")
Back to Journals » Journal of Experimental Pharmacology » Volume 13
Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy
Authors Tchounwou PB , Dasari S, Noubissi FK, Ray P, Kumar S
Received 27 December 2020
Accepted for publication 23 February 2021
Published 18 March 2021 Volume 2021:13 Pages 303—328
DOI https://doi.org/10.2147/JEP.S267383
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bal Lokeshwar
Paul B Tchounwou,1 Shaloam Dasari,1 Felicite K Noubissi,1 Paresh Ray,2 Sanjay Kumar3
1Cellomics and Toxicogenomics Research Laboratory, NIH-RCMI Center for Health Disparities Research, Jackson State University, Jackson, MS, USA; 2Department of Chemistry and Biochemistry, College of Science, Engineering and Technology, Jackson State University, Jackson, MS, USA; 3Department of Life Sciences, School of Earth, Biological, and Environmental Sciences, Central University of South Bihar, Gaya, India
Correspondence: Paul B Tchounwou
NIH-RCMI Center for Health Disparities Research, Jackson State University, 1400 JR Lynch Street, Jackson, MS, 39217, USA
Tel +1 601-979-0777
Fax +1 601-979-0570
Email [email protected]
Abstract: Cisplatin and other platinum-based chemotherapeutic drugs have been used extensively for the treatment of human cancers such as bladder, blood, breast, cervical, esophageal, head and neck, lung, ovarian, testicular cancers, and sarcoma. Cisplatin is commonly administered intravenously as a first-line chemotherapy for patients suffering from various malignancies. Upon absorption into the cancer cell, cisplatin interacts with cellular macromolecules and exerts its cytotoxic effects through a series of biochemical mechanisms by binding to Deoxyribonucleic acid (DNA) and forming intra-strand DNA adducts leading to the inhibition of DNA synthesis and cell growth. Its primary molecular mechanism of action has been associated with the induction of both intrinsic and extrinsic pathways of apoptosis resulting from the production of reactive oxygen species through lipid peroxidation, activation of various signal transduction pathways, induction of p53 signaling and cell cycle arrest, upregulation of pro-apoptotic genes/proteins, and down-regulation of proto-oncogenes and anti-apoptotic genes/proteins. Despite great clinical outcomes, many studies have reported substantial side effects associated with cisplatin monotherapy, while others have shown substantial drug resistance in some cancer patients. Hence, new formulations and several combinational therapies with other drugs have been tested for the purpose of improving the clinical utility of cisplatin. Therefore, this review provides a comprehensive understanding of its molecular mechanisms of action in cancer therapy and discusses the therapeutic approaches to overcome cisplatin resistance and side effects.
Keywords: cisplatin, molecular mechanisms of action, combination therapy, cancer treatment
Introduction
In 1844, Peyrone synthesized cisplatin for the first time. In 1893, Alfred Werner contributed towards designing the structure of cisplatin with a firm basis of coordination chemistry.1 The identification of cisplatin and its bioactivity evolved from laboratory experiments of Professor Rosenberg along with significant contributions by Loretta Van Camp, Tom Krigas, Eugene Grimley, and Andrew Thomson from the biophysics department at Michigan State University.2,3
Cisplatin is structurally a coordination compound with square planar geometry. It appears as white or deep yellow to yellow-orange crystalline powder at room temperature.4,5 Under normal temperature and pressure, cisplatin is found to be stable with a water solubility of 2.53g/L at 25°C.6
Cisplatin has been widely used against human cancers following its first usage by the National Cancer Institute for clinical trials.7 It is well established as an anti-cancer drug agent based upon its therapeutic implications. The molecular structure of cisplatin determines the anticancer activity in a dose-dependent fashion inhibiting Deoxy Ribonucleic Acid (DNA) synthesis. However, therapeutic efficacy has been challenging beyond the comprehensive drug development as neurotoxicity, ototoxicity and renal toxicity, etc., have been observed. Thus, strategies need to be developed to address cisplatin-related toxicities and drug resistance. An extensive search for less toxic analogs of cisplatin, combination therapy with other cancer drugs and nanotechnology can be used to combat the side effects. This review provides a perspective on the advances in our understanding of the molecular mechanisms of action of cisplatin for targeted cancer therapy.
Molecular Mechanisms of Action
Overview of Mechanisms
Cisplatin’s cytotoxicity has been attributed to DNA binding, tailed by single-stranded DNA breaks.8 Cisplatin is activated upon its entry into the cytoplasm by displacing chloride atoms with water molecules forming an electrophile with affinity towards sulfhydryl groups on proteins and nitrogen donor atoms on nucleic acids.9 This selective binding to 1, 2-intrastrand cross-links of purine bases with cisplatin triggers blockage in cell division and induces apoptotic cell death.10 The most common cisplatin-DNA adduct formation through covalent binding has been observed at the N7 positions of the imidazole ring of two adjacent guanines.11 Inter and intrastrand crosslinks and nonfunctional adducts have been reported to contribute to cisplatin’s toxicity.12,13 As shown in Figures 1 and 2, research conducted in our laboratory has demonstrated that cisplatin inhibits cell growth, induces DNA adducts and causes cytotoxic effects in leukemia cells in a concentration-dependent manner.14
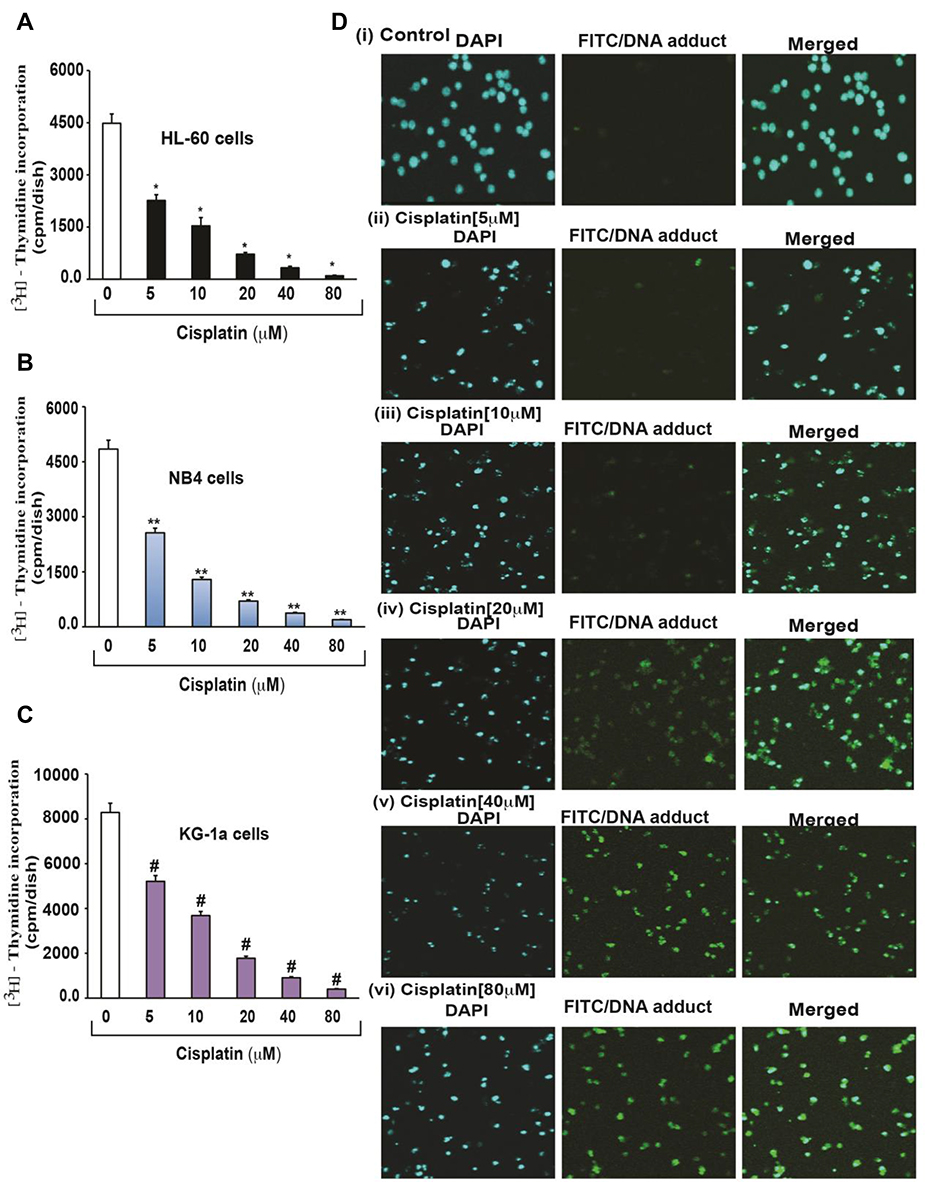 |
Figure 1 Cisplatin-induced growth inhibition and formation of DNA-adduct in APL cells.14 APL cells (HL-60, NB4 and KG-1a) were exposed to various concentrations (0, 5, 10, 20, 40, and 80 μM) of cisplatin for 24 hours and further incubated for 24 hours with tritium labeled thymidine. After incubation, cells were harvested by centrifugation and counted using liquid scintillation analyzer. 3H-methyl thymidine incorporation was expressed as cpm/dish. Data represent the means of three independent experiments ± SDs. Highly statistically significant decreases (p < 0.01) in cell proliferation were observed in all cisplatin-treated APL cells including HL-60 (A), NB4 (B) and Kg-1a (C) cells. Cisplatin–induced formation of DNA adduct was assessed by immunocytochemistry and confocal microscopy analysis. APL cells were treated with various concentrations of cisplatin for 48 hours and immunocytochemistry as well as confocal microscopy using FITC filter were performed to confirm DNA adduct formation. The results showed that cisplatin caused a significant concentration-dependent increase in DNA-adduct formation in APL cells [D (i–vi)]. Multiple symbol indicators (*, **, #) indicate highly significant reductions (p<0.01) in growth between control and cisplatin-treated cells, respectively, for HL-60, NB4, and KG1a cells. Reproduced from Kumar S, Tchounwou PB. Molecular mechanisms of cisplatin cytotoxicity in acute promyelocytic leukemia cells. Oncotarget. 2015;6(38):40734–40746. Creative Commons license and disclaimer available from: http://creativecommons.org/licenses/by/4.0/legalcode.14 |
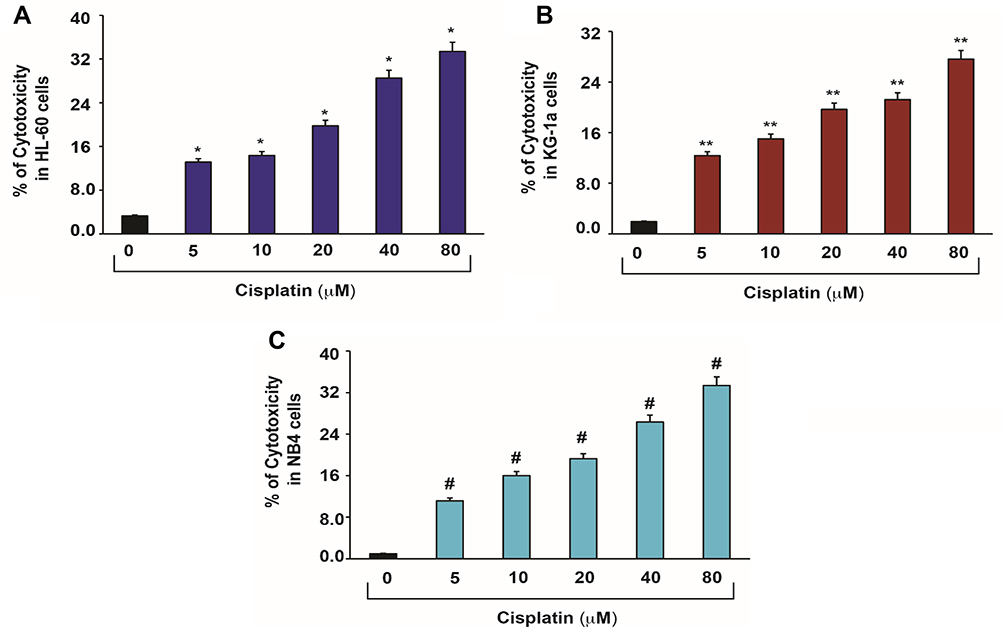 |
Figure 2 Cisplatin-induced cytotoxic effects in APL cells.14 APL cells were exposed to various concentrations (0, 5, 10, 20, 40 and 80 μM) of cisplatin for 48 hours and LDH released in medium was measured using Promega non-radioactive cytotoxicity assay technical bulletin protocol. Then, % cytotoxicity was calculated by dividing the levels of released LDH in treated cells over the total LDH released from control cells. Highly statistically significant increases (p < 0.01) in cytotoxicity were observed in all cisplatin-treated APL cells including HL-60 (A), KG-1a (B) and NB4 (C) cells in a concentration-dependent fashion. Multiple symbol indicators (*, **, #) indicate highly significant increases (p<0.01) in cytotoxicity between control and cisplatin-treated cells, respectively, for HL-60, NB4, and KG1a cells. Reproduced from Kumar S, Tchounwou PB. Molecular mechanisms of cisplatin cytotoxicity in acute promyelocytic leukemia cells. Oncotarget. 2015;6(38):40734–40746. Creative Commons license and disclaimer available from: http://creativecommons.org/licenses/by/4.0/legalcode.14 |
Cisplatin-Induced Oxidative Stress
Cancer cells exhibit high metabolism, demanding elevated reactive ROS concentrations to facilitate their propagation. ROS affect key signaling proteins that are involved in biochemical pathways for cell proliferation. Tumor development and progression are regulated by ROS production by modulating biological pathways such as activation of extracellular-regulated kinase 1/2 (ERK1/2) and ligand-independent Receptor Tyrosine Kinases (RTK), evasion of apoptosis or anoikis through activation of Src, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ƙ) and phosphatidylinositol-3 kinase (PI3K), secretion of metalloproteinase into the extracellular matrix, Met overexpression, and Rho–Rac interaction for tissue invasion, metastasis and angiogenesis by releasing vascular endothelial growth factor and angiopoietin.15
Cisplatin has shown to generate ROS directly or indirectly through mitochondria.16,17 Cisplatin cytotoxicity was related to lipid peroxidation, confirming the role of oxygen free radicals.18 This release of lipid peroxidation by-products such as malondialdehyde (MDA) increases carbonylation of proteins, inducing oxidative damage of cell membranes, and eventually triggering apoptosis.19 Similar to other published reports, we have previously demonstrated that cisplatin strongly induces oxidative stress leading to lipid peroxidation, increased MDA production, reduced GSH activity, and DNA damage in acute promyelocytic leukemia (APL) cells (Figure 3).14
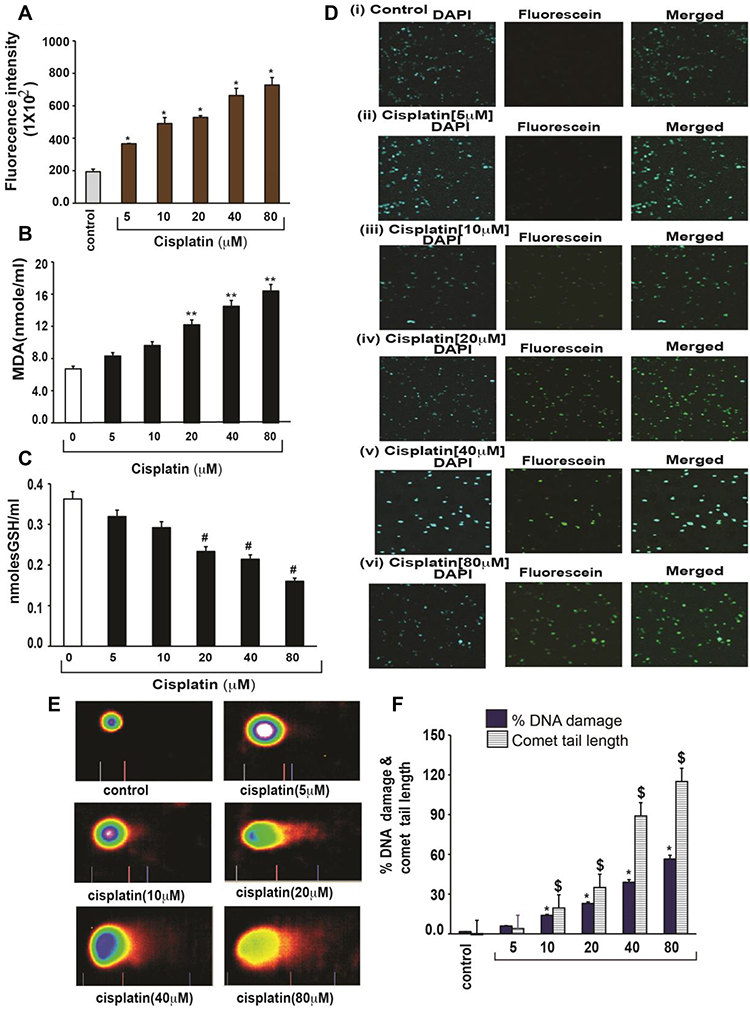 |
Figure 3 Cisplatin-induced oxidative stress and DNA damage in APL cells.14 APL cells were exposed to various concentrations (0, 5, 10, 20, 40, and 80 μM) of cisplatin for 48 hours. ROS release was assessed by spectrofluorometry based on DCF fluorescence intensity after 30 min incubation with dichlorofluorescein diacetate (DCFDA). After incubation, ROS release was measured through measuring DCF fluorescence intensity by spectrofluorometry. Malondialdehyde (MDA) and GSH concentration levels were also measured by spectrophotometry. DNA damage was analyzed by both TUNEL and alkaline gel electrophoresis (Comet) assays. (A) ROS concentrations; (B) MDA concentrations; (C) GSH concentrations; (D) TUNEL assay images of DNA damage; (E) Comet assay images of DNA damage; (F) Percentages of DNA damage, and Comet tail lengths. Multiple symbol indicators (*, **, #, $) indicate highly significant increases (p<0.01) in ROS (*), MDA (**), DNA damage (*) and Comet tail length ($), and highly significant decreases (p<0.01) in GSH ($) between control and cisplatin-treated cells. Reproduced from Kumar S, Tchounwou PB. Molecular mechanisms of cisplatin cytotoxicity in acute promyelocytic leukemia cells. Oncotarget. 2015;6(38):40734–40746. Creative Commons license and disclaimer available from: http://creativecommons.org/licenses/by/4.0/legalcode.14 |
Cisplatin Modulation of Calcium Signaling
Biological processes like gene regulation, cell proliferation, migration, death, etc., are influenced by intracellular calcium levels. However, intracellular Ca2+ homeostasis is altered in cancer cells to enhance cell proliferation, increase cell motility and invasion, fight cell death, and avoid immune-attack.20 Tumorigenic pathways are associated with altered expression level or abnormal activation of Ca2+ channels, transporters or Ca2+ ATPases.21
A recent study has shown that calcium efflux from the Endoplasmic Reticulum (ER) regulates cisplatin-induced apoptosis in human cervical cancer cells.22 Endoplasmic reticulum is a cytosolic target of cisplatin via caspase-3 activation in cytoplasts since they require calcium and the activity of the calcium-dependent protease calpain. Cisplatin treatment has been reported to increase the expression of 78-kDa glucose-regulated protein (Grp78/BiP), biomarker of ER stress.23 A dose-dependent increase of Ca2+ was observed in cisplatin-treated MCF-7 cells, suggesting the fact that the specific pathway responsible for cell death depends on the cell type and the treatment conditions.
