")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Adjuvant therapies for Parkinson’s disease: critical evaluation of safinamide
Received 16 September 2015
Accepted for publication 20 November 2015
Published 5 February 2016 Volume 2016:10 Pages 609—618
DOI https://doi.org/10.2147/DDDT.S77749
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Video abstract presented by Fabrizio Stocchi
Views: 1739
Fabrizio Stocchi, Margherita Torti
Institute for Research and Medical Care, IRCCS San Raffaele Pisana, Rome, Italy
Abstract: Safinamide (SAF) is a new drug developed for the treatment of Parkinson’s disease (PD). It is a benzylamino derivative with multiple mechanisms of action and antiparkinsonian, anticonvulsant, and neuroprotective properties. SAF inhibits monoamine oxidase B and dopamine reuptake and glutamate release, blocks voltage-dependent sodium channels, and modulates calcium channels. Although the antiparkinsonian effect can be ascribed in part to the inhibition of the monoamine oxidase B, which is complete at 50 mg, the enhanced benefit seen at the 100 mg dose is probably due to nondopaminergic mechanisms. SAF will represent an important option for patients with both early and advanced PD. In early PD patients, the addition of SAF to dopamine agonists may be an effective treatment strategy to improve motor function, prolong the use of dopamine agonists, and/or delay the introduction of levodopa. In advanced parkinsonian patients, SAF has been demonstrated to significantly increase on time with no, or nontroublesome dyskinesias. All studies performed have demonstrated its efficacy in benefiting both short-term and long-term quality-of-life outcomes in both early and advanced PD patients. SAF has been investigated in long-term (24 months), double-blind, placebo-controlled studies, where it showed a very good safety profile. SAF has not been studied in de novo PD patients, and its potential positive effect on dyskinesia deserves further dedicated studies.
Keywords: safinamide, Parkinson’s disease, add-on, dopamine agonist, L-dopa, motor fluctuations, dyskinesia, a-aminoamide
Aims
The objective of this review is to analyze the pharmacological properties that make SAF an innovative drug for the treatment of Parkinson’s disease (PD) and its role in the schedule of treatment of early and advanced parkinsonian patients. This review also focuses on clinical trials performed to evaluate the efficacy and safety of SAF as an add-on therapy in patients with Parkinson’s disease.
Research methods
A literature search was performed using Medline: the search terms used were “safinamide, Parkinson’s disease, add-on, dopamine agonist (DA), a-aminoamide.” Searches were supplemented by manually reviewing bibliographies of all selected articles.
The database includes 26 articles: six regarding biochemical properties and mechanism of action, one regarding metabolism in healthy male volunteers, 12 reviewing the impact of SAF in Parkinson’s disease, five regarding clinical trials evaluating the efficacy and safety in early and advanced Parkinson’s disease (Table 1), and two on the effect of SAF on pressure response to tyramine.
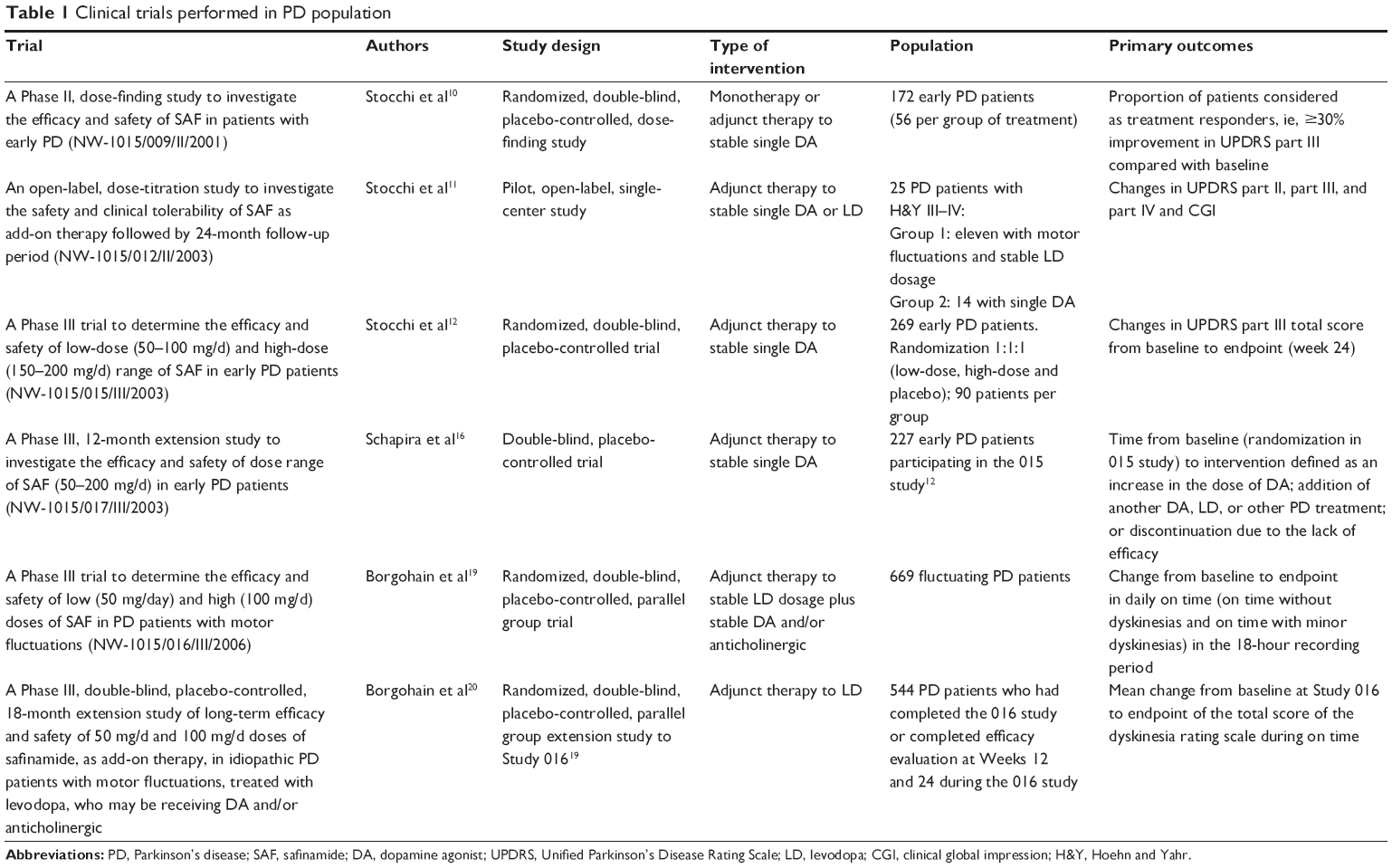 | Table 1 Clinical trials performed in PD population |
Introduction
Safinamide (SAF) is being developed as a treatment for PD. It combines several mechanisms of action that can be beneficial for this disease, including potentiation of dopaminergic neurotransmission via monoamine oxidase B (MAO-B), dopamine reuptake inhibition, and modulation of glutamatergic neurotransmission with reduction in oxidative damage, which may provide neuroprotective action and improvement of cognitive function.
