α-MSH, α-melanocyte-stimulating hormone. |
Afamelanotide mirrors the activity of α-MSH by binding to the G-protein coupled melanocortin 1 receptor (MC1R) that is present in dermal cells, including melanocytes, to activate melanogenesis.36 When the MC1R is stimulated by α-MSH, melanin synthesis transitions from the less protective yellow-red pheomelanin to more protective brown-black eumelanin.33,38–40 Eumelanin provides photoprotection by absorbing and scattering visible and ultraviolet (UV) light, scavenging free radicals and reactive oxygen species, and functioning as a neutral density filter capable of equal reduction of transmission of all wavelengths of light.33,34,39,41 Afamelanotide stimulates eumelanin synthesis independent of exposure to UV radiation, as opposed to endogenous α-MSH, which is produced in response to UV damaged skin cells to prevent further damage.40,41
First demonstrated to induce skin pigmentation and tanning in the early 1990s, afamelanotide increases eumelanin density in normal subjects as well as patients with EPP or XLPP.40–43 In normal volunteers, afamelanotide administration at a dose of 0.16 mg/kg/d subcutaneously induced tanning with an increase in the eumelanin: pheomelanin ratio in the absence of sun exposure (p=0.054), accompanied by significant increases in eumelanin at different anatomical sites.40 In a study of healthy Caucasian adult volunteers, afamelanotide administered subcutaneously at 0.16 mg/kg/d significantly increased melanin density (p<0.0001) and luminance (skin darkening; p<0.001) in all subjects, independent of sun exposure, as compared with placebo.41 Additionally, afamelanotide reduced apoptotic sunburn cell injury by greater than 50% (p<0.001).41 Consistent with these findings, in a Phase II study of five patients with EPP who received a subcutaneous afamelanotide implant designed to release the active drug slowly at a dose of 20 mg given at intervals of 60 days, skin melanin density was significantly increased from day 30 to day 120 compared to baseline (p=0.004).43 Data from a Phase III crossover trial further supports these results in that two separate treatment groups of EPP patients receiving afamelanotide (16 mg subcutaneous) implants experienced increases in absolute melanin levels of 29.1% and 28.4%, respectively.44,45
Afamelanotide is administered subcutaneously, a route known to be effective for many proteins and peptide-based drugs, as the tridecapeptide does not penetrate the epidermis and it is rapidly degraded in the gastrointestinal tract when taken orally.46,47
Data regarding the pharmokinetics of afamelanotide are limited, and are lacking for its active or inactive metabolites.48 With the current formulation of afamelanotide by Clinuvel Pharmaceuticals, once implanted, the majority of the active substance is released within 2 days and over 90% is released within 5 days. In most studies, afamelanotide plasma levels were below the level of quantitation by 10 days.48 Although it has a half-life of between 30 and 50 minutes, the effect of the drug is seen for up to 60 days, albeit with reduction in efficacy often noted by approximately day 40–45, and the implant is fully resorbed within this time.36,48 Data regarding the use of afamelanotide in patients with renal or hepatic impairment, special populations (pediatric, adolescent, elderly), or in pregnancy or lactation are limited.48
Efficacy, safety, and tolerability of afamelanotide
A five-patient pilot study in Switzerland was the first to suggest the potential therapeutic benefit of afamelanotide for patients with EPP.49 In subsequent Phase II, randomized, placebo-controlled studies conducted in the United States and Europe, afamelanotide significantly increased both patients’ ability to experience pain-free sun exposure and their quality of life.50 Additionally, these trials demonstrated favorable risk–safety profile and patient tolerability.