Cisplatin-Induced Cell Apoptosis
Induction of apoptosis in cancer cells through intrinsic and extrinsic pathways, involves regulation of caspase-dependent proteolysis of several cellular proteins, membrane blebbing and endonucleolytic cleavage of chromosomal DNA.24 In response to various apoptotic stimuli, initiator caspases such as caspase-2, −8, −9, or −10 are activated, in turn activating zymogenic forms of executioner caspases (caspase-3 or −7). These cascades of events result in proteolytic cleavage of explicit cellular substrates and, therefore, lead to cell death.25 Apoptosis can be executed through either extrinsic or intrinsic pathway. In the extrinsic pathway, initiator caspases are activated via death-inducing signal complex (DISC) concerning the engagement of extracellular ligands like Fas or Tumor necrosis factor (TNF). In the intrinsic pathway of apoptosis, mitochondrial outer membrane permeabilization leads to activation of certain pro-apoptotic proteins like cytochrome c and mitochondria-derived activator of caspases. In the cytoplasm, binding of cytochrome c to the adaptor protein Apoptotic Protease Activating Factor-1 (APAF-1) forms apoptosome.25 Since the balance between pro- and anti-apoptotic B-Cell Lymphoma 2 (BCL-2) family proteins is a fundamental determinant for the initiation of mitochondrial outer membrane permeabilization, cancer cells may escape apoptosis by modulating the apoptotic pathways transcriptionally, translationally, and post-translationally.25
The apoptotic mechanism via cisplatin therapy varies by cell type. The molecular mechanism of cisplatin-induced apoptosis involves activation of tumor protein 53 (p53), phosphorylation of activator protein (AP-1) component leading to cell cycle arrest through stimulation of p21 and downregulation of cyclins and cyclin-dependent kinases.14 Studies have been done to show that cisplatin induces cell death by mechanisms mediated by p53, p38 mitogen-activated protein kinase (p38 MAPKs) and/or the c-jun N-terminal kinases (JNK) activation suggesting activation of the intrinsic pathway of apoptosis through alteration of the mitochondrial membrane potential, release of cytochrome C, and up-regulation of caspase 3 activity.26–28 However, research studies suggest that cisplatin-induced cell death not only relies on the event of apoptosis but also on necrosis29 or autophagy.30
Cisplatin and Protein Kinase C
Protein kinase C (PKC) is a family of lipid-dependent serine/threonine kinases that have been subdivided into 3 groups such as conventional (α, β1, βII andγ), novel (δ, ε, η and θ), and atypical (ζ, 1/λ).31,32 PKC shows a fundamental role in signal transduction involved in the regulation of cell proliferation, differentiation and apoptosis. Mitochondrial dysfunction via depolarization of the mitochondrial inner membrane (decreases the active Na+ transport) leads to cisplatin-induced apoptosis and these events are mediated by activation of caspase-3 by acting downstream of cytochrome c release from mitochondria.33–35
Cisplatin and Mitogen-Activated Protein Kinase (MAPK)
Mitogen-activated protein kinases (MAPK) are a family of structurally related serine/threonine kinases regulating cell proliferation, differentiation, and cell survival.36–38 The MAPK subfamilies consist of the extracellular signal-regulated kinases (ERK), the c-jun N-terminal kinases (JNK)/stress-activated protein kinases (SAPK), and the p38 MAP kinases.39 Studies have been conducted showing that cisplatin-induced DNA damage results in the activation of ERK1/2 via PKCδ.40 ERK is activated via a signaling pathway involving Ras, Raf, MEK, and ERK, transducing signals from the plasma membrane to the cell nucleus.36
Cisplatin and Jun Amino-Terminal Kinase (JNK)
The c-Jun NH2-terminal kinases (JNKs) are an evolutionarily conserved sub-group of mitogen-activated protein (MAP) kinases. Phosphorylation of c-Jun activates activator protein-1 (AP-1) transcription factor which is a major target for JNK signaling pathway.41,42 c-Jun N-terminal kinase regulates numerous essential cellular functions like cell proliferation, differentiation, survival and apoptosis while activating and inhibiting substrates for phosphorylation transcription factors consequently inducing expression of pro-apoptosis and pro-survival factors. Studies establish that the JNK/c-Jun pathway is activated by cisplatin-induced DNA damage in a dose-dependent manner.43–45 Therefore, it is proposed that JNK signal pathway is very crucial in cisplatin treatment, regulating pro-apoptosis factor and improving resistance to cisplatin-based chemotherapy.46
Cisplatin and p38 Mitogen-Activated Protein Kinase (MAPK)
p38 MAPK is a member of mitogen-activated protein kinases regulating migration and proliferation of tumor cells, epithelial-mesenchymal transition, cell intravasation, and colonization of distant sites signaling.47 Previous studies reported that inhibition of p38 induced apoptosis mediated by ROS and JNK in cells resistant to cisplatin, irinotecan and 5-fluorouracil.48 Hence, p38 MAPK inhibition results in ROS upregulation, which in turn activates the JNK pathway via inactivation of phosphatases, sensitizing tumor cells to cisplatin-induced apoptosis.49 High p38 MAPK phosphorylation has been correlated with poor life expectancy in patients with HER‐2 negative breast cancer.50
Cisplatin and AKT
AKT is one of the major downstream effectors of Phosphoinositide 3-kinases (PI3K).51 Upon PI3K activation, AKT is translocated to the inner membrane, where it is phosphorylated by PDK1.52 This AKT modification activates the mammalian target of rapamycin complex 1 (mTORC1). Studies reveal that complete AKT activation leads to additional substrate-specific phosphorylation events in both cytoplasm and nucleus, like inhibitory phosphorylation of the pro-apoptotic forkhead box transcription factors (FOXO) proteins.53
Akt/mTOR survival pathway plays an important role in cisplatin treatment in human cancer cells. Cisplatin activates Akt in lung cancer cells mediated by Epidermal Growth Factor (EGFR), Src and PI3-kinase and reduces apoptosis by survivin upregulation, the inhibitor of apoptosis protein that is overexpressed in many tumors but is absent in most normal adult tissues.54 However, cisplatin treatment reduces survivin expression, thereby inhibiting Akt in few cancer cells.55
Cisplatin and Signaling for DNA Damage
DNA is considered as the primary target of cisplatin.56,57 Studies show that covalent bonding of cisplatin with the N7 position of purine bases to form 1, 2- or 1, 3-intrastrand and interstrand crosslinks interferes with DNA replication and transcription.58 Since nucleotide excision repair (NER) system plays major role in DNA repair, low levels of XP complementation group A (XPA) and excision repair cross-complementation group I (ERCCI) have been noticed.59,60 On the other hand, mismatch repair (MMR) system proteins (MSH2 and MLH1) have been reported to uncouple from their function to restore lost genetic information.61–63
p53 and DNA Damage Response to Cisplatin Treatment
p53 is a sequence-specific transcriptional activator, playing a vital role in apoptosis, inhibition of cell cycle progression, senescence, differentiation and acceleration of DNA repair.64 If a cell undergoes stress to become cancerous, p53 is triggered to either repair the DNA damage (eg, Growth Arrest and DNA Damage-inducible 45 (GADD45)) or induce programmed cell death through (Bcl-2-associated X protein) Bax activation.65 However, most of the cancer signaling pathways inactivate p53 for its protective tumor-suppressor ability, and hence its activation paves a way for novel chemotherapy.
Recognition of DNA adducts induced by cisplatin is the first step towards triggering apoptosis. Molecular mechanisms of cisplatin-induced cell death via p53 include activation of FADD-like interleukin-1β converting enzyme (FLICE)-like inhibitory protein ubiquitination.66,67 The intra-strand lesions generated by cisplatin-induced DNA crosslinks activate the MMR system and in turn trigger tyrosine kinase c-Abl, leading to apoptosis.68 So, studies show that cisplatin-induced ERK activation is an upstream regulator of the p53 response to DNA damage caused by cisplatin.69 Though p53 plays a vital role in cisplatin-induced DNA damage response, p53-negative cells also respond, proposing alternate pathways.58 Increased accumulation of p53 has been found in cisplatin-resistant cell lines with dysfunctional p53.70
Cyclobutanedicarboxylate (c-Abl) and DNA Damage Response to Cisplatin Treatment
c-Abl is a non-receptor tyrosine kinase localized in the nucleus and cytoplasm involved in the regulation of cell growth, survival and morphogenesis. C-Abl is activated in response to cisplatin activating JNK/stress-activated protein kinase (SAPK).71 Activation of c-Abl in response to cisplatin led to phosphorylation and stabilization of p73, a member of the p53 family, to trigger apoptosis.72
In addition, studies demonstrate that c-Abl can activate the p38 MAPK signaling pathway not only via tyrosine kinase mechanism but also through MKK6 in cisplatin-based therapy. This serves as an interesting evidence of a novel signaling mechanism that fails to inhibit the p38 MAPK pathway alone or in combination with cisplatin.73
Cisplatin Modulation of Gene Expression
It has been reported that several genes are modulated and work collectively to induce apoptosis/autophagy of cancer cells upon cisplatin treatment. Genes related to drug transportation (CTR1, ATP7A, ATP7B, GSH, Metallothioneins), DNA repair system (ERCC1, MLHI, MSH2, POLH, REV3, BRCA1, BRCA2, VDAC) and pro-apoptotic and apoptotic proteins (Bax, BCl-2, surviving, caspases, MAP Kinases, TP53) have shown altered expression.74 Genome-wide studies through Affymetrix Gene Chip Human Exon 1.0 Sequence Tag Array have identified six, two, and nine representative SNPs that contribute to cisplatin-induced cytotoxicity through their effects on 8, 2, and 16 gene expressions. These genetic variants contribute to 27%, 29%, and 45% of the overall variation, respectively, in cell sensitivity to cisplatin.75 cDNA microarray analysis of gene expression profiles showed overexpression of GST-pi mRNA, DNA repair associated genes ((X-ray repair complementing defective repair in Chinese hamster cells) XRCC5, XRCC6, ERCC5, MutL homolog 1 (hMLH1)), apoptosis inhibitors (Insulin-Like Growth Factor (IGFR) type I and II), while apoptosis inducer (caspase 3 and Bcl-2 homologous antagonist killer (BAK)) were underexpressed.76
Computational Studies with Cisplatin
The anticancer activity of cisplatin has led to several experimental and theoretical studies to further understand its structural and spectroscopic properties. Various computational tools are used to study molecular mechanisms in cancer biology. Genome-scale metabolic model (GSMM) has been used to understand the accurate prediction of numerous metabolic phenotypes, including growth rates, nutrient uptake rates, gene essentiality, etc.,77 Another model called embedded multiscale method has been used to analyze hyperthermic treatment of cancer based on systemically injected vascular magnetic nanoconstructs carrying super-paramagnetic iron oxide nanoparticles.78
Solvation is the critical step in activating cisplatin to its active forms, [Pt (NH3)2(OH2) Cl] − and [Pt (NH3)2(OH2)2]2+, wherein chloride ions are displaced by water molecules.79 The thermodynamic and kinetic aspects of hydration reactions of cisplatin were studied using polarizable continuum model revealing the fact that all the hydration reactions are slightly endothermic while the Gibbs energies of cisplatin hydration amount to 7.0 for the chloride and 14.2 kcal/mol for ammonium replacement.80 The equilibrium constants of individual pKa have been shown by many laboratories to be pKa1 = 5.5, pKa21 = 7.3 and pKa22 = 6.6 (Figure 4).
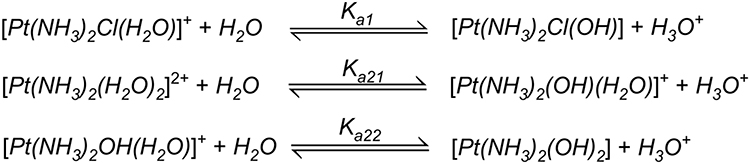 |
Figure 4 Chemical reactions showing hydrolysis of cisplatin. Reproduced with permission from Burda JV, Zeizinger M, Leszczynski J. Hydration process as an activation of trans- and cisplatin complexes in anticancer treatment. DFT and Ab initio computational study of thermodynamic and kinetic parameters. J Comput Chem. 2005;26(9):907–914. Copyright © 2005 Wiley Periodicals, Inc.81 |
Cisplatin undergoes substitution reactions by an associative mechanism in the case of Pt (II), a common mechanism for the transition metals.81 Rate constants for dechlorination for the first and second steps and both cis- and trans-conformers at T = 318.2 K, pH 2.8–3.4 and 0.1 M NaClO4 solution were also determined. The concentration of Cl− anions, when passing from blood is about 100 mM, and to cellular environment is about 10–80 mM.80 Cisplatin is studied to be very hydrophilic, with an octanol–water partition coefficient (log Poct) of less than −2.0.82 This leads to poor intestinal absorption of cisplatin, and therefore, intravenous administration is suggested.
The high ligand binding affinity of activated cisplatin with biomolecules in the cellular environment, like guanines in DNA, forms an intrastrand 1,2- d(GpG) complex, peptides (glutathione), or peptides with sulphur-containing amino acids, cysteine, and methionine, or RNAs, introduce the prospect of their use in chemopreventive application. The preference of cisplatin’s affinity towards guanine at the N7-position is established experimentally explaining formation of weak hydrogen bond between the chloride ligand of cisplatin and the H2N−C6 group of adenine and stronger molecular orbital interaction for guanine compared to adenine.83 Free energy reactions of cisplatin derivatives with cysteine showed strong thermodynamic stability and exhibited clear constant preferences in the whole pH range.84 Further studies were conducted to describe the physicochemical properties of cisplatin through a model capable of quantitatively describing and interpreting the structural and vibrational properties.85
Taken together, cisplatin-induced cancer cell death is mediated through a series of biochemical effects and molecular mechanisms that include the formation of DNA adducts, induction of reactive oxygen species, genotoxic damage, p53 activation, MAPK and JNK signaling, overexpression of p38, p21 and cell cycle arrest, activation of apoptotic genes/proteins, and down-regulation of proto-oncogenes and related proteins. Figure 5 presents the specific molecular mechanisms of action of cisplatin resulting from experimental studies conducted in molecular pharmacology laboratories including ours.14
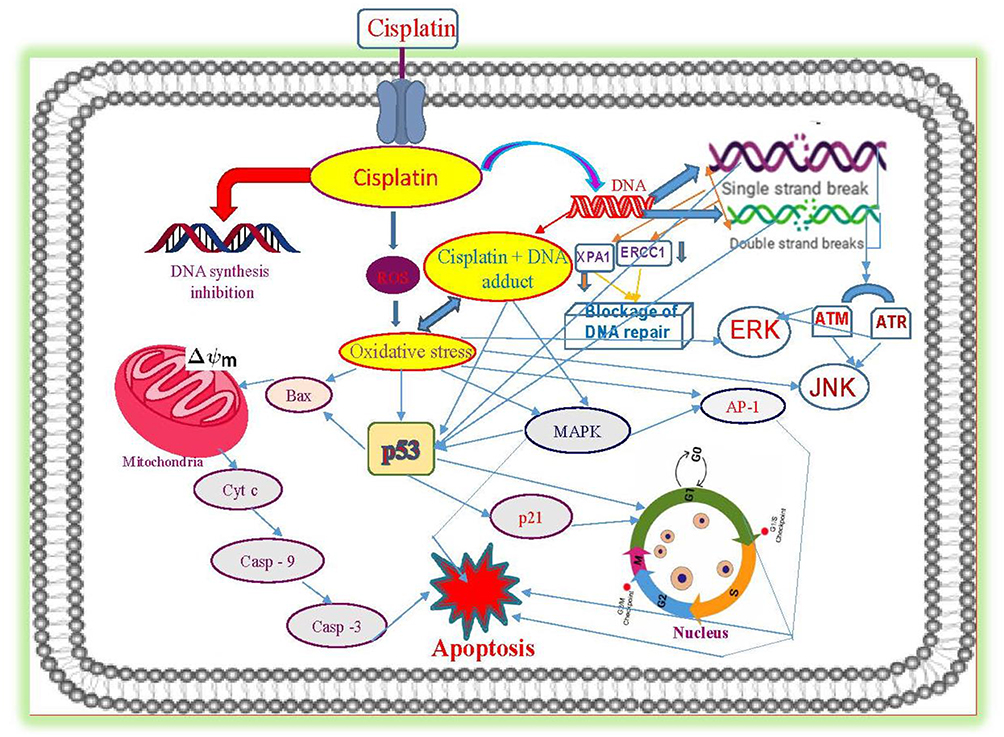 |
Figure 5 Molecular mechanisms of cisplatin-induced cytotoxicity in APL cells. Reproduced from Kumar S, Tchounwou PB. Molecular mechanisms of cisplatin cytotoxicity in acute promyelocytic leukemia cells. Oncotarget. 2015;6(38):40734–40746. Creative Commons license and disclaimer available from: http://creativecommons.org/licenses/by/4.0/legalcode.14 |
Toxicological Effects of Cisplatin
Overview
Modulation of intracellular changes controlling the regulation of few signaling pathways and depletion of bioavailable Cu pool leads to sensitization of cisplatin transport and triggers apoptosis.86 Toxic effects of cisplatin are varied and have been an area of concern for many oncologists, practitioners and researchers. Cisplatin is a potent chemotherapeutic drug associated with several side effects such as myelotoxicity, gastrointestinal toxicity, neurotoxicity, ototoxicity, cardiotoxicity and nephrotoxicity.87 Common side effects of cisplatin include nausea and vomiting.88 Therapeutic strategies such as cell cycle inhibitors,89 heavy metal chelating agents,90 bioflavonoids,91 algae,92 and natural products93,94 have been documented to manage the toxicological implications without compromising oncolytic action.