Since then, three Phase III trials have evaluated the effectiveness of afamelanotide as a therapeutic tool in the management of EPP (Table 4). In CUV017, one Australian and seven European centers performed a 12-month, randomized, double-blind, placebo-controlled Phase III crossover study that evaluated afamelanotide as a photoprotectant.44,45,48 A total of 91 patients with EPP aged 18–70 years received alternating subcutaneous implants of either 16 mg afamelanotide or placebo once every 2 months for a total of six implants over 12 months (three afamelanotide and three placebo).45 Treatment with afamelanotide was associated with a significant reduction in total number of days during which patients experienced cutaneous pain (p=0.0023), reduced average pain severity (p=0.0017), and significantly increased sun exposure (p<0.0001).45
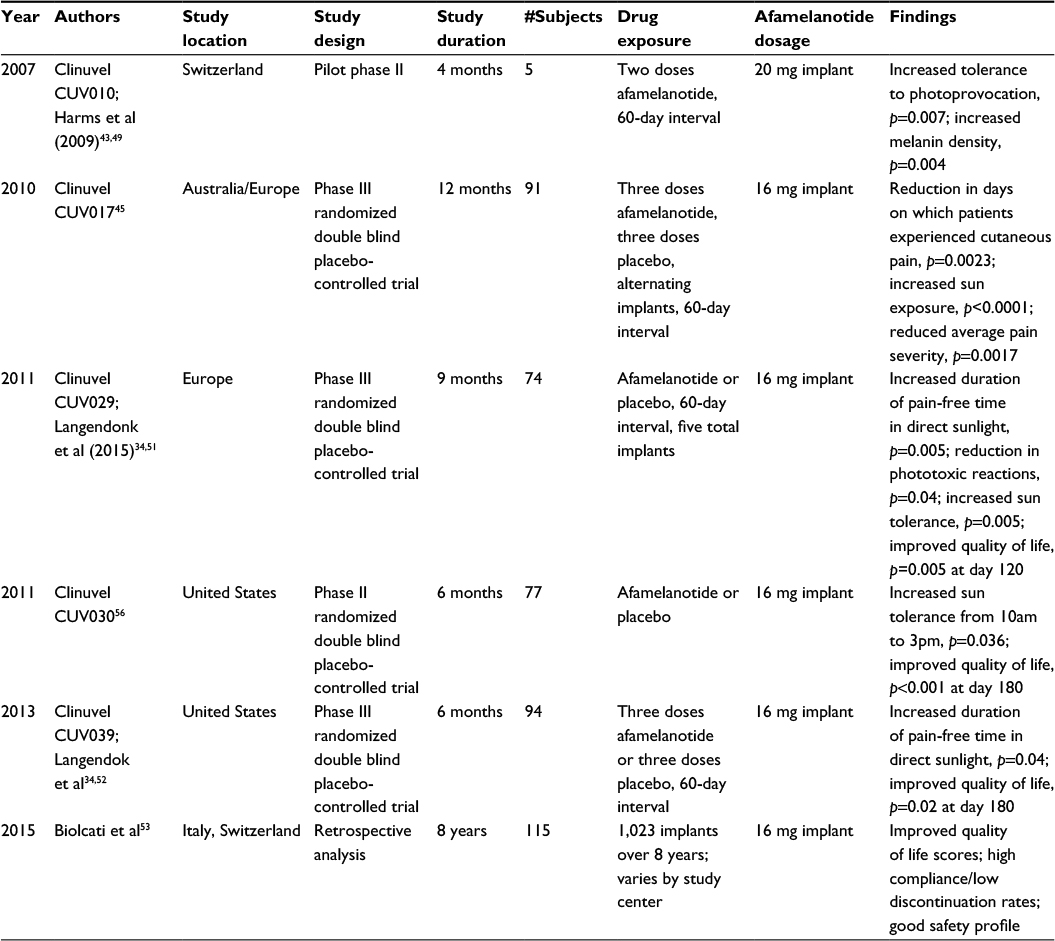 | Table 4 Summary of clinical trials of afamelanotide Note: Selected key features of the clinical trials of afamelanotide from 2007 to 2015 are summarized. |
In CUV029, eight European centers performed a 9-month, randomized, double-blind, placebo-controlled trial consisting of two parallel treatment arms evaluating afamelanotide as a photoprotectant for patients with EPP.34,51 The primary end point was the number of hours of direct exposure to sunlight without pain, with a secondary end point evaluating quality of life. A total of 74 adult patients with EPP aged ≥18 years were randomly assigned to receive a subcutaneous implant of afamelanotide 16 mg (n=38) or placebo (n=36) every 60 days for a total of five implants. A significant increase in the duration of pain-free time in direct sunlight was seen in the afamelanotide group compared to the placebo group (median 6.0 hours vs 0.8 hours; p=0.005). Additionally, the patients who received afamelanotide had a significant reduction in phototoxic reactions (77 vs 146; p=0.04), a lower median and maximum pain score per phototoxic episode (p=0.035 and p=0.018), a faster recovery time from phototoxic reactions (p=0.04) and were able to spend more time in direct sunlight (p=0.005) without experiencing pain.34,51 Quality of life improved with afamelanotide therapy, with a significant difference (as measured by the EPP quality of life questionnaire) on days 120 (p=0.005) and 270 (p=0.011).34,51
In CUV039, seven centers in the United States performed a 6-month randomized, double-blind, placebo-controlled trial consisting of two parallel treatment arms evaluating afamelanotide as a photoprotectant for patients with EPP.34,52 The primary end point was the number of hours of direct exposure to sunlight without pain, with a secondary end point evaluating quality of life. A total of 94 adult patients (age >18 years) with EPP were enrolled; one withdrew prior to receiving any study drug. Among the remaining 93 subjects, 89 subjects in the intent-to-treat population were randomly assigned to receive a subcutaneous implant of afamelanotide 16 mg (n=46) or placebo (n=43) every 60 days for a total