Hepatotoxicity
Clinical studies have shown cisplatin-induced liver damage either alone or in combination with other drugs.95 Oxidative stress played an important role in cisplatin-induced hepatotoxicity.96 Cisplatin-induced hepatotoxicity resulted in elevated expression of cytochrome P450 leading to reduction of cell viability, through reduced GSH, and higher ROS production.97 Clinical manifestations of liver damage from a histopathology standpoint include high levels of serum alanine transaminase (ALT) and Aminotransferase (AST), liver caspase-3 activity, and increased positive staining in Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.98
Cardiotoxicity
Several experimental and clinical studies support the fact that increased oxidative stress and apoptosis have been involved in the cisplatin-mediated cardiotoxicity.99 Acute cardiovascular toxicity by cisplatin has been observed by direct damage of the vascular endothelium because of an increase of von Willebrand factor released by endothelial cells during chemotherapy.100 Cisplatin-induced vascular toxicity is becoming an increasing concern affecting up to 12% of the patients.101 Clinical manifestations of cardiotoxicity due to cisplatin chemotherapy include heart failure, angina, acute myocardial infarction, thromboembolic events, autonomic cardiovascular dysfunction, both hypertension and hypotension, myocarditis, pericarditis, and severe congestive cardiomyopathy, supraventricular tachycardia, ventricular arrhythmias, atrial fibrillation, occasional sinus bradycardia and occasional complete atrioventricular block reported in the literature.102
Nephrotoxicity
The concentration of cisplatin in proximal tubular epithelial cells of kidney is roughly 5 times the serum concentration leading to cisplatin-induced nephrotoxicity.91 Clinical and experimental literature on cisplatin nephrotoxicity states that unbound cisplatin is freely filtered at the glomerulus and transported into renal tubular cells of kidney.103 Clinical manifestations include tubular damage and dysfunction leading to decrease in glomerular filtration, segmental degeneration, necrosis, and desquamation of the epithelial cells in the pars convoluta and pars recta of the proximal tubules and the distal tubules.104 Long-term cisplatin treatment and injury may cause cyst formation and interstitial fibrosis.105 Mechanisms of cisplatin nephrotoxicity include high expression of calcitonin receptor (Ctr1) and its localization to the basolateral membrane of the proximal tubule, suggesting that Ctr1 is an important cisplatin uptake mechanism in these cells.106 Another investigation showed knockout of the Octamer-binding protein 2 (OCT2) gene significantly reduced urinary cisplatin excretion and nephrotoxicity.107 Low concentrations of cisplatin resulted in apoptotic cell death while at higher concentrations necrosis has been reported.17,108
Other Organ Toxicity
Neurotoxicity is the third and most serious cisplatin toxicity.109 Cisplatin treatment caused high levels of Pt–DNA binding and apoptosis of dorsal root ganglion neurons. Studies report changes in the circulation of nerve growth factor could be responsible to the dorsal root ganglia sensory neuron damage induced by cisplatin and return to normal levels after recovery from the neurotoxic damage.110
Cisplatin-induced ototoxicity affects up to 93% of the patients receiving cisplatin chemotherapy. Clinical symptoms include progressive and irreversible sensorineural hearing loss.111 It is hypothesized that transporters interfere in cisplatin influx, leading to cisplatin-induced ototoxicity. Several transporters like copper transporters, organic cation transporters, the transient receptor potential channel family, calcium channels, multidrug resistance-associated proteins, mechanotransduction channels and chloride channels have been studied to understand the pathophysiology of cisplatin-induced ototoxicity.112
Testicular toxicity has been linked to cisplatin treatment with damaging effects on spermatogenesis and testicular endocrine function.113 Histopathological studies have shown spermatogenic damage, germ cell apoptosis, Leydig cell dysfunction and testicular steroidogenic disorder.114,115
Retinal toxicity has also been observed after high-dose cisplatin treatment.116 Clinical manifestations such as secondary glaucoma, internal ophthalmoplegia, retinopathy, cavernous sinus syndrome have been observed after administration of cisplatin.117
Cisplatin Resistance
Overview of Mechanisms of Resistance
Molecular mechanisms of cisplatin resistance leading to therapeutic failure include abnormal gene expression, reduced intracellular drug accumulation, enhanced DNA damage repair, efficient drug efflux, altered cell regulatory pathways, abnormal formation of blood and lymphatic vasculature promoting tumor growth, progression, and metastasis, etc.74 Here below, we discuss several events occurring either before or after cisplatin interacts with the target leading to drug resistance.
Drug Uptake and Efflux for Cisplatin
Reduced uptake and increased efflux are characteristic features of cells exhibiting cisplatin resistance. While CTR1 plays a major role in reduced uptake, ATP7A/ATP7B and MRP2 regulate increased efflux of the drug and metallothioneins may bind and inactivate cisplatin.118
Reduced Uptake
The uptake of cisplatin by a cell is a major contributing factor for its resistance. It is estimated that about 50% of cisplatin uptake is regulated by CTRs while other half is transported via passive diffusion or by other transporters.119 Several studies have been conducted to confirm the roles of copper transporters (CTR) in cancer resistance to cisplatin.120 In vitro studies in leukemia proved that resistant cells had 36–60% reduction in uptake compared to sensitive cells.121 Copper, the main CTR1 substrate, homeostasis regulates the up-take of cisplatin.122,123 CTR1 is happened to be a major protein leading cisplatin uptake.9 Cisplatin uptake is mediated by the copper transporter 1, distributed to various intracellular compartments by the copper chaperones, and exported from tumor cells with the help of ATP7A and ATP7B.124 Cisplatin binds to the metal-binding domain of ATPase in a similar way to that in the Atox1, such copper-induced interaction between Atox1 and ATPase is the basis of the copper transportation in cells.125 Hence, the link between Ctr1 and cisplatin transport may clarify cisplatin resistance to some extent in humans and suggests ways of regulating sensitivity and toxicity to cisplatin.
Increased Efflux
Previous studies conducted on gene expression of Multi-Drug Resistance (MDR) proteins found that MRP2 (MDR protein 2) as the major ATPase responsible for an increased efflux of cisplatin in resistant cells.126 P-glycoprotein, MRP1 (Multi-Drug Protein 1) and MRP2 (Multi-Drug Protein 2) belong to ATP-binding cassette (ABC) transporter proteins leading to multi-drug resistance via reduced cellular drug accumulation.127
P-Glycoprotein (P-gp) is usually overexpressed in most of the cancer types contributing towards chemoresistance.128 Structurally, P-gp consists of 12 transmembrane regions and two ATP-binding sites resulting in efflux of every effective drug. P-gp is an ABCB1 member of the ABC (ATP binding Cassette) drug transporter family.129 Knockdown of P-gp or using P-gp inhibitors (Dofequidar fumarate) have sensitized the cells towards cisplatin resistance further confirming their role in effective chemotherapy.130 Even though cisplatin is not a substrate for P-gp, studies show that P-gp depresses cisplatin-induced caspase-3 activation, reduces cisplatin-induced Bcl-2 suppression and substitutes cisplatin-induced apoptosis progression, thereby, modulating changes in cell regulatory pathways leading to loss of cisplatin sensitivity.131 A study conducted on cisplatin-resistant cells showed that upregulation of miR130a might be associated with MDR1/P-glycoprotein-mediated drug resistance. Their results from real-time qRT-PCR for miR-130a expression level and Western blot analysis for comparison of P-gp expression in both cisplatin-resistant and parent cells validated the contribution of P-gp in cisplatin responsiveness. Therefore, miR130a might modulate the expression levels of MDR1 mRNA and P-gp.132 Using natural products like Asiatic acid, Hesperetin, etc., as inhibitors for P-gp has proved to be effective by modulating NF-kB and MAPK–ERK pathways leading to apoptosis, thereby sensitizing cells for cisplatin therapy.133,134
Increased Inactivation
Upon cisplatin administration, the platinum atom binds to endogenous thiols glutathione (GSH) and metallothioneins (MTs) forming complex that is effluxed from the cell in an ATP-dependent fashion by the glutathione transporter family.135 Moreover, prolonged exposure to cisplatin leads to overexpression of MT, GSH, and other cellular thiols, leading to cell’s resistance to cisplatin.136 Additionally, their reactivity seems to be faster than that of glutathione.137
Several studies have been conducted to establish the relationship between metallothionein and cisplatin resistance in variety of cancers. Poor outcome of cisplatin chemotherapy has been linked to regulation of metallothionein gene expression.138–141 Gene regulation studies via RNA interference targeting expression of MT inhibited cisplatin resistance.142 Translocation of metallothionein from cytoplasm to nucleus seems to be the possible event leading to cisplatin resistance by protecting DNA from cytotoxic effect.143 Hence, MTs have been shown to play an important role in carcinogenesis since they regulate the supply of zinc for proteins and the activity of zinc-dependent transcription factors to modulate tumor cell growth and proliferation. Therefore, chemotherapy targeting metallothionein can be a novel proposal to increase the efficacy of cisplatin treatment.
Studies have been conducted to identify the role of glutathione transferase (GST) P1-1 that is often overexpressed in drug-resistant tumors, as a cis-DDP-binding protein.144 The control mechanism involves GST P1-1 binding to the c-Jun N-terminal kinase (JNK) to inhibit JNK phosphorylation, triggering downstream apoptosis signaling. In addition, other studies have reported that GST P1-1 knockout mice are sensitive to cisplatin treatment than their wild-type counterparts.145
Increase in Repair of DNA Damage
Formation of DNA adducts leading to cell death signaling pathway is often impaired in cisplatin-resistant cells. Not only DNA damage repair system is improved but also translesion synthesis mediates cisplatin responsiveness.
Nucleotide Excision Repair (NER) System
Usually, NER protects DNA integrity by chopping damaged nucleotides followed by DNA synthesis. ERCC1-XPF endonuclease is essential for NER of DNA lesions performing interstrand crosslink repair.146 However, early reports suggest that sensitivity to cisplatin treatment has been identified with low expression of NER proteins, like Xeroderma Pigmentosum group A (XPA) and Excision repair cross-complementing-1 (ERCC1), lacking NER repair mechanism in several cancers.60 Overexpression of human ERCC-1 may inhibit a pathway specific to the repair leading to cisplatin sensitivity.147 Hence, ERCC1 expression is considered as a biomarker for the prediction of cisplatin resistance.
Mismatch Repair (MMR) System
Genomic stability is maintained by MMR system involves the correction of mismatched nucleotides in the complementary DNA strands via insertions or deletions, arising from DNA replication errors. hMLH1 is the human homologue of the yeast mismatch repair gene MLH1.148 Loss of expression of the hMLH1 and hPMS2 of MMR complex after administration of cisplatin has been observed in cisplatin resistance types.149 Such loss of hMLH1 protein has been linked to methylation of the hMLH1 promoter.148
Translesion Synthesis
Translesion synthesis (TLS) is a DNA damage tolerance process by which DNA replicates past DNA lesions. Cisplatin adduct resulting in a delayed replication complex will trigger DNA polymerases including POLH, POLI, POLK, REV1, REV3 and REV7.150 This will result in bypass of the lesion by translesion synthesis so that replication can resume. In addition, defects in MLH1 and MSH6 (the MMR proteins) are associated with increased level of translesion synthesis, the phenomenon whereby DNA synthesis is not blocked but proceeds beyond cisplatin adducts.151 Increased translesion synthesis occurs in cisplatin-resistant cells due to overexpression of Human Homolog of the Saccharomyces cerevisiae 3 (REV3).152 REV3 is the catalytic subunit of TLS polymerase ζ, plays an important role in DNA damage response induced by cisplatin. Combined knockdown of REV3 and Ataxia Telangiectasia and Rad3-related protein (ATR) enhanced cytotoxicity in cisplatin-sensitive and ‐resistant cell line.153
Homologous Recombinational Repair
Homologous recombination system repairs breaks in the double-stranded DNA. Cisplatin adducts are usually recognized in the S Phase of cell cycle and treated by HRR. However, HR-deficient cells (mutated BRCA1/2) are more sensitive to cisplatin treatment establishing the role of BRCA1 and BRCA2 to cisplatin resistance.154 Moreover, it has been reported that cisplatin resistance develops in sensitive tumors because of secondary mutations that compensate for BRCA1/2 deficiency.155
Cytoplasmic Proteins
Studies show that cytoplasmic proteins like voltage-dependent anion channel (VDAC) are found to be responsible for the cisplatin-resistant phenotype.156 VDAC is a mitochondrial protein with vital and lethal functions. Cisplatin binds to mitochondrial DNA as well leading to cell death possible direct effect of cisplatin on mitochondria independent of nuclear DNA interaction.157
Control Mechanisms for Cisplatin Resistance
Although clinical data show high initial response to platinum-based chemotherapy, over time neoplasms become refractory, increasing mortality due to chemoresistance. Cisplatin resistance has been studied extensively for better understanding in drug designing and treatment. Previous studies showed the evidence of different mechanisms responsible for resistance to cisplatin treatment.
Nrf2 Signaling
Cisplatin decreases mitochondrial membrane potential reducing ATP content with an increase in ROS.158 Another study introduced HIF-independent, CD133-mediated mechanism of cisplatin resistance.159 CD133 is a biomarker for glioblastoma (GBM). The molecular mechanism of CD133-dependent resistance could be upregulation of CD133 under hypoxic conditions that may lead to activation of the anti-apoptotic Akt pathway, resulting in drug resistance.160 Further molecular studies have revealed the role of nuclear factor erythroid 2‐related factor 2 (Nrf2) as the sensor of oxidative stress, and has been shown to be involved in cisplatin resistance and chemoprotection.161–164 Under normal physiological conditions, Kelch-like ECH-associated protein 1 (Keap1)-Nrf2 blocks Nrf2 in the cytoplasm, however, oxidative stress activates Nrf2 through the PI3K/Akt signaling pathway and translocate into the nucleus activating the small musculoaponeurotic fibrosarcoma oncogene homologue (sMAF) heterodimerization.165,166 This leads to the activation of the genes containing the antioxidant response element (ARE), activating the transcription of antioxidant enzymes.167 It has been shown that natural products such as melatonin and berberine have cytoprotective effect from oxidant injuries through activation of the Nrf2 pathway.161–163 Studies show a regression in cisplatin-mediated cytotoxicity in Nrf2-deficient cells via reduction in total GSH levels leading to cisplatin resistance.168 A recent study has been conducted on micro RNAs (lncRNAs, circRNAs, NF-κB, SOX2 and TRIM65) and their regulation in cisplatin chemotherapy of lung cancer via targeting various pathways such as Wnt/β-catenin, Rab6, CASP2, PTEN, and Apaf-1 and STAT3 to suppress cisplatin resistance.169 Further investigations indicated that microRNA-mediated regulation of Nrf2 signaling pathway restoring cisplatin sensitivity in human cancer cells is considered as a novel therapy.170
Epithelial–Mesenchymal Transition (EMT)
EMT is a physiological process necessary for embryonic development. However, cancer cells apply this mechanism significantly contributing to chemoresistance. The mechanism involves converting the immobile epithelial cells into mobile mesenchymal cells and therefore regulating cell–cell adhesion and cellular extracellular matrix.171 This cascade of events leads to tumor cell invasion. Cisplatin stimulates macrophages to secrete chemokine ligand 20 (CCL20) for recruiting T helper cells to maintain the immunosuppressive tumor microenvironment facilitating cancer cell progression and chemokine receptor 6 (CCR6) enhancing tumor cell migration and induces the EMT mechanism, causing EMT-mediated drug resistance.172–176
Autophagy
Autophagy is a “self-eating” mechanism that is important for balancing out energy demands during growth and also stress. Therefore, autophagy serves as a survival mechanism, where toxic and aged organelles and macromolecules are degraded. Studies have shown that prolonged exposure to cisplatin impedes autophagy leading to cisplatin resistance.177 A recent study has demonstrated that under hypoxia, cisplatin-induced apoptosis is significantly reduced suggesting autophagy-mediated cisplatin resistance.178 Recent studies have also pointed out that hypoxia-inducible factor (HIF)-1α and histone deacetylases 4 (HDAC4) facilitate the interaction between p53 and rat sarcoma (RAS) signaling in cisplatin-resistant cell lines through dysregulation of apoptosis and autophagy.179 GAS5 downregulation is linked to cisplatin resistance by inhibiting autophagy resulting in cisplatin sensitivity in Non-small cell lung carcinoma (NSCLC) cells.180 Reports show that cisplatin-treated ovarian cancer cells showed decreased putative serine/threonine protein phosphatase (PRPA1)-medicated autophagy.181 In addition, autophagy is studied as a cytoprotective process during cisplatin-induced injury.182,183
Combination Therapy of Cisplatin with Other Cancer Drugs
Overview
Drug resistance has been observed in many patients who have relapsed from cisplatin treatment due to its cellular uptake and efflux of cisplatin, increased biotransformation and detoxification in the liver, and increase in DNA repair and anti-apoptotic mechanisms. To overcome drug resistance, cisplatin has been commonly used in combination with some other drugs in treating a wide variety of cancers.
Cisplatin and Paclitaxel
Paclitaxel (Taxol®) chemically synthesized from bark of western yew acts as an effective treatment for various cancers especially ovarian and breast cancer.184 However, because of its low therapeutic index, an improved delivery system is deemed necessary. Therefore, a combination therapy of intravenous paclitaxel plus intraperitoneal cisplatin has been employed to improve the survival of ovarian cancer patients.185 Numerous clinical studies have been conducted to test the efficacy of this combination in various cancers.186,187 The literature search disclosed that there exists an inverse relationship between platinum and taxane with about 26 genes responsible for this effect.188
Cisplatin and Tegafur-Uracil (UFT)
Tegafur-uracil (UFT) is an oral fluorouracil (FU) composed of tegafur and uracil. Antitumor effects of UFT are low in most malignant tumors.189 But UFT is widely used in Japan as a postoperative adjuvant chemotherapy because of its mild toxicity characteristics.190 Few studies have reported that the combination of cisplatin with UFT has a significant effect in patients.191–194 While other research groups have pointed out that fluorouracil alone is a better regimen than in combination with cisplatin.195
Cisplatin and Doxorubicin
In 1960, doxorubicin (DOX) was isolated from Streptomyces peucetius. DOX is widely used antineoplastic drugs alone or in combination due to its cytotoxic activity.196 The combination effect studied to be an effective therapy with modest antitumor activity with tolerable adverse effects in patients.197,198 Chemotherapy with cisplatin-doxorubicin significantly enhanced overall survival for endometrial cancer patients.199 However, compared with cisplatin plus doxorubicin, cisplatin monotherapy yielded similar survival among children with standard-risk of hepatoblastoma.200
Cisplatin and Gemcitabine
Gemcitabine [2′- deoxy- 2′, 2′- difluorocytidine monohydrochloride (β isomer); dFdC] is a novel nucleoside analogue with antiviral effects and efficient for anticancer therapy with mild toxicity profile.201 Its mode of action associated with the incorporation of gemcitabine diphosphate (dFdCTP) into DNA is most likely considered as being the major mechanism by which gemcitabine causes cell death.202 Clinical studies have shown that cisplatin plus gemcitabine has a significant survival advantage without the addition of substantial toxicity.203,204
Cisplatin and Vitamin D
Not only as a steroid hormone playing critical role in mineral homeostasis and bone metabolism, but Vitamin D [1α, 25-dihydroxycholecalciferol] also had broad-spectrum anti-tumor activities.205 Calcitriol acts through both genomic and non-genomic mechanisms.206 Vitamin D compounds are found to be effective against colorectal, breast, prostate, ovarian, bladder, lung, skin cancers and leukemia.207 The molecular mechanisms behind their cytotoxicity involve inhibition of cancer cell proliferation and invasiveness, induction of differentiation and apoptosis, and promotion of angiogenesis.205 Combination therapy of Vitamin D with cisplatin or carboplatin inhibited cell proliferation in breast cancer cell lines.208 Studies on SCC showed synergy between cisplatin and Vitamin D via regulating Lipocalin-2 (LCN2)-modulated NF-κB pathway.209 Furthermore, Glutathione Peroxidase 1 (GPX1) expression has been studied to be crucial in cisplatin-resistance but can be reduced by vitamin D.210 The combination therapy with calcitriol offers evidence and support for the further study of calcitriol and its implications in cancer chemotherapy.
Other Possible Drug Combinations
Scientific evidence shows that combination therapies of cisplatin with other drugs have been tested in cancer cell lines for more efficacy. Some have shown additive while few other responses have been found to be synergistic. Table 1 presents the different drug combinations used to treat various cancers in combination with cisplatin.
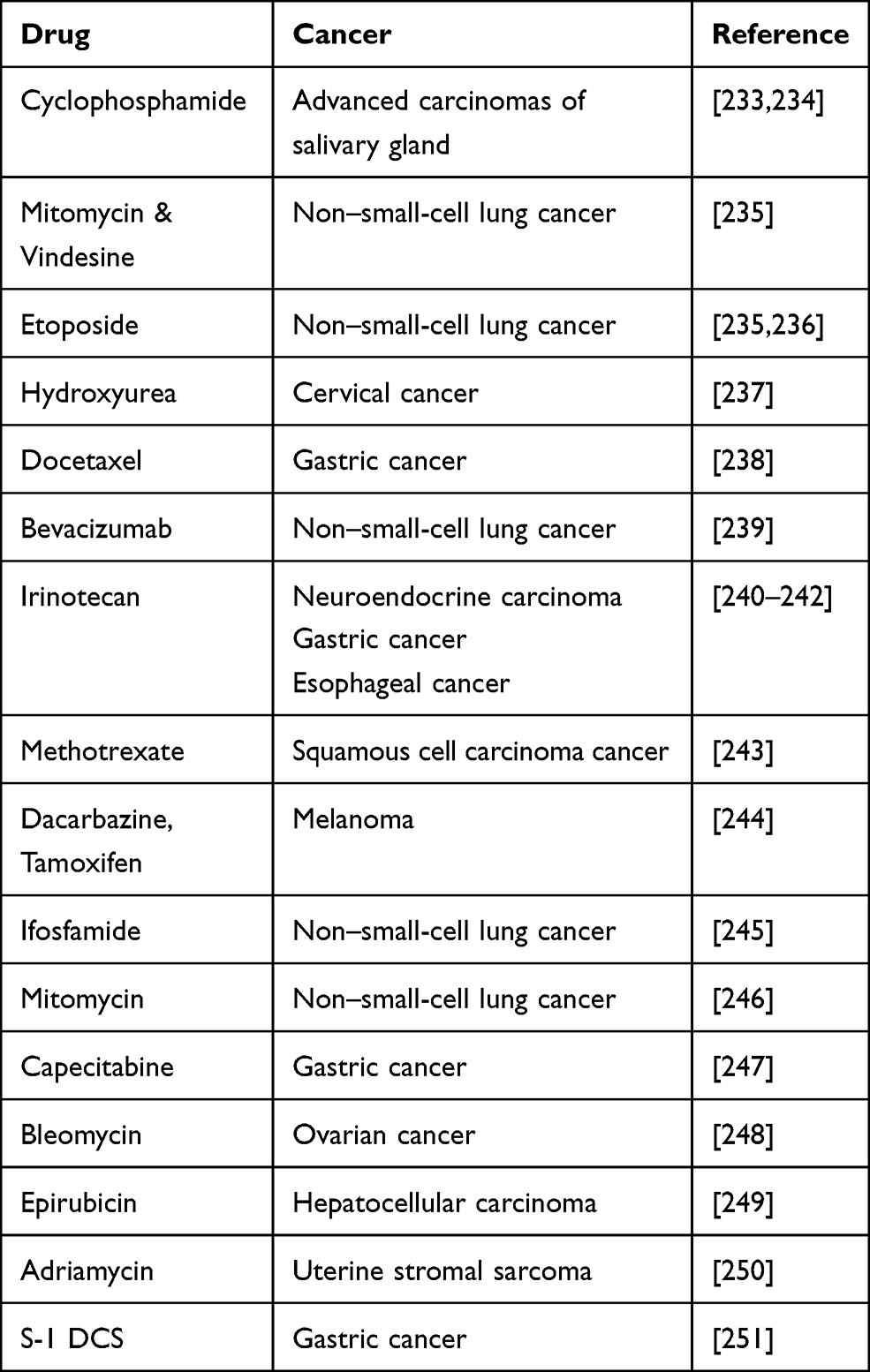 |
Table 1 Other Potential Cancer Drug Combinations with Cisplatin |
Natural Products
Chemoresistance has become a serious challenge in the cancer management. However, natural products, in the form of chemo sensitizers have attracted the attention of researchers. Natural products for cancer therapy have been very promising after thorough clinical and pre-clinical investigations. The bioactive components derived from these natural products have been tested for anti-cancer activity. These compounds work either alone or in combination with conventional chemotherapeutic drugs to alleviate the side effects of the drug administered and also protecting healthy cells in vicinity. The molecular target for individual product varies, but leading to dose and time-dependent cytotoxicity. The majority of natural products effectively target cancer cells specific ROS-sensing signaling pathways, thus attacking tumor development and progression. We have compiled the updated literature available on natural products in a tabular form below, describing the role of natural products in overcoming cisplatin chemo-resistance (Table 2).
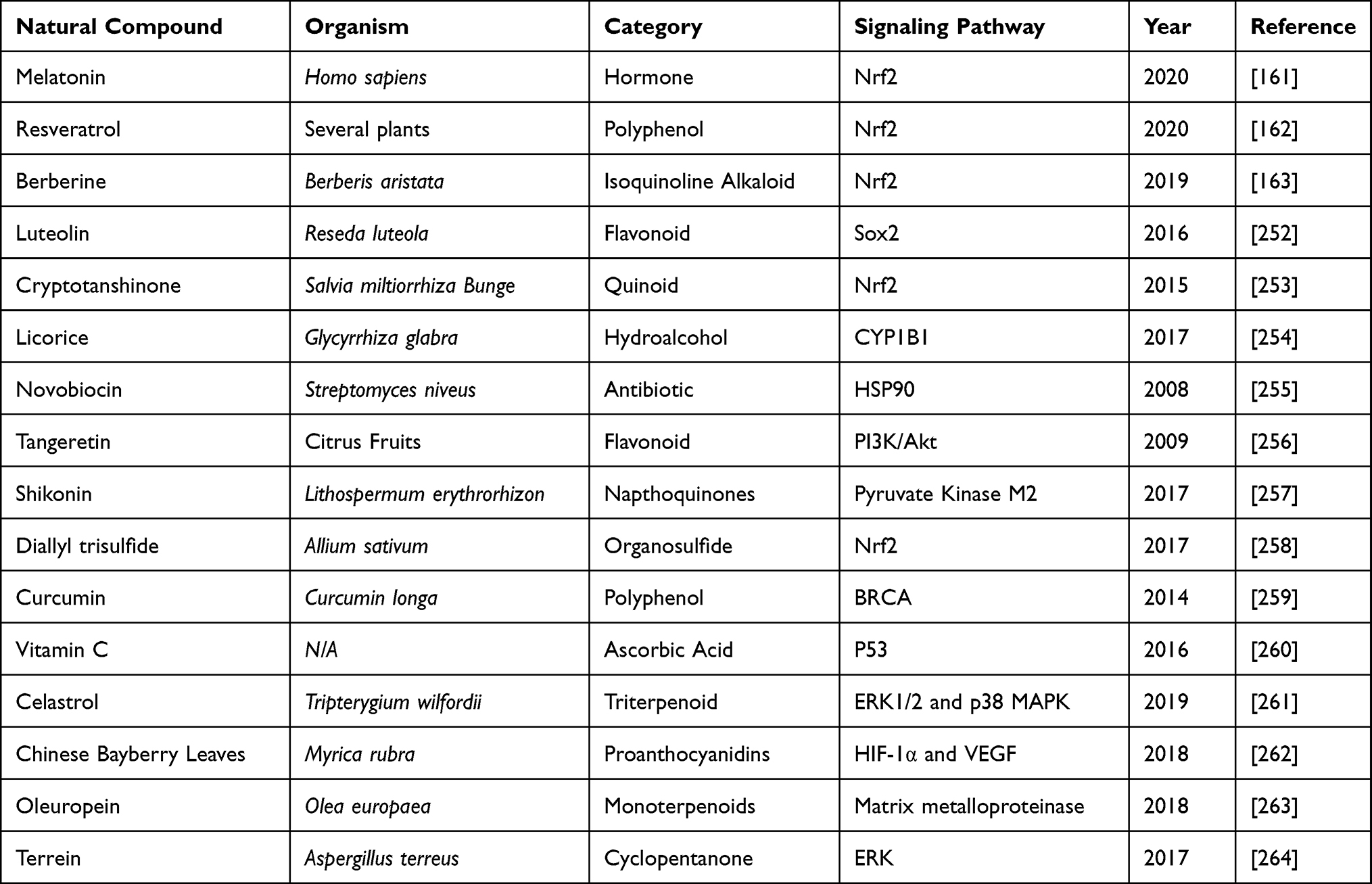 |
Table 2 Natural Products Used in Reducing Cisplatin Toxicity and Suppressing Its Resistance |
Nanotechnologic Formulations
Effective drug delivery and tumor targeting are of paramount importance in clinical oncology. However, the pharmacokinetics of chemotherapy drugs is often affected by factors like poor solubility, loss of bioactive structure, inadequate cellular uptake, short plasma half-lives due to rapid excretion from kidneys or enzymatic degradation, drug resistance, and side effects due to non-specific binding.211 Several nanoparticle formulations have been developed to address the problems encountered by conventional chemotherapy. More than a few nanoparticle platforms are designed with polymers and targeting agents enhancing colloidal stability and successful delivery of the bioactive compound in cancer cells. Clinically relevant therapeutic and diagnostic nanoparticles include two categories, Organic (liposomes, protein-based, polymeric/micelle) and inorganic (iron oxide, silica, and gold). Inorganic NPs are mainly used for cancer imaging applications along with thermal ablation of tumors while organic NPs are approved for vaccination, hemostasis, drug delivery systems, and topical dermal delivery.212
Nanocarriers
Clinical manifestations of a typical tumor site include leaky blood vessels and poor lymphatic drainage.213 Hence, multiple drug resistance occurs because of lack of control on drug delivery in conventional chemotherapy.129 In spite of non-specific diffusion, a nanocarrier can escape into the tumor tissue through leaky vasculature via EPR effect offering many advantages over free drugs. Studies have shown that nanoparticle diameter size <200 nm is more effective for successful drug delivery.214,215 Not only nanocarriers protect the drug from premature degradation, they also enhance absorption of the drugs into a selected tissue and improve intracellular penetration.216 Targeted drug delivery is possible through nanocarriers by conjugating a ligand molecule that binds to specific receptors on cell surface.217 A recent study has shown that cisplatin-loaded human epidermal growth factor receptor 2 (HER2) targeted poly(lactic-co-glycolic) nano platform system increases the therapeutic efficacy by taking advantage of overexpressed (HER2) receptors, using them as docks for targeted chemotherapy.218 Such specific-binding leads to receptor-mediated internalization, a requisite for drug release inside the cell. A successful liposomal formulation of encapsulated into liposome has been clinically investigated as lipoplatin.219 The therapeutic efficacy of lipoplatin is attributed to its primary action targeting tumors and hence, causing a greater damage to tumor tissues compared to normal tissues.220 In addition, preclinical studies with lipoplatin have shown less nephrotoxicity compared to cisplatin, contributing cytoprotective function to adjacent cells.221,222 Also, for the first time, lipid-coated cisplatin nanoparticles administered through microneedle in a mice xenograft model significantly induced cytotoxicity and apoptosis (apoptotic index of 58.6%). This cisplatin-nanoparticle microneedle system did not elicit other organ toxicity in the animal model, indicating that it is bio-safe.223 Another study on human melanoma tumor cells treated with cisplatin nanoparticle showed significant cytotoxicity through the “neighboring effect” upon administration of 1 mg/kg dosage.224 We reviewed recent clinical investigations of cisplatin nanoparticle formulations in a tabular form showing their importance as a disruptive technology for effective chemotherapy (Table 3).