of three implants and completed the study. Four subjects dropped out due to diary card end points; three subjects were terminated early for not returning any diaries (one afamelanotide subject and two placebo subjects), and one subject was terminated who received all three doses but did not return any diaries or attend visit 4 (afamelanotide subject). A 70% increase in the duration of pain-free time in direct sunlight was seen in the patients who received afamelanotide (69.4 hours vs 40.8 hours; p=0.04).34 However, there was no significant change in the total number of phototoxic reactions in patients who received afamelanotide as compared to placebo after 6 months (46 vs 43; p=0.60).34 Quality of life improved with afamelanotide therapy, with a significant difference (as measured by the EPP quality of life questionnaire) on days 60 (p<0.001), 120 (p<0.001), and 180 (p=0.02).34
Afamelanotide has been well tolerated in patients with EPP. In CUV029 and CUV039, the majority of adverse events that occurred during the study period were mild to moderate in severity; the most common side effects were headache, nausea, nasopharyngitis, and back pain.34 Between-group differences in adverse events were unremarkable, save for mild hyperpigmentation at the site of afamelanotide implants (33% of patients) and a small number of patients reported moles that darkened while on afamelanotide.34 There were no deaths in the study, and the four serious adverse events in the afamelanotide groups and two in the placebo groups were considered to be unrelated to the study drug. A longitudinal observational study supported a favorable safety profile.53 The study followed 115 patients with EPP treated with a total of 1,023 afamelanotide implants over a period of up to 8 years at two porphyria centers (Italy and Switzerland).53 Only minor adverse events were reported that were attributed to afamelanotide, consisting mainly of nausea and headache.53 Two patients noted a new melanocytic nevus; one of them was removed by excisional biopsy and was negative for malignancy.53
Concern has been raised regarding the potential for it to promote the development of melanoma.34,36 Several in vivo and in vitro studies have not shown a correlation between afamelanotide use and malignancy, and, in fact, have shown that melanoma cell proliferation is inhibited.36 The primary risk factor for melanoma is ultraviolet B (UVB) radiation, which can induce DNA damage leading to a pro-proliferative state.36 α-MSH increases eumelanin production and provides protection against melanoma by reducing exposure to UVB irradiation, scavenging oxygen free radicals produced after exposure to UVB radiation, and by stimulating DNA repair via MC1R signaling in response to α-MSH.34,36 Additionally, melanoma stem cells do not express the MC1R, and it is important to note that the elevated levels of melanin seen with melanoma are not the cause of melanoma.36 Thus, it seems likely that afamelanotide will decrease the risk of development of malignant melanoma.