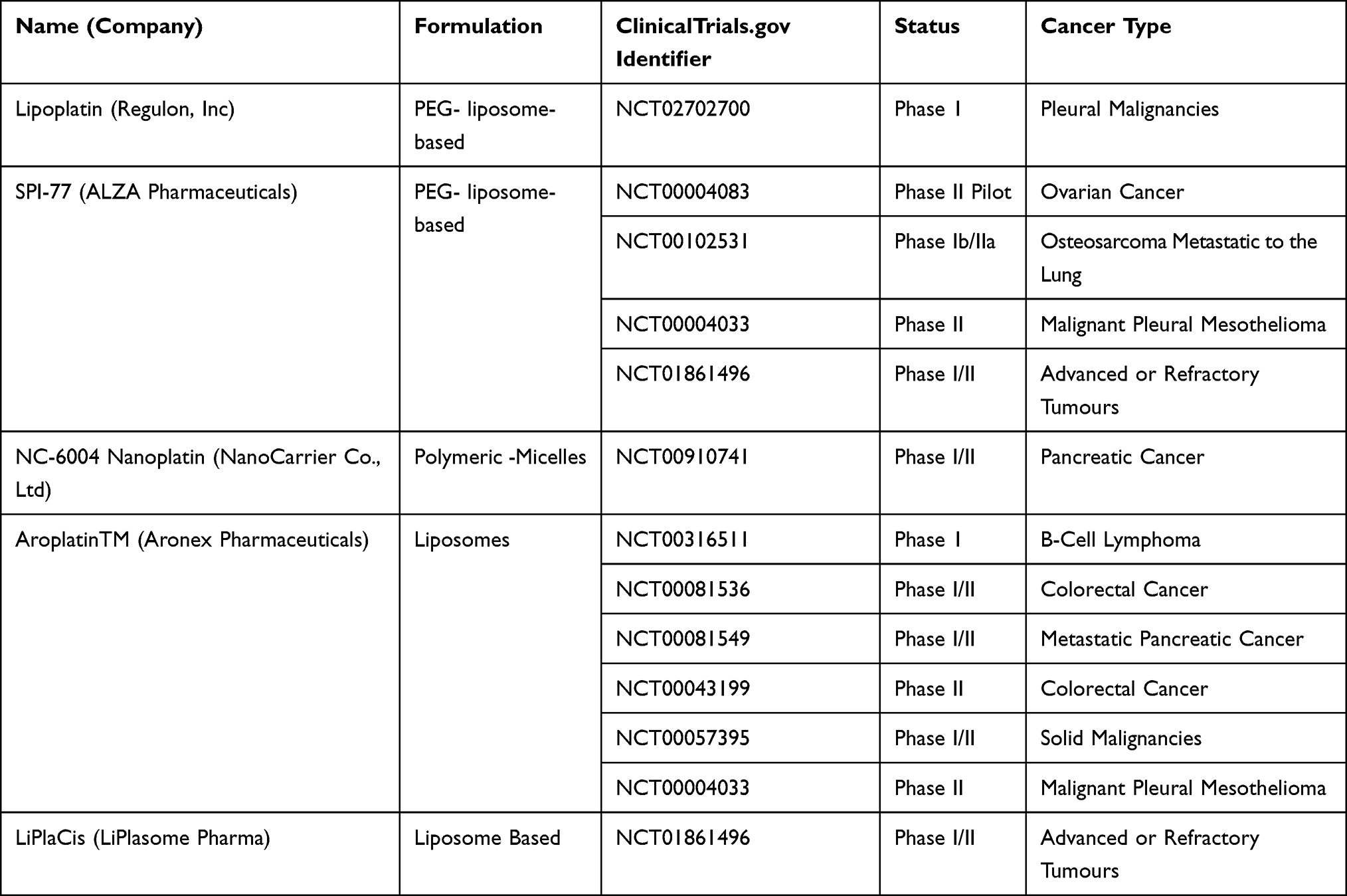 |
Table 3 Cisplatin-Nanoparticle Combinations Under Clinical Trials |
Combination Chemotherapy with Nanoparticles
The combination of drugs with nanoparticles is advantageous over single drug administration. Those benefits include absolute delivery of a correct ratio of each drug, synergistic therapeutic effects, suppressed drug resistance, and the ability to control drug exposure temporally.225 Combination therapy of doxycycline and cisplatin-loaded polysaccharide-based nanovehicles was found to be synergistic for breast cancer treatment.226 This nanoparticle-based drug delivery was made possible from HER2 antibody-decorated nanoparticles assembled from aldehyde hyaluronic acid and hydroxyethyl chitosan with spherical shape, having an average size of ~160 nm and a zeta potential of −28 mV, and showing a pH-responsive surface charge reversal and drug release with significant enhancement on the cellular uptake.226 Another interesting study has shown the delivery of doxorubicin and cisplatin via a single polymer-caged nanobin prepared with doxorubicin-encapsulated liposomal core protected by a pH-responsive cisplatin prodrug-loaded polymer shell.227 The cytotoxicity of each drug at low doses exhibits higher synergy than combinations of either the free drugs or separately nano-packaged drugs. A synergistic cytotoxic effect of doxorubicin and cisplatin has been accomplished by designing polymeric nanogels of about <100 nm in size based on polyacrylic acid. The experimental results have shown efficacy for the multidrug-resistant MCF-7/ADR tumor with reduced side effects.228 Furthermore, systemic co-delivery of cisplatin and doxorubicin through hyaluronic acid-modified amine-terminated fourth-generation polyamidoamine dendrimer nanoparticles was shown to be effective treatment for breast cancer treatment.229
Combination of cisplatin and a protein phosphatase 2A inhibitor (4-(3-carboxy-7-oxa-bicyclo[2.2.1]heptane-2-carbonyl)piperazine-1-carboxylic acid tert-butyl ester) using PEG-b-PLGA micelles has facilitated to overcome tumor resistance to cisplatin.230 This combination has shown to prolong drug residence in the blood while minimizing the side effects. In vitro studies with mesoporous silica nanoparticles loaded with cisplatin and phthalocyanine has been evaluated for its potential treatment efficacy in HeLa cells as nanocarriers.231 The confocal microscopy experiments presented that the silica nanomaterials can be effectively internalized in HeLa cells. The in vitro and in vivo studies with oligonucleotides assembled Au nanorod for combination remote-controlled drug delivery of doxorubicin and cisplatin have reported significant cell toxicity and controlled solid tumor growth, respectively. The design included a complementary DNA (cDNA) with the 5′ amine functional group assisted to chain cisplatin-Dox with amide bond.232 We discussed updated literature on nanoparticle formulations, in combination with cisplatin in Table 4.
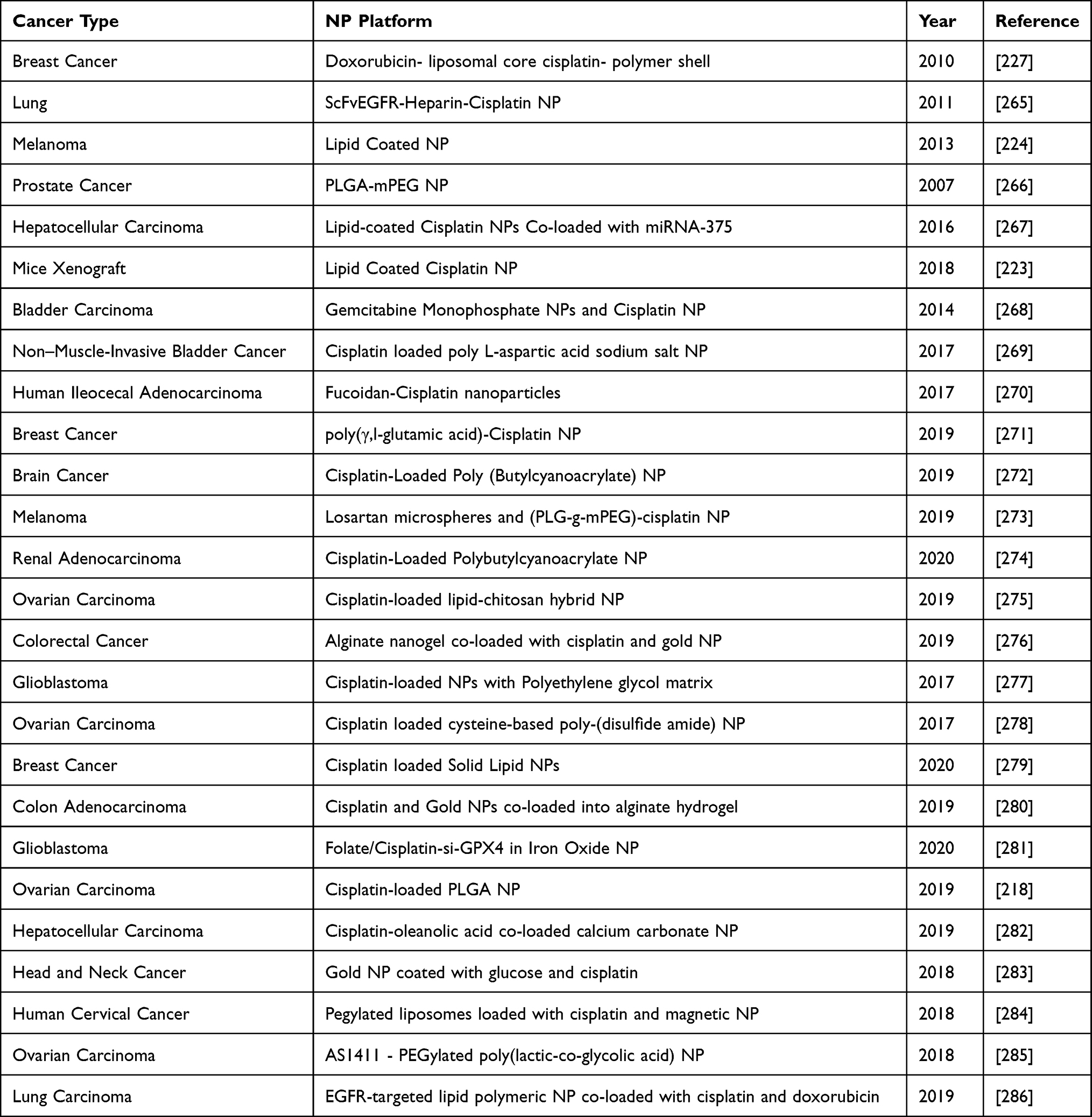 |
Table 4 Nanoparticles Used in Combination with Cisplatin |
Conclusions
Cisplatin is a commonly used metal coordination compound that was approved for clinical treatment of various cancers. Its molecular mechanism of action primarily involves formation of DNA adducts leading to onset of programmed cell death through initiation of major signaling pathways. Production of free radicals from ruptured cell membrane due to disruption of calcium homeostasis, lipid peroxidation, DNA damage, p53 activation, overexpression of p38, MAPK and JNK signaling pathways, caspase 3 and caspase 9 activation, and down-regulation of oncogenes, collectively trigger apoptosis of cancer cells. These molecular mechanisms of toxic action constitute that hallmarks of cisplatin bio-activity. However, its clinical utilization for cancer treatment has also raised safety concerns associated with a number of side effects including hepatotoxicity, cardiotoxicity, neurotoxicity, retinal toxicity and other systemic effects. Another cause for concern is related to drug resistance associated with the alterations in cisplatin uptake and efflux processes; increased drug biotransformation and excretion, and enhanced DNA repair mechanisms to reduce cisplatin toxicity to cancer cells. Both drug resistance and safety issues in cancer monotherapy with cisplatin have led to the development and implementation of novel therapeutic strategies that include combination treatments with other cancer drugs such as doxorubicin, gemcitabine, paclitaxel, vitamin D, and tegafur-uracil, as well as the application of nanotechnology-based formulations to enhance cisplatin drug delivery to cancer patients.
Acknowledgments
The research described in this publication was made possible by a grant from the National Institutes of Health (Grant No. 1U54MD015929-01-RCMI Center for Health Disparities Research) and by a grant from the National Science Foundation (Grant #HRD 1547754-Center for Nanotoxicity Studies) at Jackson State University. The content of this paper is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health or the National Science Foundation.
Disclosure
All authors have declared that they do not have any competing and/or financial interests for this work.
References
1. Alderden RA, Hall MD, Hambley TW. The discovery and development of cisplatin. J Chem Educ. 2006;83(5):728. doi:10.1021/ed083p728
2. Muggia FM, Bonetti A, Hoeschele JD, Rozencweig M, Howell SB. Platinum antitumor complexes: 50 Years since Barnett Rosenberg’s discovery. J Clin Oncol. 2015;33(35):4219–4226. doi:10.1200/JCO.2015.60.7481
3. Rosenberg B, Van Camp L, Krigas T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode [17]. Nature. 1965;205(4972):698–699. doi:10.1038/205698a0
4. IARC, International Agency for Research on Cancer. IARC monographs on the evaluation of carcinogenic risks to humans overall evaluations of carcinogenicity: an updating of IARC monographs volumes 1 to 42; SUPPLEMENT 7. IARC Monogr Eval Carcinog RISKS TO HUMANS - Overall Eval Carcinog An Update IARC Monographs; 1987.
5. The Department of Chemistry at the University of Akron. Akron the chemical database; 2009. Available from: https://pubchem.ncbi.nlm.gov/compound/cis-Platin . Accessed March 04, 2021.
6. HSDB. Hazardous substances data bank; 2009. Available from: https://www.nlm.nih.gov/toxnet/index.html. Accessed November 24, 2020.
7. Lippert B. In: Lippert B, editor. Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug. John Wiley & Sons; 2006. doi:10.1002/9783906390420
8. Cepeda V, Fuertes M, Castilla J, Alonso C, Quevedo C, Perez J. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem. 2008. doi:10.2174/187152007779314044
9. Ishida S, Lee J, Thiele DJ, Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci U S A. 2002. doi:10.1073/pnas.162491399
10. Brown A, Kumar S, Tchounwou PB. Cisplatin-based chemotherapy of human cancers. J Cancer Sci Ther. 2019;11(4).
11. Fichtinger-Schepman AMJ, Lohman PHM, van der Veer JL, den Hartog JHJ, Reedijk J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry. 1985;24(3):707–713. doi:10.1021/bi00324a025
12. Beck DJ, Brubaker RR. Effect of cis platinum (II) diamminodichloride on wild type and deoxyribonucleic acid repair deficient mutants of Escherichia coli. J Bacteriol. 1973;116(3):1247–1252. doi:10.1128/jb.116.3.1247-1252.1973
13. Fraval HNA, Rawlings CJ, Roberts JJ. Increased sensitivity of UV-repair-deficient human cells to DNA bound platinum products which unlike thymine dimers are not recognized by an endonuclease extracted from Micrococcus luteus. Mutat Res Fundam Mol Mech Mutagen. 1978;51(1):121–132. doi:10.1016/0027-5107(78)90014-3
14. Kumar S, Tchounwou PB. Molecular mechanisms of cisplatin cytotoxicity in acute promyelocytic leukemia cells. Oncotarget. 2015;6(38):40734–40746. doi:10.18632/oncotarget.5754
15. Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H, LLeonart ME. Oxidative stress and cancer: an overview. Ageing Res Rev. 2013;12(1):376–390. doi:10.1016/j.arr.2012.10.004
16. Ueda N, Kaushal GP, Shah SV. Apoptotic mechanisms in acute renal failure. Am J Med. 2000;108(5):403–415. doi:10.1016/S0002-9343(00)00311-9
17. Lieberthal W, Triaca V, Levine J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: apoptosis vs. necrosis. Am J Physiol. 1996. doi:10.1152/ajprenal.1996.270.4.f700
18. Matsushima H, Yonemura K, Ohishi K, Hishida A. The role of oxygen free radicals in cisplatin-induced acute renal failure in rats. J Lab Clin Med. 1998;131(6):518–526. doi:10.1016/S0022-2143(98)90060-9
19. Shaloam RD. Preclinical assessment of low doses of cisplatin in the management of acute promyelocytic leukemia. Int J Cancer Res Mol Mech. 2015. doi:10.16966/2381-3318.113
20. Cui C, Merritt R, Fu L, Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. 2017;7(1):3–17. doi:10.1016/j.apsb.2016.11.001
21. Zhu H, Zhang H, Jin F, et al. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget. 2014;5(11):3455–3471. doi:10.18632/oncotarget.1903
22. Shen L, Wen N, Xia M, et al. Calcium efflux from the endoplasmic reticulum regulates cisplatin-induced apoptosis in human cervical cancer HeLa cells. Oncol Lett. 2016;11(4):2411–2419. doi:10.3892/ol.2016.4278
23. Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003. doi:10.1074/jbc.M210284200
24. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020. doi:10.1038/s41571-020-0341-y
25. Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol. 2013;23(12):620–633. doi:10.1016/j.tcb.2013.07.006
26. Perfettini JL, Castedo M, Nardacci R, et al. Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J Exp Med. 2005;201(2):279–289. doi:10.1084/jem.20041502
27. Benhar M, Dalyot I, Engelberg D, Levitzki A. Enhanced ROS production in oncogenically transformed cells potentiates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase activation and sensitization to genotoxic stress. Mol Cell Biol. 2001;21(20):6913–6926. doi:10.1128/mcb.21.20.6913-6926.2001
28. Jeong HG, Cho HJ, Chang IY, et al. Rac1 prevents cisplatin-induced apoptosis through down-regulation of p38 activation in NIH3T3 cells. FEBS Lett. 2002. doi:10.1016/S0014-5793(02)02674-1
29. Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol. 2001. doi:10.1124/mol.59.4.657
30. Ashrafizadeh M, Ahmadi Z, Farkhondeh T, Samarghandian S. Autophagy as a molecular target of quercetin underlying its protective effects in human diseases. Arch Physiol Biochem. 2019. doi:10.1080/13813455.2019.1671458
31. Basu A. The potential of protein kinase C as A target for anticancer treatment. Pharmacol Ther. 1993;59(3):257–280. doi:10.1016/0163-7258(93)90070-T
32. Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258(5082):607–614. doi:10.1126/science.1411571
33. Erhardt P, Schremser EJ, Cooper GM. B-RAF inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol. 1999. doi:10.1128/mcb.19.8.5308
34. Tashker JS, Olson M, Kornbluth S. Post-cytochrome c protection from apoptosis conferred by a MAPK pathway in Xenopus egg extracts. Mol Biol Cell. 2002. doi:10.1091/mbc.01-06-0291
35. Nowak G. Protein kinase C-α and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J Biol Chem. 2002. doi:10.1074/jbc.M206373200
36. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298(5600):1911–1912. doi:10.1126/science.1072682
37. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi:10.1038/35065000
38. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80(2):179–185. doi:10.1016/0092-8674(95)90401-8
39. Ueda Y, Hirai SI, Osada SI, Suzuki A, Mizuno K, Ohno S. Protein kinase C δ activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem. 1996. doi:10.1074/jbc.271.38.23512
40. Basu A, Tu H. Activation of ERK during DNA damage-induced apoptosis involves protein kinase Cδ. Biochem Biophys Res Commun. 2005. doi:10.1016/j.bbrc.2005.06.199
41. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi:10.1016/S0092-8674(00)00116-1
42. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12(1):14–21. doi:10.1016/S0959-437X(01)00258-1
43. Potapova O, Haghighi A, Bost F, et al. The Jun kinase/stress-activated protein kinase pathway functions to regulate DNA repair and inhibition of the pathway sensitizes tumor cells to cisplatin. J Biol Chem. 1997;272(22):14041–14044. doi:10.1074/jbc.272.22.14041
44. Hayakawa J, Ohmichi M, Kurachi H, et al. Inhibition of extracellular signal-regulated protein kinase or c-Jun N- terminal protein kinase cascade, differentially activated by cisplatin, sensitizes human ovarian cancer cell line. J Biol Chem. 1999;274(44):31648–31654. doi:10.1074/jbc.274.44.31648
45. Potapova O, Basu S, Mercola D, Holbrook NJ. Protective role for c-Jun in the cellular response to DNA damage. J Biol Chem. 2001. doi:10.1074/jbc.M102075200
46. Yan D, An GY, Kuo MT. c-jun N-terminal kinase signalling pathway in response to cisplatin. J Cell Mol Med. 2016. doi:10.1111/jcmm.12908
47. Gupta J, Nebreda AR. Roles of p38α mitogen-activated protein kinase in mouse models of inflammatory diseases and cancer. FEBS J. 2015. doi:10.1111/febs.13250
48. Grossi V, Peserico A, Tezil T, Simone C. p38α MAPK pathway: a key factor in colorectal cancer therapy and chemoresistance. World J Gastroenterol. 2014;20(29):9744. doi:10.3748/wjg.v20.i29.9744
49. Pereira L, Igea A, Canovas B, Dolado I, Nebreda AR. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol Med. 2013;5(11):1759–1774. doi:10.1002/emmm.201302732
50. Nahta R, Hung MC, Esteva FJ. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 2004;64(7):2343–2346. doi:10.1158/0008-5472.CAN-03-3856
51. Burgering BMT, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376(6541):599–602. doi:10.1038/376599a0
52. Wick MJ, Dong LQ, Riojas RA, Ramos FJ, Liu F. Mechanism of phosphorylation of protein kinase B/Akt by a constitutively active 3-phosphoinositide-dependent protein kinase-1. J Biol Chem. 2000;275(51):40400–40406. doi:10.1074/jbc.M003937200
53. Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Ann Med. 2014;46(6):372–383. doi:10.3109/07853890.2014.912836
54. Peng DJ, Wang J, Zhou JY, Wu GS. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem Biophys Res Commun. 2010;394(3):600–605. doi:10.1016/j.bbrc.2010.03.029
55. Belyanskaya LL, Hopkins-Donaldson S, Kurtz S, et al. Cisplatin activates Akt in small cell lung cancer cells and attenuates apoptosis by survivin upregulation. Int J Cancer. 2005;117(5):755–763. doi:10.1002/ijc.21242
56. Sedletska Y, Giraud-Panis MJ, Malinge JM. Cisplatin is a DNA-damaging antitumour compound triggering multifactorial biochemical responses in cancer cells: importance of apoptotic pathways. Curr Med Chem Anticancer Agents. 2005;5(3):251–265. doi:10.2174/1568011053765967
57. Jamieson ER, Lippard SJ. Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev. 1999;99(9):2467–2498. doi:10.1021/cr980421n
58. Basu A, Krishnamurthy S. Cellular responses to cisplatin-induced DNA damage. J Nucleic Acids. 2010;2010:1–16. doi:10.4061/2010/201367
59. Welsh C, Day R, McGurk C, Masters JRW, Wood RD, Köberle B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int J Cancer. 2004;110(3):352–361. doi:10.1002/ijc.20134
60. Köberle B, Grimaldi KA, Sunters A, Hartley JA, Kelland LR, Masters JRW. DNA repair capacity and cisplatin sensitivity of human testis tumour cells. Int J Cancer. 1997;70(5):551–555. doi:10.1002/(SICI)1097-0215(19970304)70:5<551::AID-IJC10>3.0.CO;2-G
61. Avdievich E, Reiss C, Scherer SJ, et al. Distinct effects of the recurrent Mlh1G67R mutation on MMR functions, cancer, and meiosis. Proc Natl Acad Sci U S A. 2008;105(11):4247–4252. doi:10.1073/pnas.0800276105
62. Yang G, Scherer SJ, Shell SS, et al. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6(2):139–150. doi:10.1016/j.ccr.2004.06.024
63. Lin DP, Wang Y, Scherer SJ, et al. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004;64(2):517–522. doi:10.1158/0008-5472.CAN-03-2957
64. Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10(4):431–442. doi:10.1038/sj.cdd.4401183
65. De Laurenzi V, Melino G. Evolution of functions within the p53/p63/p73 family. Ann N Y Acad Sci. 2000;926:90–100. doi:10.1111/j.1749-6632.2000.tb05602.x
66. Abedini MR, Muller EJ, Brun J, Bergeron R, Gray DA, Tsang BK. Cisplatin induces p53-dependent FLICE-like inhibitory protein ubiquitination in ovarian cancer cells. Cancer Res. 2008. doi:10.1158/0008-5472.CAN-08-0673
67. Sirvent A, Benistant C, Roche S. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol Cell. 2008. doi:10.1042/bc20080020
68. Kerr JFR, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer Therapy. Cancer. 1994;73(8):2013–2026. doi:10.1002/1097-0142(19940415)73:8<2013::AID-CNCR2820730802>3.0.CO;2-J
69. Persons DL, Yazlovitskaya EM, Pelling JC. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J Biol Chem. 2000;275(46):35778–35785. doi:10.1074/jbc.M004267200
70. Brown R, Clugston C, Edlin A, et al. Increased accumulation of p53 protein in cisplatin‐resistant ovarian cell lines. Int J Cancer. 1993;55(4):678–684. doi:10.1002/ijc.2910550428
71. Kharbanda S, Ren R, Pandey P, et al. Activation of the c-Abl tyrosine kinase in the stress response to DMA-damaging agents. Nature. 1995;376(6543):785–788. doi:10.1038/376785a0
72. Tsai KKC, Yuan ZM. c-Abl stabilizes p73 by a phosphorylation-augmented interaction. Cancer Res. 2003.
73. Galan-Moya EM, Hernandez-Losa J, Aceves Luquero CI, et al. C-Abl activates p38 MAPK independently of its tyrosine kinase activity: implications in cisplatin-based therapy. Int J Cancer. 2008. doi:10.1002/ijc.23063
74. Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–1883. doi:10.1038/onc.2011.384
75. Huang RS, Duan S, Shukla SJ, et al. Identification of genetic variants contributing to cisplatin-induced cytotoxicity by use of a genomewide approach. Am J Hum Genet. 2007;81(3):427–437. doi:10.1086/519850
76. Sakamoto M, Kondo A, Kawasaki K, et al. Analysis of gene expression profiles associated with cisplatin resistance in human ovarian cancer cell lines and tissues using cDNA microarray. Hum Cell. 2001.
77. Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429(6987):92–96. doi:10.1038/nature02456
78. Nabil M, Zunino P. A computational study of cancer hyperthermia based on vascular magnetic nanoconstructs. R Soc Open Sci. 2016;3(9):160287. doi:10.1098/rsos.160287
79. Aprile F, Martin DS. Chlorotriammineplatinum(II) Ion. Acid hydrolysis and isotopic exchange of chloride ligand. Inorg Chem. 1962;1(3):551–557. doi:10.1021/ic50003a021
80. Burda JV, Zeizinger M, Leszczynski J. Hydration process as an activation of trans- and cisplatin complexes in anticancer treatment. DFT and Ab initio computational study of thermodynamic and kinetic parameters. J Comput Chem. 2005;26(9):907–914. doi:10.1002/jcc.20228
81. Henderson RA. Kinetics and mechanism of reactions of transition metal complexes. J Organomet Chem. 1992. doi:10.1016/0022-328x(92)83075-s
82. Platts JA, Hibbs DE, Hambley TW, Hall MD. Calculation of the hydrophobicity of platinum drugs. J Med Chem. 2001. doi:10.1021/jm001080k
83. Baik MH, Friesner RA, Lippard SJ. Theoretical study of cisplatin binding to purine bases: why does cisplatin prefer guanine over adenine? J Am Chem Soc. 2003;125(46):14082–14092. doi:10.1021/ja036960d
84. Zimmermann T, Burda JV. Reactions of cisplatin with cysteine and methionine at constant pH; A computational study. Dalton Trans. 2010. doi:10.1039/b913803j
85. Tasinato N, Puzzarini C, Barone V. Correct modeling of cisplatin: a paradigmatic case. Angew Chem Int Ed. 2017. doi:10.1002/anie.201707683
86. Chen HHW, Song IS, Hossain A, et al. Elevated glutathione levels confer cellular sensitization to cisplatin toxicity by up-regulation of copper transporter hCtr1. Mol Pharmacol. 2008;74(3):697–704. doi:10.1124/mol.108.047969
87. Bano N, Najam R, Qazi F. Adverse cardiac manifestations of cisplatin - A review. Int J Pharm Sci Rev Res. 2013.
88. Jefford M, Michael M, Rosenthal MA, et al. A novel combination of cisplatin, irinotecan, and capecitabine in patients with advanced cancer. Invest New Drugs. 2004;22(2):185–192. doi:10.1023/B:DRUG.0000011796.20332.a9
89. Price PM, Safirstein RL, Megyesi J. Protection of renal cells from cisplatin toxicity by cell cycle inhibitors. Am J Physiol Ren Physiol. 2004. doi:10.1152/ajprenal.00192.2003
90. Berry JM, Jacobs C, Sikic B, Halsey J, Borch RF. Modification of cisplatin toxicity with diethyldithiocarbamate. J Clin Oncol. 1990;8(9):1585–1590. doi:10.1200/JCO.1990.8.9.1585
91. Kuhlmann MK, Burkhardt G, Köhler H. Insights into potential cellular mechanisms of cisplatin nephrotoxicity and their clinical application. Nephrol Dial Transplant. 1997. doi:10.1093/ndt/12.12.2478
92. Bhattacharyya S, Mehta P. The hepatoprotective potential of Spirulina and vitamin C supplemention in cisplatin toxicity. Food Funct. 2012;3(2):164–169. doi:10.1039/c1fo10172b
93. Amin A, Hamza AEA. Effects of Roselle and Ginger on cisplatin-induced reproductive toxicity in rats. Asian J Androl. 2006. doi:10.1111/j.1745-7262.2006.00179.x
94. Ibrahim A, Eldaim MAA, Abdel-Daim MM. Nephroprotective effect of bee honey and royal jelly against subchronic cisplatin toxicity in rats. Cytotechnology. 2016;68(4):1039–1048. doi:10.1007/s10616-015-9860-2
95. Cersosimo RJ. Hepatotoxicity associated with cisplatin chemotherapy. Ann Pharmacother. 1993;27(4):438–441. doi:10.1177/106002809302700408
96. Liu J, Liu Y, Habeebu SSM, Klaassen CD. Metallothionein (MT)-null mice are sensitive to cisplatin-induced hepatotoxicity. Toxicol Appl Pharmacol. 1998. doi:10.1006/taap.1997.8325
97. Lu Y, Cederbaum AI. Cisplatin-induced hepatotoxicity is enhanced by elevated expression of cytochrome P450 2E1. Toxicol Sci. 2006;89(2):515–523. doi:10.1093/toxsci/kfj031
98. Pezeshki Z, Khosravi A, Nekuei M, et al. Time course of cisplatin-induced nephrotoxicity and hepatotoxicity. J Nephropathol. 2017;6(3):163–167. doi:10.15171/jnp.2017.28
99. El-Awady ESE, Moustafa YM, Abo-Elmatty DM, Radwan A. Cisplatin-induced cardiotoxicity: mechanisms and cardioprotective strategies. Eur J Pharmacol. 2011;650(1):335–341. doi:10.1016/j.ejphar.2010.09.085
100. Dieckmann KP, Struss WJ, Budde U. Evidence for acute vascular toxicity of cisplatin-based chemotherapy in patients with germ cell tumour. Anticancer Res. 2011;31(12):4501–4505.
101. Jafri M, Protheroe A. Cisplatin-associated thrombosis. Anticancer Drugs. 2008;19(9):927–929. doi:10.1097/CAD.0b013e3283100e9c
102. Patanè S. Cardiotoxicity: cisplatin and long-term cancer survivors. Int J Cardiol. 2014. doi:10.1016/j.ijcard.2014.04.238
103. Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: a review. Am J Med Sci. 2007;334(2):115–124. doi:10.1097/MAJ.0b013e31812dfe1e
104. Tanaka H, Ishikawa E, Teshima S, Shimizu E. Histopathological study of human cisplatin nephropathy. Toxicol Pathol. 1986;14(2):247–257. doi:10.1177/019262338601400215
105. Cornelison TL, Reed E. Nephrotoxicity and hydration management for cisplatin, carboplatin, and ormaplatin. Gynecol Oncol. 1993;50(2):147–158. doi:10.1006/gyno.1993.1184
106. Pabla N, Murphy RF, Liu K, Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Ren Physiol. 2009;296(3):F505–F511. doi:10.1152/ajprenal.90545.2008
107. Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel). 2010;2(11):2490–2518. doi:10.3390/toxins2112490
108. Hwa Lee R, Mi Song J, Young Park M, Kyung Kang S, Keun Kim Y, Sup Jung J. Cisplatin-induced apoptosis by translocation of endogenous Bax in mouse collecting duct cells. Biochem Pharmacol. 2001. doi:10.1016/S0006-2952(01)00748-1
109. Aldossary SA. Review on pharmacology of cisplatin: clinical use, toxicity and mechanism of resistance of cisplatin. Biomed Pharmacol J. 2019;12(1):07–15. doi:10.13005/bpj/1608
110. Cavaletti G, Pezzoni G, Pisano C, et al. Cisplatin-induced peripheral neurotoxicity in rats reduces the circulating levels of nerve growth factor. Neurosci Lett. 2002;322(2):103–106. doi:10.1016/S0304-3940(02)00091-5
111. Rybak LP, Whitworth CA, Mukherjea D, Ramkumar V. Mechanisms of cisplatin-induced ototoxicity and prevention. Hear Res. 2007;226(1–2):157–167. doi:10.1016/j.heares.2006.09.015
112. Waissbluth S, Daniel SJ. Cisplatin-induced ototoxicity: transporters playing a role in cisplatin toxicity. Hear Res. 2013;299:37–45. doi:10.1016/j.heares.2013.02.002
113. Colpi GM, Contalbi GF, Nerva F, Sagone P, Piediferro G. Testicular function following chemo-radiotherapy. Eur J Obstet Gynecol Reprod Biol. 2004;113:S2–S6. doi:10.1016/j.ejogrb.2003.11.002
114. Malarvizhi D, Mathur PP. Effects of cisplatin on testicular functions in rats. Indian J Exp Biol. 1996.
115. Cherry SM, Hunt PA, Hassold TJ. Cisplatin disrupts mammalian spermatogenesis, but does not affect recombination or chromosome segregation. Mutat Res Genet Toxicol Environ Mutagen. 2004;564(2):115–128. doi:10.1016/j.mrgentox.2004.08.010
116. Wilding G, Caruso R, Lawrence TS, et al. Retinal toxicity after high-dose cisplatin therapy. J Clin Oncol. 1985;3(12):1683–1689. doi:10.1200/JCO.1985.3.12.1683
117. Miller DF, Bay JW, Lederman RJ, Purvis JD, Rogers LR, Tomsak RL. Ocular and orbital toxicity following intracarotid injection of BCNU (Carmustine) and cisplatinum for malignant gliomas. Ophthalmology. 1985;92(3):402–406. doi:10.1016/S0161-6420(85)34036-8
118. Zisowsky J, Koegel S, Leyers S, et al. Relevance of drug uptake and efflux for cisplatin sensitivity of tumor cells. Biochem Pharmacol. 2007;73(2):298–307. doi:10.1016/j.bcp.2006.10.003
119. Lee J, Prohaska JR, Thiele DJ. Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc Natl Acad Sci U S A. 2001;98(12):6842–6847. doi:10.1073/pnas.111058698
120. Kuo MT, Chen HHW, Song IS, Savaraj N, Ishikawa T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007;26(1):71–83. doi:10.1007/s10555-007-9045-3
121. Hromas RA, North JA, Burns CP. Decreased cisplatin uptake by resistant L1210 leukemia cells. Cancer Lett. 1987. doi:10.1016/0304-3835(87)90091-7
122. Katano K, Safaei R, Samimi G, Holzer A, Rochdi M, Howell SB. The copper export pump ATP7B modulates the cellular pharmacology of carboplatin in ovarian carcinoma cells. Mol Pharmacol. 2003;64(2):466–473. doi:10.1124/mol.64.2.466
123. Samimi G, Safaei R, Katano K, et al. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin Cancer Res. 2004;10(14):4661–4669. doi:10.1158/1078-0432.CCR-04-0137
124. Safaei R, Otani S, Larson BJ, Rasmussen ML, Howell SB. Transport of cisplatin by the copper efflux transporter ATP7B. Mol Pharmacol. 2008;73(2):461–468. doi:10.1124/mol.107.040980
125. Xu D, Xi Z, Zhao L, Liu Y. Transporting platinum drugs from a copper chaperone to ATPase: the mechanistic implication of drug efflux mediated cisplatin resistance. Inorg Chem Front. 2014. doi:10.1039/c3qi00068k
126. Koike K, Kawabe T, Tanaka T, et al. A canalicular multispecific organic anion transporter (cMOAT) antisense cDNA enhances drug sensitivity in human hepatic cancer cells. Cancer Res. 1997;57(24):5475–5479.
127. Leslie EM, Deeley RG, Cole SPC. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005;204(3):216–237. doi:10.1016/j.taap.2004.10.012
128. Fojo AT, Ueda K, Slamon DJ, Poplack DG, Gottesman MM, Pastan I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc Natl Acad Sci U S A. 1987;84(1):265–269. doi:10.1073/pnas.84.1.265
129. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. doi:10.1038/nrc706
130. He C, Sun Z, Hoffman RM, et al. P-glycoprotein overexpression is associated with cisplatin resistance in human osteosarcoma. Anticancer Res. 2019;39(4):1711–1718. doi:10.21873/anticanres.13277
131. Gibalová L, Šereš M, Rusnák A, et al. P-glycoprotein depresses cisplatin sensitivity in L1210 cells by inhibiting cisplatin-induced caspase-3 activation. Toxicol Vitro. 2012;26(3):435–444. doi:10.1016/j.tiv.2012.01.014
132. Yang L, Li N, Wang H, Jia X, Wang X, Luo J. Altered microRNA expression in cisplatin-resistant ovarian cancer cells and upregulation of miR-130a associated with MDR1/P-glycoprotein-mediated drug resistance. Oncol Rep. 2012;28(2):592–600. doi:10.3892/or.2012.1823
133. Kong W, Ling X, Chen Y, et al. Hesperetin reverses P-glycoprotein-mediated cisplatin resistance in DDP-resistant human lung cancer cells via modulation of the nuclear factor-κB signaling pathway. Int J Mol Med. 2020. doi:10.3892/ijmm.2020.4485
134. Cheng Q, Liao M, Hu H, Li H, Wu L. Asiatic Acid (AA) sensitizes multidrug-resistant human lung adenocarcinoma A549/DDP cells to cisplatin (DDP) via downregulation of P-Glycoprotein (MDR1) and its targets. Cell Physiol Biochem. 2018;47(1):279–292. doi:10.1159/000489806
135. Ishikawa T, Ali-Osman F. Glutathione-associated cis-diamminedichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia cells. Molecular characterization of glutathione-platinum complex and its biological significance. J Biol Chem. 1993;268(27):20116–20125. doi:10.1016/S0021-9258(20)80702-9
136. Hagrman D, Goodisman J, Dabrowiak JC, Souid AK. Kinetic study on the reaction of cisplatin with metallothionein. Drug Metab Dispos. 2003;31(7):916–923. doi:10.1124/dmd.31.7.916
137. Chu G. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. J Biol Chem. 1994;269(2):787–790. doi:10.1016/S0021-9258(17)42175-2
138. Kelley SL, Basu A, Teicher BA, Hacker MP, Hamer DH, Lazo JS. Overexpression of metallothionein confers resistance to anticancer drugs. Science. 1988;241(4874):1813–1815. doi:10.1126/science.3175622
139. Siegsmund MJ, Marx C, Seemann O, et al. Cisplatin-resistant bladder carcinoma cells: enhanced expression of metallothioneins. Urol Res. 1999;27(3):157–163. doi:10.1007/s002400050103
140. Komiya K, Matsuda T, Nishio K, Ohmori T, Sugimoto Y, Saijo N. Metallothionein content correlates with the sensitivity of human small cell lung cancer cell lines to cisplatin. Cancer Res. 1991;51(12):3237–3242. doi:10.1016/0169-5002(92)90185-m
141. Wülfing C, van Ahlen H, Eltze E, Piechota H, Hertle L, Schmid KW. Metallothionein in bladder cancer: correlation of overexpression with poor outcome after chemotherapy. World J Urol. 2007;25(2):199–205. doi:10.1007/s00345-006-0141-8
142. Lee JH, Chae JW, Kim JK, Kim HJ, Chung JY, Kim YH. Inhibition of cisplatin-resistance by RNA interference targeting metallothionein using reducible oligo-peptoplex. J Control Release. 2015;215:82–90. doi:10.1016/j.jconrel.2015.07.015
143. Kondo Y, Kuo SM, Watkins SC, Lazo JS. Metallothionein localization and cisplatin resistance in human hormone-independent prostatic tumor cell lines. Cancer Res. 1995.