Afamelanotide has not been associated with immunogenicity during long-term administration in patients with EPP.54 In 26 patients with EPP receiving long-term administration of afamelanotide (16–20 mg subcutaneous implants), anti-afamelanotide IgG antibody levels were undetectable in 23 patients after up to 6 years of use with cumulative dosing between 32 and 408 mg.54 Three patients had anti-α-MSH antibody levels (as measured by enzyme-linked immunosorbent assay [ELISA]) above the predetermined cutoff in this study, although the optical density values did not change remarkably after afamelanotide administration.54
Conclusion
In conclusion, EPP and XLPP are rare, usually inherited disorders of porphyrin and heme synthesis that are characterized biochemically by excessive production of PP. The cardinal clinical features are severe and near immediate photosensitivity following exposure to sunlight or other strong light in the visible spectrum. Development of symptomatic gallstones early in life is common. Because excess PP is potentially hepatotoxic, and is removed from the body by uptake into hepatocytes and secretion into the bile, a minority (1%–3%) of patients develop liver disease, which may lead to pigmentary cirrhosis, for which liver transplantation may be required. Cure of the disease requires bone marrow or stem cell transplantation, because developing red blood cells are the principal source of overproduction of PP. Management has previously depended upon preventing cholestatic disorders, such as viral hepatitis, alcoholic liver disease, drug-induced liver injury, as well as avoidance and protection from sunlight or other strong light. β-carotene and other oral agents have not proven of much benefit. Iron deficiency is common in EPP/XLPP, and therapy with iron has been reported to be of benefit in some patients, although there also have been claims of adverse effects of iron therapy. The role of iron replacement is in need of further careful study. Afamelanotide has proven effective to decrease photosensitivity and has been approved for use in the treatment of EPP/XLPP in the European Union and Switzerland. Clinuvel is currently seeking US Food and Drug Administration approval for use of afamelanotide in the United States. Based upon recent reports and the clinical experience of the senior author and other clinicians who have used afamelanotide, it represents a positive therapeutic advancement in the management of EPP/XLPP, and we are hopeful that it soon will be approved for use in the United States and other non-European countries.
Acknowledgments
Supported in part by a cooperative agreement (DK 083909) and grant (HL117199) from the US National Institutes of Health and funds from the US Rare Diseases Clinical Research Network, awarded to HLB.
Disclosure
Within the past 3 years, Dr Bonkovsky has received research funding from Alnylam Pharma. He has served as a consultant to Alnylam Pharma, Clinuvel, Recordati Rare Chemicals, and Mitsubishi-Tanabe. The authors report no other conflicts of interest in this work.