144. De Luca A, Parker LJ, Ang WH, et al. A structure-based mechanism of cisplatin resistance mediated by glutathione transferase P1-1. Proc Natl Acad Sci U S A. 2019;116(28):13943–13951. doi:10.1073/pnas.1903297116
145. Nebert DW, Vasiliou V. Analysis of the glutathione S-transferase (GST) gene family. Hum Genomics. 2004;1(6):460. doi:10.1186/1479-7364-1-6-460
146. Ahmad A, Robinson AR, Duensing A, et al. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol Cell Biol. 2008;28(16):5082–5092. doi:10.1128/mcb.00293-08
147. Bramson J, Panasci LC. Effect of ERCC-1 overexpression on sensitivity of Chinese hamster ovary cells to DNA damaging agents. Cancer Res. 1993.
148. Strathdee G, MacKean MJ, Illand M, Brown R. A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene. 1999;18(14):2335–2341. doi:10.1038/sj.onc.1202540
149. Brown R, Hirst GL, Gallagher WM, et al. hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene. 1997;15(1):45–52. doi:10.1038/sj.onc.1201167
150. Shachar S, Ziv O, Avkin S, et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009;28(4):383–393. doi:10.1038/emboj.2008.281
151. Bassett E, Vaisman A, Tropea KA, et al. Frameshifts and deletions during in vitro translesion synthesis past Pt-DNA adducts by DNA polymerases β and η. DNA Repair (Amst). 2002;1(12):1003–1016. doi:10.1016/S1568-7864(02)00150-7
152. Wang W, Sheng W, Yu C, et al. REV3L modulates cisplatin sensitivity of non-small cell lung cancer H1299 cells. Oncol Rep. 2015;34(3):1460–1468. doi:10.3892/or.2015.4121
153. Jiang HG, Chen P, Su JY, et al. Knockdown of REV3 synergizes with ATR inhibition to promote apoptosis induced by cisplatin in lung cancer cells. J Cell Physiol. 2017;232(12):3433–3443. doi:10.1002/jcp.25792
154. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108(2):171–182. doi:10.1016/S0092-8674(02)00615-3
155. Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–17084. doi:10.1073/pnas.0806092105
156. Tajeddine N, Galluzzi L, Kepp O, et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene. 2008;27(30):4221–4232. doi:10.1038/onc.2008.63
157. Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Quo Z, Cullen KJ. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin Cancer Res. 2006;12(19):5817–5825. doi:10.1158/1078-0432.CCR-06-1037
158. Choi YM, Kim HK, Shim W, et al. Mechanism of cisplatin-induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PLoS One. 2015. doi:10.1371/journal.pone.0135083
159. Ahmed EM, Bandopadhyay G, Coyle B, Grabowska A. A HIF-independent, CD133-mediated mechanism of cisplatin resistance in glioblastoma cells. Cell Oncol. 2018;41(3):319–328. doi:10.1007/s13402-018-0374-8
160. Dong L, Qi N, Ge RM, Cao CL, Lan F, Shen L. Overexpression of CD133 promotes the phosphorylation of Erk in U87MG human glioblastoma cells. Neurosci Lett. 2010;484(3):210–214. doi:10.1016/j.neulet.2010.08.057
161. Ahmadi Z, Ashrafizadeh M. Melatonin as a potential modulator of Nrf2. Fundam Clin Pharmacol. 2020;34(1):11–19. doi:10.1111/fcp.12498
162. Farkhondeh T, Folgado SL, Pourbagher-Shahri AM, Ashrafizadeh M, Samarghandian S. The therapeutic effect of resveratrol: focusing on the Nrf2 signaling pathway. Biomed Pharmacother. 2020;127:110234. doi:10.1016/j.biopha.2020.110234
163. Ashrafizadeh M, Fekri HS, Ahmadi Z, Farkhondeh T, Samarghandian S. Therapeutic and biological activities of berberine: the involvement of Nrf2 signaling pathway. J Cell Biochem. 2020;121(2):1575–1585. doi:10.1002/jcb.29392
164. Ashrafizadeh M, Ahmadi Z, Farkhondeh T, Samarghandian S. Back to nucleus: combating with a toxicity using Nrf2 signaling pathway as a promising therapeutic target. Biol Trace Elem Res. 2020;197(1):52–62. doi:10.1007/s12011-019-01980-4
165. Tonelli C, Chio IIC, Tuveson DA. Transcriptional Regulation by Nrf2. Antioxid Redox Signal. 2018;29(17):1727–1745. doi:10.1089/ars.2017.7342
166. Kansanen E, Kuosmanen SM, Leinonen H, Levonenn AL. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1(1):45–49. doi:10.1016/j.redox.2012.10.001
167. Dong J, Sulik KK, Chen SY. Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: implications for the prevention of fetal alcohol spectrum disorders. Antioxid Redox Signal. 2008;10(12):2023–2033. doi:10.1089/ars.2007.2019
168. Cho JM, Manandhar S, Lee HR, Park HM, Kwak MK. Role of the Nrf2-antioxidant system in cytotoxicity mediated by anticancer cisplatin: implication to cancer cell resistance. Cancer Lett. 2008;260(1–2):96–108. doi:10.1016/j.canlet.2007.10.022
169. Ashrafizadeh M, Zarrabi A, Hushmandi K, et al. Lung cancer cells and their sensitivity/resistance to cisplatin chemotherapy: role of microRNAs and upstream mediators. Cell Signal. 2021. doi:10.1016/j.cellsig.2020.109871
170. Ashrafizadeh M, Ahmadi Z, Samarghandian S, et al. MicroRNA-mediated regulation of Nrf2 signaling pathway: implications in disease therapy and protection against oxidative stress. Life Sci. 2020;244:117329. doi:10.1016/j.lfs.2020.117329
171. Ashrafizadeh M, Zarrabi A, Hushmandi K, et al. Association of the epithelial–mesenchymal transition (EMT) with cisplatin resistance. Int J Mol Sci. 2020;21(11):4002. doi:10.3390/ijms21114002
172. Kryczek I, Lin Y, Nagarsheth N, et al. IL-22+CD4+ T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity. 2014;40(5):772–784. doi:10.1016/j.immuni.2014.03.010
173. Liu W, Wang W, Wang X, Xu C, Zhang N, Di W. Cisplatin-stimulated macrophages promote ovarian cancer migration via the CCL20-CCR6 axis. Cancer Lett. 2020. doi:10.1016/j.canlet.2019.12.024
174. Chen KJ, Lin SZ, Zhou L, et al. Selective recruitment of regulatory T cell through CCR6-CCL20 in hepatocellular carcinoma fosters tumor progression and predicts poor prognosis. PLoS One. 2011;6(9). doi:10.1371/journal.pone.0024671
175. Walch-Rückheim B, Mavrova R, Henning M, et al. Stromal fibroblasts induce CCL20 through IL6/C/EBPβ to support the recruitment of Th17 cells during cervical cancer progression. Cancer Res. 2015;75(24):5248–5259. doi:10.1158/0008-5472.CAN-15-0732
176. Ranasinghe R, Eri R. Modulation of the CCR6-CCl20 axis: a potential therapeutic target in inflammation and cancer. Med. 2018. doi:10.3390/medicina54050088
177. Sirichanchuen B, Pengsuparp T, Chanvorachote P. Long-term Cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Mol Cell Biochem. 2012;364(1–2):11–18. doi:10.1007/s11010-011-1199-1
178. Wu HM, Jiang ZF, Ding PS, Shao LJ, Liu RY. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci Rep. 2015. doi:10.1038/srep12291
179. Zhang X, Qi Z, Yin H, Yang G. Interaction between p53 and ras signaling controls cisplatin resistance via HDAC4- and HIF-1α-mediated regulation of apoptosis and autophagy. Theranostics. 2019. doi:10.7150/thno.29673
180. Zhang N, Yang GQ, Shao XM, Wei L. GAS5 modulated autophagy is a mechanism modulating cisplatin sensitivity in NSCLC cells. Eur Rev Med Pharmacol Sci. 2016.
181. Liu Q, Zhu D, Hao B, Zhang Z, Tian Y. Luteolin promotes the sensitivity of cisplatin in ovarian cancer by decreasing PRPA1-medicated autophagy. Cell Mol Biol. 2018;64(6):17. doi:10.14715/cmb/2018.64.6.4
182. Takahashi A, Kimura T, Takabatake Y, et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol. 2012;180(2):517–525. doi:10.1016/j.ajpath.2011.11.001
183. Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74(5):631–640. doi:10.1038/ki.2008.214
184. Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235(1–2):179–192. doi:10.1016/S0378-5173(01)00986-3
185. Armstrong DK, Bundy B, Wenzel L, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354(1):34–43. doi:10.1056/nejmoa052985
186. Gibson MK, Li Y, Murphy B, et al. Randomized Phase III evaluation of cisplatin plus fluorouracil versus cisplatin plus paclitaxel in advanced head and neck cancer (E1395): an Intergroup Trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2005;23(15):3562–3567. doi:10.1200/JCO.2005.01.057
187. Ilson DH, Forastiere A, Arquette M, et al. A Phase II trial of paclitaxel and cisplatin in patients with advanced carcinoma of the esophagus. Cancer J. 2000;6(5):316–323.
188. Stordal B, Davey R, Systematic A. Review of genes involved in the inverse resistance relationship between cisplatin and paclitaxel chemotherapy: role of BRCA1. Curr Cancer Drug Targets. 2009;9(3):354–365. doi:10.2174/156800909788166592
189. Taguchi T. Clinical application of biochemical modulation in cancer chemotherapy: biochemical modulation for 5-FU. Oncol. 1997;54(1):12–18. doi:10.1159/000227740
190. Hamada C, Tanaka F, Ohta M, et al. Meta-analysis of postoperative adjuvant chemotherapy with tegafur-uracil in non-small-cell lung cancer. J Clin Oncol. 2005;23(22):4999–5006. doi:10.1200/JCO.2005.09.017
191. Jeen YT, Yoon SY, Shin SW, et al. Phase II trial of epirubicin, cisplatin, oral uracil and tegafur, and leucovorin in patients with advanced gastric carcinoma. Cancer. 2001;91(12):2288–2293. doi:10.1002/1097-0142(20010615)91:12<2288::AID-CNCR1260>3.0.CO;2-8
192. Kubota H, Fukuta K, Yamada K, et al. Feasibility of metronomic chemotherapy with tegafur-uracil, cisplatin, and dexamethasone for docetaxel-refractory prostate cancer. J Rural Med. 2017;12(2):112–119. doi:10.2185/jrm.2938
193. Tsuchiya A, Imai M, Kamimura H, et al. Successful treatment of multiple lung metastases of hepatocellular carcinoma by combined chemotherapy with docetaxel, cisplatin and tegafur/uracil. World J Gastroenterol. 2009;15(14):1779. doi:10.3748/wjg.15.1779
194. Suga S, Iwase H, Shimada M, et al. Neoadjuvant chemotherapy in scirrhous cancer of the stomach using uracil and tegafur and cisplatin. Intern Med. 1996;35(12):930–936. doi:10.2169/internalmedicine.35.930
195. Ohtsu A, Shimada Y, Shirao K, et al. Randomized phase III trial of fluorouracil alone versus fluorouracil plus cisplatin versus uracil and tegafur plus mitomycin in patients with unresectable, advanced gastric cancer: the Japan clinical oncology group study (JCOG9205). J Clin Oncol. 2003;21(1):54–59. doi:10.1200/JCO.2003.04.130
196. Carvalho C, Santos R, Cardoso S, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16(25):3267–3285. doi:10.2174/092986709788803312
197. Tyagi AK, Agarwal C, Chan DCF, Agarwal R. Synergistic anti-cancer effects of silibinin with conventional cytotoxic agents doxorubicin, cisplatin and carboplatin against human breast carcinoma MCF-7 and MDA-MB468 cells. Oncol Rep. 2004. doi:10.3892/or.11.2.493
198. Park SH, Lee Y, Han SH, et al. Systemic chemotherapy with doxorubicin, cisplatin and capecitabine for metastatic hepatocellular carcinoma. BMC Cancer. 2006;6(1). doi:10.1186/1471-2407-6-3
199. Randall ME, Filiaci VL, Muss H, et al. Randomized phase III trial of whole-abdominal irradiation versus doxorubicin and cisplatin chemotherapy in advanced endometrial carcinoma: a gynecologic oncology group study. J Clin Oncol. 2006;24(1):36–44. doi:10.1200/JCO.2004.00.7617
200. Pappo A. Cisplatin versus cisplatin plus doxorubicin for standard-risk hepatoblastoma. Chin J Clin Oncol. 2010. doi:10.1056/nejmoa0810613
201. Noble S, Goa KL. Gemcitabine. A review of its pharmacology and clinical potential in non-small cell lung cancer and pancreatic cancer. Drugs. 1997. doi:10.2165/00003495-199754030-00009
202. Aapro MS, Martin C, Hatty S. Gemcitabine - A safety review. Anticancer Drugs. 1998;9(3):191–202. doi:10.1097/00001813-199803000-00001
203. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273–1281. doi:10.1056/nejmoa0908721
204. Peters GJ, Bergman AM, Ruiz Van Haperen VWT, Veerman G, Kuiper CM, Braakhuis BJM. Interaction between cisplatin and gemcitabine in vitro and in vivo. Semin Oncol. 1995.
205. Ma Y, Trump DL, Johnson CS. Vitamin D in combination cancer treatment. J Cancer. 2010;101–107. doi:10.7150/jca.1.101
206. Sutton ALM, MacDonald PN. Vitamin D: more than a “bone-a-fide” hormone. Mol Endocrinol. 2003;17(5):777–791. doi:10.1210/me.2002-0363
207. Brown AJ, Slatopolsky E. Vitamin D analogs: therapeutic applications and mechanisms for selectivity. Mol Aspects Med. 2008;29(6):433–452. doi:10.1016/j.mam.2008.04.001
208. Cho YL, Deppe G, Malviya VK, Malone JM, Saunders DE. Combined effects of 1,25-dihydroxyvitamin D3 and platinum drugs on the growth of MCF-7 cells. Cancer Res. 1991;51(11):2848–2853.
209. Huang Z, Zhang Y, Li H, et al. Vitamin D promotes the cisplatin sensitivity of oral squamous cell carcinoma by inhibiting LCN2-modulated NF-κB pathway activation through RPS3. Cell Death Dis. 2019;10(12). doi:10.1038/s41419-019-2177-x
210. Gan X, Chen B, Shen Z, et al. High GPX1 expression promotes esophageal squamous cell carcinoma invasion, migration, proliferation and cisplatin-resistance but can be reduced by vitamin D. Int J Clin Exp Med. 2014.
211. Miao T, Wang J, Zeng Y, Liu G, Chen X. Polysaccharide-based controlled release systems for therapeutics delivery and tissue engineering: from bench to bedside. Adv Sci. 2018. doi:10.1002/advs.201700513
212. Anselmo AC, Mitragotri S. Nanoparticles in the clinic. Bioeng Transl Med. 2016;1(1):10–29. doi:10.1002/btm2.10003
213. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986.
214. Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–3756.
215. Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95(8):4607–4612. doi:10.1073/pnas.95.8.4607
216. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi:10.1038/nnano.2007.387
217. Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. doi:10.1038/nrd1632
218. Domínguez-Ríos R, Sánchez-Ramírez DR, Ruiz-Saray K, et al. Cisplatin-loaded PLGA nanoparticles for HER2 targeted ovarian cancer therapy. Colloids Surfaces B Biointerfaces. 2019. doi:10.1016/j.colsurfb.2019.03.011
219. Boulikas T. Clinical overview on Lipoplatin™: a successful liposomal formulation of cisplatin. Expert Opin Investig Drugs. 2009;18(8):1197–1218. doi:10.1517/13543780903114168
220. Boulikas T, Stathopoulos GP, Volakakis N, Vougiouka M. Systemic lipoplatin infusion results in preferential tumor uptake in human studies. Anticancer Res. 2005;25(4):3031–3039.
221. Devarajan P, Tarabishi R, Mishra J, et al. Low renal toxicity of lipoplatin compared to cisplatin in animals. Anticancer Res. 2004;24(4):2193–2200.
222. Fedier A, Poyet C, Perucchini D, Boulikas T, Fink D. MLH1-deficient tumor cells are resistant to lipoplatin, but retain sensitivity to lipoxal. Anticancer Drugs. 2006;17(3):315–323. doi:10.1097/00001813-200603000-00010
223. Lan X, She J, Lin DA, et al. Microneedle-mediated delivery of lipid-coated cisplatin nanoparticles for efficient and safe cancer therapy. ACS Appl Mater Interfaces. 2018. doi:10.1021/acsami.8b12926
224. Guo S, Wang Y, Miao L, et al. Lipid-coated cisplatin nanoparticles induce neighboring effect and exhibit enhanced anticancer efficacy. ACS Nano. 2013;7(11):9896–9904. doi:10.1021/nn403606m
225. Kolishetti N, Dhar S, Valencia PM, et al. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci U S A. 2010;107(42):17939–17944. doi:10.1073/pnas.1011368107
226. Wang Y, Qian J, Yang M, et al. Doxorubicin/cisplatin co-loaded hyaluronic acid/chitosan-based nanoparticles for in vitro synergistic combination chemotherapy of breast cancer. Carbohydr Polym. 2019;225:115206. doi:10.1016/j.carbpol.2019.115206
227. Lee SM, O’Halloran TV, Nguyen ST. Polymer-caged nanobins for synergistic cisplatin-doxorubicin combination chemotherapy. J Am Chem Soc. 2010. doi:10.1021/ja107333g
228. Wu H, Jin H, Wang C, et al. Synergistic cisplatin/doxorubicin combination chemotherapy for multidrug-resistant cancer via polymeric nanogels targeting delivery. ACS Appl Mater Interfaces. 2017. doi:10.1021/acsami.6b16844
229. Guo XL, Kang XX, Wang YQ, et al. Co-delivery of cisplatin and doxorubicin by covalently conjugating with polyamidoamine dendrimer for enhanced synergistic cancer therapy. Acta Biomater. 2019. doi:10.1016/j.actbio.2018.12.007
230. Zhou D, Cong Y, Qi Y, et al. Overcoming tumor resistance to cisplatin through micelle-mediated combination chemotherapy. Biomater Sci. 2015;3(1):182–191. doi:10.1039/c4bm00305e
231. Vivero-Escoto JL, Elnagheeb M. Mesoporous silica nanoparticles loaded with cisplatin and phthalocyanine for combination chemotherapy and photodynamic therapy in vitro. Nanomaterials. 2015;5(4):2302–2316. doi:10.3390/nano5042302
232. Shanmugam V, Chien YH, Cheng YS, et al. Oligonucleotides-assembled au nanorod-assisted cancer photothermal ablation and combination chemotherapy with targeted dual-drug delivery of doxorubicin and cisplatin prodrug. ACS Appl Mater Interfaces. 2014;6(6):4382–4393. doi:10.1021/am5000905
233. Dreyfuss AI, Clark JR, Fallon BG, Posner MR, Norris CM, Miller D. Cyclophosphamide, doxorubicin, and cisplatin combination chemotherapy for advanced carcinomas of salivary gland origin. Cancer. 1987;60(12):2869–2872. doi:10.1002/1097-0142(19871215)60:12<2869::AID-CNCR2820601203>3.0.CO;2-Y
234. Sohn JW, Lee SY, Lee SJ, et al. MDR1 polymorphisms predict the response to etoposide-cisplatin combination chemotherapy in small cell lung cancer. Jpn J Clin Oncol. 2006;36(3):137–141. doi:10.1093/jjco/hyi231
235. Segawa Y, Kiura K, Takigawa N, et al. Phase III trial comparing docetaxel and cisplatin combination chemotherapy with mitomycin, vindesine, and cisplatin combination chemotherapy with concurrent thoracic radiotherapy in locally advanced non-small-cell lung cancer: OLCSG 0007. J Clin Oncol. 2010. doi:10.1200/JCO.2009.24.7577
236. Giaccone G, Ardizzoni A, Kirkpatrick A, Clerico M, Sahmoud T, Van Zandwijk N. Cisplatin and etoposide combination chemotherapy for locally advanced or metastatic thymoma: a Phase II Study of the European Organization for Research and Treatment of Cancer Lung Cancer Cooperative Group. J Clin Oncol. 1996;14(3):814–820. doi:10.1200/JCO.1996.14.3.814
237. Rose PG, Ali S, Watkins E, et al. Long-term follow-up of a randomized trial comparing concurrent single agent cisplatin, cisplatin-based combination chemotherapy, or hydroxyurea during pelvic irradiation for locally advanced cervical cancer: a gynecologic oncology group study. J Clin Oncol. 2007. doi:10.1200/JCO.2006.09.4532
238. Sato Y, Takayama T, Sagawa T, et al. Phase II study of S-1, docetaxel and cisplatin combination chemotherapy in patients with unresectable metastatic gastric cancer. Cancer Chemother Pharmacol. 2010;66(4):721–728. doi:10.1007/s00280-009-1215-2
239. Duan J, Yang Z, Liu D, Zhang Z, Shi Y. Clinical efficacy of bevacizumab combined with gemcitabine and cisplatin combination chemotherapy in the treatment of advanced non-small cell lung cancer. J BUON. 2018;23(5):1402–1406.
240. Niho S, Kenmotsu H, Sekine I, et al. Combination chemotherapy with irinotecan and cisplatin for large-cell neuroendocrine carcinoma of the lung: a multicenter phase II study. J Thorac Oncol. 2013;8(7):980–984. doi:10.1097/JTO.0b013e31828f6989
241. Kim M, Keam B, Kim TM, et al. Phase II study of irinotecan and cisplatin combination chemotherapy in metastatic, unresectable esophageal cancer. Cancer Res Treat. 2017. doi:10.4143/crt.2016.121
242. Boku N, Ohtsu A, Shimada Y, et al. Phase ii study of a combination of irinotecan and cisplatin against metastatic gastric cancer. J Clin Oncol. 1999;17(1):319. doi:10.1200/jco.1999.17.1.319
243. Dexeus FH, Logothetis CJ, Sella A, et al. Combination chemotherapy with methotrexate, bleomycin and cisplatin for advanced squamous cell carcinoma of the male genital tract. J Urol. 1991;146(5):1284–1287. doi:10.1016/S0022-5347(17)38069-2
244. Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Prospective randomized trial of the treatment of patients with metastatic melanoma using chemotherapy with cisplatin, dacarbazine, and tamoxifen alone or in combination with interleukin-2 and interferon alfa-2b. J Clin Oncol. 1999;17(3):968. doi:10.1200/jco.1999.17.3.968
245. Fujita A, Fukuoka S, Takabatake H, Tagaki S, Sekine K. Combination chemotherapy of cisplatin, ifosfamide, and irinotecan with rhG-CSF support in patients with brain metastases from non-small cell lung cancer. Oncology. 2000. doi:10.1159/000012185
246. Cartei G, Cartei F, Cantone A, et al. Cisplatin-cyclophosphamide-mitomycin combination chemotherapy with supportive care versus supportive care alone for treatment of metastatic non-small-cell lung cancer. J Natl Cancer Inst. 1993;85(10):794–800. doi:10.1093/jnci/85.10.794
247. Yun J, Lee J, Park SH, et al. A randomised phase II study of combination chemotherapy with epirubicin, cisplatin and capecitabine (ECX) or cisplatin and capecitabine (CX) in advanced gastric cancer. Eur J Cancer. 2010;46(5):885–891. doi:10.1016/j.ejca.2009.12.015
248. Kang H, Kim TJ, Kim WY, et al. Outcome and reproductive function after cumulative high-dose combination chemotherapy with bleomycin, etoposide and cisplatin (BEP) for patients with ovarian endodermal sinus tumor. Gynecol Oncol. 2008;111(1):106–110. doi:10.1016/j.ygyno.2008.05.033
249. Lee JE, Bae SH, Choi JY, Yoon SK, You YK, Lee MA. Epirubicin, Cisplatin, 5-FU combination chemotherapy in sorafenib-refractory metastatic hepatocellular carcinoma. World J Gastroenterol. 2014;20(1):235–241. doi:10.3748/wjg.v20.i1.235
250. Peters WA, Rivkin SE, Smith MR, Tesh DE. Cisplatin and Adriamycin combination chemotherapy for uterine stromal sarcomas and mixed mesodermal tumors. Gynecol Oncol. 1989. doi:10.1016/0090-8258(89)90166-2
251. Ohnuma H, Sato Y, Hirakawa M, et al. Docetaxel, cisplatin and S-1 (DCS) combination chemotherapy for gastric cancer patients with peritoneal metastasis: a retrospective study. Cancer Chemother Pharmacol. 2018;81(3):539–548. doi:10.1007/s00280-018-3523-x
252. Liu L, Peng Z, Huang H, Xu Z, Wei X. Luteolin and apigenin activate the Oct-4/Sox2 signal via NFATc1 in human periodontal ligament cells. Cell Biol Int. 2016;40(10):1094–1106. doi:10.1002/cbin.10648
253. Xia C, Bai X, Hou X, et al. Cryptotanshinone reverses cisplatin resistance of human lung carcinoma A549 cells through down-regulating Nrf2 pathway. Cell Physiol Biochem. 2015;37(2):816–824. doi:10.1159/000430398
254. Sharma R, Gatchie L, Williams IS, et al. Glycyrrhiza glabra extract and quercetin reverses cisplatin resistance in triple-negative MDA-MB-468 breast cancer cells via inhibition of cytochrome P450 1B1 enzyme. Bioorg Med Chem Lett. 2017;27(24):5400–5403. doi:10.1016/j.bmcl.2017.11.013
255. Donnelly A, Blagg B. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem. 2008;15(26):2702–2717. doi:10.2174/092986708786242895
256. Arafa ESA, Zhu Q, Barakat BM, et al. Tangeretin sensitizes cisplatin-resistant human ovarian cancer cells through downregulation of phosphoinositide 3-kinase/Akt signaling pathway. Cancer Res. 2009;69(23):8910–8917. doi:10.1158/0008-5472.CAN-09-1543
257. Wang X, Zhang F, Wu XR. Inhibition of pyruvate kinase M2 markedly reduces chemoresistance of advanced bladder cancer to cisplatin. Sci Rep. 2017. doi:10.1038/srep45983
258. Jiang XY, Zhu XS, Xu HY, et al. Diallyl trisulfide suppresses tumor growth through the attenuation of Nrf2/Akt and activation of p38/JNK and potentiates cisplatin efficacy in gastric cancer treatment. Acta Pharmacol Sin. 2017;38(7):1048–1058. doi:10.1038/aps.2016.176
259. Chen P, Li J, Jiang HG, Lan T, Chen YC. Curcumin reverses cisplatin resistance in cisplatin-resistant lung cancer cells by inhibiting FA/BRCA pathway. Tumor Biol. 2015. doi:10.1007/s13277-014-2996-4
260. Leekha A, Gurjar BS, Tyagi A, Rizvi MA, Verma AK. Vitamin C in synergism with cisplatin induces cell death in cervical cancer cells through altered redox cycling and p53 upregulation. J Cancer Res Clin Oncol. 2016;142(12):2503–2514. doi:10.1007/s00432-016-2235-z
261. Hsieh MJ, Wang CW, Lin JT, et al. Celastrol, a plant-derived triterpene, induces cisplatin-resistance nasopharyngeal carcinoma cancer cell apoptosis through ERK1/2 and p38 MAPK signaling pathway. Phytomedicine. 2019;58:152805. doi:10.1016/j.phymed.2018.12.028
262. Zhang Y, Chen S, Wei C, et al. Dietary compound proanthocyanidins from Chinese bayberry (Myrica rubra Sieb. et Zucc.) leaves inhibit angiogenesis and regulate cell cycle of cisplatin-resistant ovarian cancer cells via targeting Akt pathway. J Funct Foods. 2018. doi:10.1016/j.jff.2017.11.045
263. Sherif IO, Al-Gayyar MMH. Oleuropein potentiates anti-tumor activity of cisplatin against HepG2 through affecting proNGF/NGF balance. Life Sci. 2018;198:87–93. doi:10.1016/j.lfs.2018.02.027
264. Wu Y, Zhu Y, Li S, et al. Terrein performs antitumor functions on esophageal cancer cells by inhibiting cell proliferation and synergistic interaction with cisplatin. Oncol Lett. 2017. doi:10.3892/ol.2017.5758
265. Peng XH, Wang Y, Huang D, et al. Targeted delivery of cisplatin to lung cancer using ScFvEGFR-heparin- cisplatin nanoparticles. ACS Nano. 2011;5(12):9480–9493. doi:10.1021/nn202410f
266. Gryparis EC, Hatziapostolou M, Papadimitriou E, Avgoustakis K. Anticancer activity of cisplatin-loaded PLGA-mPEG nanoparticles on LNCaP prostate cancer cells. Eur J Pharm Biopharm. 2007;67(1):1–8. doi:10.1016/j.ejpb.2006.12.017
267. Yang T, Zhao P, Rong Z, et al. Anti-tumor efficiency of lipid-coated cisplatin nanoparticles co-loaded with microRNA-375. Theranostics. 2016;6(1):142–154. doi:10.7150/thno.13130
268. Zhang J, Miao L, Guo S, et al. Synergistic anti-tumor effects of combined gemcitabine and cisplatin nanoparticles in a stroma-rich bladder carcinoma model. J Control Release. 2014;182:90–96. doi:10.1016/j.jconrel.2014.03.016
269. Kates M, Date A, Yoshida T, et al. Preclinical evaluation of intravesical cisplatin nanoparticles for non–muscle-invasive bladder cancer. Clin Cancer Res. 2017;23(21):6592–6601. doi:10.1158/1078-0432.CCR-17-1082
270. Hwang PA, Lin XZ, Kuo KL, Hsu FY. Fabrication and cytotoxicity of fucoidan-cisplatin nanoparticles for macrophage and tumor cells. Materials (Basel). 2017;10(3):291. doi:10.3390/ma10030291
271. Xu Y, Han X, Li Y, et al. Sulforaphane mediates glutathione depletion via polymeric nanoparticles to restore cisplatin chemosensitivity. ACS Nano. 2019;13(11):13445–13455. doi:10.1021/acsnano.9b07032
272. Chiani M, Milani AT, Nemati M, et al. Anticancer effect of cisplatin-loaded poly (Butylcyanoacrylate) nanoparticles on A172 brain cancer cells line. Asian Pac J Cancer Prev. 2019. doi:10.31557/APJCP.2019.20.1.303
273. Yu M, Zhang C, Tang Z, Tang X, Xu H. Intratumoral injection of gels containing losartan microspheres and (PLG-g-mPEG)-cisplatin nanoparticles improves drug penetration, retention and anti-tumor activity. Cancer Lett. 2019. doi:10.1016/j.canlet.2018.11.011
274. Ghaferi M, Amari S, Mohrir BV, Raza A, Shahmabadi HE, Alavi SE. Preparation, characterization, and evaluation of cisplatin-loaded polybutylcyanoacrylate nanoparticles with improved in vitro and in vivo anticancer activities. Pharmaceuticals. 2020;13(3):44. doi:10.3390/ph13030044
275. Khan MM, Madni A, Torchilin V, et al. Lipid-chitosan hybrid nanoparticles for controlled delivery of cisplatin. Drug Deliv. 2019;26(1):765–772. doi:10.1080/10717544.2019.1642420
276. Mirrahimi M, Abed Z, Beik J, et al. A thermo-responsive alginate nanogel platform co-loaded with gold nanoparticles and cisplatin for combined cancer chemo-photothermal therapy. Pharmacol Res. 2019;143:178–185. doi:10.1016/j.phrs.2019.01.005
277. Zhang C, Nance EA, Mastorakos P, et al. Convection enhanced delivery of cisplatin-loaded brain penetrating nanoparticles cures malignant glioma in rats. J Control Release. 2017. doi:10.1016/j.jconrel.2017.03.007
278. Ling X, Chen X, Riddell IA, et al. Glutathione-Scavenging Poly(disulfide amide) nanoparticles for the effective delivery of Pt(IV) prodrugs and reversal of cisplatin resistance. Nano Lett. 2018;18(7):4618–4625. doi:10.1021/acs.nanolett.8b01924
279. Aldawsari HM, Singh S. Rapid microwave-assisted cisplatin-loaded solid lipid nanoparticles: synthesis, characterization and anticancer study. Nanomaterials. 2020;10(3):510. doi:10.3390/nano10030510
280. Mirrahimi M, Khateri M, Beik J, et al. Enhancement of chemoradiation by co-incorporation of gold nanoparticles and cisplatin into alginate hydrogel. J Biomed Mater Res Part B Appl Biomater. 2019;107(8):2658–2663. doi:10.1002/jbm.b.34356
281. Zhang Y, Fu X, Jia J, et al. Glioblastoma therapy using codelivery of cisplatin and glutathione peroxidase targeting siRNA from iron oxide nanoparticles. ACS Appl Mater Interfaces. 2020. doi:10.1021/acsami.0c12042
282. Ma X, Khan MW, Zhao P, et al. Synergism of cisplatin-oleanolic acid co-loaded calcium carbonate nanoparticles on hepatocellular carcinoma cells for enhanced apoptosis and reduced hepatotoxicity. Int J Nanomedicine. 2019. doi:10.2147/IJN.S196651
283. Davidi ES, Dreifuss T, Motiei M, et al. Cisplatin-conjugated gold nanoparticles as a theranostic agent for head and neck cancer. Head Neck. 2018. doi:10.1002/hed.24935
284. Toro-Cordova A, Flores-Cruz M, Santoyo-Salazar J, et al. Liposomes loaded with cisplatin and magnetic nanoparticles: physicochemical characterization, pharmacokinetics, and in-vitro efficacy. Molecules. 2018;23(9):2272. doi:10.3390/molecules23092272
285. Vandghanooni S, Eskandani M, Barar J, Omidi Y. AS1411 aptamer-decorated cisplatin-loaded poly(lactic-co-glycolic acid) nanoparticles for targeted therapy of miR-21-inhibited ovarian cancer cells. Nanomedicine. 2018;13(21):2729–2758. doi:10.2217/nnm-2018-0205
286. Nan Y. Lung carcinoma therapy using epidermal growth factor receptor-targeted lipid polymeric nanoparticles co-loaded with cisplatin and doxorubicin. Oncol Rep. 2019. doi:10.3892/or.2019.7323
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.