References
Todd DJ. Erythropoietic protoporphyria. Br J Dermatol. 1994;131(6):751–766. | ||
Bonkovsky HL, Guo JT, Hou W, Li T, Narang T, Thapar M. Porphyrin and heme metabolism and the porphyrias. Compr Physiol. 2013;3(1):365–401. | ||
Balwani M, Bloomer J, Desnick R; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Erythropoietic Protoporphyria, Autosomal Recessive. 2012 Sep 27 [Updated 2014 Oct 16]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. Available from: https://www.ncbi.nlm.nih.gov/books/NBK100826/. | ||
Lecha M, Puy H, Deybach JC. Erythropoietic protoporphyria. Orphanet J Rare Dis. 2009;4:19. | ||
Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375(9718):924–937. | ||
Holme SA, Anstey AV, Finlay AY, Elder GH, Badminton MN. Erythropoietic protoporphyria in the U.K.: clinical features and effect on quality of life. Br J Dermatol. 2006;155(3):574–581. | ||
Scholnick P, Marver HS, Schmid R. Erythropoietic protoporphyria: evidence for multiple sites of excess protoporphyrin formation. J Clin Invest. 1971;50(1):203–207. | ||
Besur S, Hou W, Schmeltzer P, Bonkovsky HL. Clinically important features of porphyrin and heme metabolism and the porphyrias. Metabolites. 2014;4(4):977–1006. | ||
Schmitt C, Ducamp S, Gouya L, Deybach JC, Puy H. La protoporphyrie érythropoïétique : une maladie, deux gènes et trois mécanismes moléculaires [Inheritance in erythropoietic protoporphyria]. Pathol Biol (Paris). 2010;58(5):372–380. French. | ||
Whatley SD, Mason NG, Holme SA, Anstey AV, Elder GH, Badminton MN. Molecular epidemiology of erythropoietic protoporphyria in the U.K. Br J Dermatol. 2010;162(3):642–646. | ||
Goodwin RG, Kell WJ, Laidler P, et al. Photosensitivity and acute liver injury in myeloproliferative disorder secondary to late-onset protoporphyria caused by deletion of a ferrochelatase gene in hematopoietic cells. Blood. 2006;107(1):60–62. | ||
Sarkany RP, Ross G, Willis F. Acquired erythropoietic protoporphyria as a result of myelodysplasia causing loss of chromosome 18. Br J Dermatol. 2006;155(2):464–466. | ||
Landefeld C, Kentouche K, Gruhn B, et al. X-linked protoporphyria: iron supplementation improves protoporphyrin overload, liver damage and anaemia. Br J Haematol. 2016;173(3):482–484. | ||
Do KD, Banner BF, Katz E, Szymanski IO, Bonkovsky HL. Benefits of chronic plasmapheresis and intravenous heme-albumin in erythropoietic protoporphyria after orthotopic liver transplantation. Transplantation. 2002;73(3):469–472. | ||
Reichheld JH, Katz E, Banner BF, Szymanski IO, Saltzman JR, Bonkovsky HL. The value of intravenous heme-albumin and plasmapheresis in reducing postoperative complications of orthotopic liver transplantation for erythropoietic protoporphyria. Transplantation. 1999;67(6):922–928. | ||
McGuire BM, Bonkovsky HL, Carithers RL Jr, et al. Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl. 2005;11(12):1590–1596. | ||
Mathews-Roth MM, Pathak MA, Fitzpatrick TB, Harber LC, Kass EH. Beta-carotene as a photoprotective agent in erythropoietic protoporphyria. N Engl J Med. 1970;282(22):1231–1234. | ||
Mathews-Roth MM, Pathak UA, Fitzpatrick TB, Harber LC, Kass EH. Beta-carotene as an oral photoprotective agent in erythropoietic protoporphyria. JAMA. 1974;228(8):1004–1008. | ||
Tishler PV, Rosner B. Treatment of erythropoietic protoporphyria with the oral sorbent colestipol: a proof-of-concept clinical trial. J Am Acad Dermatol. 2014;70(2):391–392. | ||
Tu JH, Sheu SL, Teng JM. Novel treatment using cimetidine for erythropoietic protoporphyria in children. JAMA Dermatol. 2016. doi: 10.1001/jamadermatol.2016.2303. Epub ahead of print. | ||
Horie Y, Udagawa M, Hirayama C. Clinical usefulness of cimetidine for the treatment of acute intermittent porphyria – a preliminary report. Clin Chim Acta. 1987;167(3):267–271. | ||
Baccino E, Lan Cheong Wah LS, Bressollette L, Mottier D. Cimetidine in the treatment of acute intermittent porphyria. JAMA. 1989;262(21):3000. | ||
Horie Y, Norimoto M, Tajima F, Sasaki H, Nanba E, Kawasaki H. Clinical usefulness of cimetidine treatment for acute relapse in intermittent porphyria. Clin Chim Acta. 1995;234(1–2):171–175. | ||
Cherem JH, Malagon J, Nellen H. Cimetidine and acute intermittent porphyria. Ann Intern Med. 2005;143(9):694–695. | ||
Langendonk J, Wilson J. Insufficient evidence to conclude that cimetidine is beneficial in protoporphyria. JAMA Dermatol. In press. | ||
Holme SA, Thomas CL, Whatley SD, Bentley DP, Anstey AV, Badminton MN. Symptomatic response of erythropoietic protoporphyria to iron supplementation. J Am Acad Dermatol. 2007;56(6):1070–1072. | ||
Bentley DP, Meek EM. Clinical and biochemical improvement following low-dose intravenous iron therapy in a patient with erythropoietic protoporphyria. Br J Haematol. 2013;163(2):289–291. | ||
Barman-Aksozen J, Minder EI, Schubiger C, Biolcati G, Schneider-Yin X. In ferrochelatase-deficient protoporphyria patients, ALAS2 expression is enhanced and erythrocytic protoporphyrin concentration correlates with iron availability. Blood Cells Mol Dis. 2015;54(1):71–77. | ||
Milligan A, Graham-Brown RA, Sarkany I, Baker H. Erythropoietic protoporphyria exacerbated by oral iron therapy. Br J Dermatol. 1988;119(1):63–66. | ||
Gordeuk VR, Brittenham GM, Hawkins CW, Mukhtar H, Bickers DR. Iron therapy for hepatic dysfunction in erythropoietic protoporphyria. Ann Intern Med. 1986;105(1):27–31. | ||
McClements BM, Bingham A, Callender ME, Trimble ER. Erythropoietic protoporphyria and iron therapy. Br J Dermatol. 1990;122(3):423–424. | ||
Bossi K, Lee J, Schmeltzer P, et al. Homeostasis of iron and hepcidin in erythropoietic protoporphyria. Eur J Clin Invest. 2015;45(10):1032–1041. | ||
Minder EI. Afamelanotide, an agonistic analog of alpha-melanocyte-stimulating hormone, in dermal phototoxicity of erythropoietic protoporphyria. Expert Opin Investig Drugs. 2010;19(12):1591–1602. | ||
Langendonk JG, Balwani M, Anderson KE, et al. Afamelanotide for erythropoietic protoporphyria. N Engl J Med. 2015;373(1):48–59. | ||
Sawyer TK, Sanfilippo PJ, Hruby VJ, et al. 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone: a highly potent alpha-melanotropin with ultralong biological activity. Proc Natl Acad Sci U S A. 1980;77(10):5754–5758. | ||
Fabrikant J, Touloei K, Brown SM. A review and update on melanocyte stimulating hormone therapy: afamelanotide. J Drugs Dermatol. 2013;12(7):775–779. | ||
Hadley ME, Abdel Malek ZA, Marwan MM, Kreutzfeld KL, Hruby VJ. [Nle4, D-Phe7]-alpha-MSH: a superpotent melanotropin that “irreversibly” activates melanoma tyrosinase. Endocr Res. 1985;11(3–4):157–170. | ||
Hunt G, Kyne S, Wakamatsu K, Ito S, Thody AJ. Nle4DPhe7 alpha-melanocyte-stimulating hormone increases the eumelanin:phaeomelanin ratio in cultured human melanocytes. J Invest Dermatol. 1995;104(1):83–85. | ||
Abdel-Malek ZA, Ruwe A, Kavanagh-Starner R, et al. alpha-MSH tripeptide analogs activate the melanocortin 1 receptor and reduce UV-induced DNA damage in human melanocytes. Pigment Cell Melanoma Res. 2009;22(5):635–644. | ||
Dorr RT, Dvorakova K, Brooks C, et al. Increased eumelanin expression and tanning is induced by a superpotent melanotropin [Nle4-D-Phe7]-alpha-MSH in humans. Photochem Photobiol. 2000;72(4):526–532. | ||
Barnetson RS, Ooi TK, Zhuang L, et al. [Nle4-D-Phe7]-alpha-melanocyte-stimulating hormone significantly increased pigmentation and decreased UV damage in fair-skinned Caucasian volunteers. J Invest Dermatol. 2006;126(8):1869–1878. | ||
Levine N, Sheftel SN, Eytan T, et al. Induction of skin tanning by subcutaneous administration of a potent synthetic melanotropin. JAMA. 1991;266(19):2730–2736. | ||
Harms JH, Lautenschlager S, Minder CE, Minder EI. Mitigating photosensitivity of erythropoietic protoporphyria patients by an agonistic analog of alpha-melanocyte stimulating hormone. Photochem Photobiol. 2009;85(6):1434–1439. | ||
Biolcati G, Deybach JC, Hanneken S. A randomized Phase III trial of afamelanotide (Scenesse), an agonistic α-melanocyte stimulating hormone analogue in the treatment of protoporphyria-induced phototoxicity. Br J Dermatol. 2011;164(5):1. | ||
Clinuvel. Clinuvel reports positive results in Phase III porphyria (EPP) clinical trial. [media release]. 2010; Available at: http://www.clinuvel.com. Accessed June 12, 2016. | ||
Hadley ME, Dorr RT. Melanocortin peptide therapeutics: historical milestones, clinical studies and commercialization. Peptides. 2006;27(4):921–930. | ||
Luger TA, Bohm M. An alpha-MSH analog in erythropoietic protoporphyria. J Invest Dermatol. 2015;135(4):929–931. | ||
Scenesse (afamelanotide) implant: EU summary of product characteristics. 2015. Available at: http://www.ema.europa.eu. Accessed June 12, 2016. | ||
Harms J, Lautenschlager S, Minder CE, Minder EI. An alpha-melanocyte-stimulating hormone analogue in erythropoietic protoporphyria. N Engl J Med. 2009;360(3):306–307. | ||
Minder EI, Schneider-Yin X. Afamelanotide (CUV1647) in dermal phototoxicity of erythropoietic protoporphyria. Expert Rev Clin Pharmacol. 2015;8(1):43–53. | ||
Clinuvel. Clinuvel announces positive results from pivotal European Phase III study: endpoints demonstrate clinically relevant treatment effect of afamelanotide [media release]. 2011. Available at: http://www.clinuvel.com. Accessed June 12, 2016. | ||
Clinuvel. Clinical significance and benefit from Scenesse in US Phase III EPP study. [media release]. 2011. Available at: http://www.clinuvel.com. Accessed June 12, 2016. | ||
Biolcati G, Marchesini E, Sorge F, Barbieri L, Schneider-Yin X, Minder EI. Long-term observational study of afamelanotide in 115 patients with erythropoietic protoporphyria. Br J Dermatol. 2015;172(6):1601–1612. | ||
Lengweiler S, Kreim S, Barman-Aksozen J, Maurer M, Minder EI. Evaluation of the immunogenicity of the synthetic alpha-melanocyte-stimulating hormone (alpha-MSH) analogue afamelanotide ([Nle4-D-Phe7]-alpha-MSH, Scenesse(R)) in erythropoietic protoporphyria patients by ELISA detecting both anti-afamelanotide and anti-alpha-MSH antibodies. Skin Pharmacol Physiol. 2015;28(2):103–113. | ||
Ponka P. Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells. Blood. 1997;89(1):1–25. | ||
Clinuvel. Clinuvel demonstrates positive treatment effect of afamelanotide in US Phase II study [media release]. 2011. Available at: http://www.clinuvel.com. Accessed June 12, 2016. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.