Mechanism of action
SAF ((S)-(+)-2-(4-(3-fluorobenzyloxy)benzylamino)propanamide) is a benzylamino derivative with multiple mechanisms of action and antiparkinsonian, anticonvulsant, and neuroprotective properties. SAF inhibits MAO-B and dopamine reuptake and glutamate release, blocks voltage-dependent sodium channels, and modulates calcium channels.1,2
MAO is responsible for the inactivation of catecholamine; MAO-A deaminates noradrenaline and serotonin, whereas MAO-B deaminates dopamine and phenylethylamine. In human beings, 80% of dopamine is metabolized by MAO-B and the selective inhibition of MAO-B results in a therapeutic effect in the treatment of PD.3 SAF is a potent, highly selective, and reversible MAO-B inhibitor; in in vitro studies, it has demonstrated 5,000 times greater selectivity for MAO-B than for MAO-A, making the need for dietary restriction unnecessary. Its selectivity for MAO-B is superior to selegiline and rasagiline;4,5 the opposite of these two molecules, inhibition of MAO-B is also completely reversible, making treatment with other drugs possible without any interactions, in case of suspension due to side effects.
Through MAO-B inhibition, SAF, as well as rasagiline and selegiline, can prevent the formation of toxins or free radical products during oxidative processes, as demonstrated in in vivo studies on 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine models of PD.6
It has been demonstrated that SAF inhibits N-type calcium currents concurring at the inhibition of neurotransmitter presynaptic release. In particular, SAF inhibits glutamate release, one of the most relevant excitotoxic inputs to neuronal death. At high K+ concentrations, the release of neurotransmitter is Ca++ mediated; therefore, SAF inhibits the glutamate release by blocking N-type Ca++ mobilization. In the studies performed on rat cortical membranes, SAF has shown high affinity for the sodium channel binding site II: it inhibits the fast sodium currents in a concentration and in a state-dependent manner. A higher number of sodium channels are kept in the inactivated state and are prevented from the activation by SAF. This results in a depression of the high firing activity. Therefore, SAF has a normalizing effect on sodium channel abnormal activity, leaving physiologic activity unaffected.6
Pharmacokinetics and pharmacodynamics
SAF is water soluble and rapidly absorbed after an oral dose, reaching a peak plasma concentration in 1.8–2.8 hours. It has an elimination half-life of 21–24 hours, permitting a once-a-day administration. Cmax increases linearly in a dose-proportional manner, demonstrating a first-order kinetic. Food intake (high fat meal) delays the rate of absorption of the drug, reaching the Tmax in approximately 5 hours without affecting the extent of absorption.7 To assess the risk of inducing the “cheese effect,” the effect of SAF and placebo on the pressor response to tyramine was investigated in a group of healthy male volunteers. The study was an open-label, single-dose, placebo-controlled trial with the two treatments in sequence. An increase of 30 mmHg systolic blood pressure was obtained by intravenous tyramine administered by 0.5 mg incremental boluses injected at 15-minute intervals. The amount of tyramine necessary to achieve such a blood pressure increase was the same after the SAF 2 mg/kg oral load compared with placebo.8
Another randomized, double-blind, placebo-, comparator- (selegiline 10 mg/d), and positive (phenelzine 30 mg/d)-controlled study investigated the pressor response to oral tyramine under fasting conditions after the administration of SAF at therapeutic (100 mg/d) and supratherapeutic (350 mg/d) dosing regimens in healthy volunteers for the purpose of assessing the need for dietary restrictions. SAF induced a mild increase in tyramine sensitivity factor, defined as the ratio of Tyr30 at screening to Tyr30 under the treatment. However, the effect on each of the doses was numerically lower than that of the comparators (geometric mean tyramine sensitivity factors: placebo, 1.52; SAF 100 mg, 2.15; SAF 350 mg, 2.74; selegiline, 3.12; phenelzine, 9.98). This study confirmed that SAF is a highly selective MAO-B inhibitor, even at supratherapeutic doses, and suggests that it can be administered without tyramine-related dietary restrictions.9
As demonstrated by studies performed on rats, mice, and monkeys, brain levels of SAF are higher than the corresponding plasma concentrations.6 SAF is largely biotransformed as shown by the very low dosage of unchanged SAF found in urine and feces; the plasma protein binding is 92%, and only a small proportion is excreted unchanged. The apparent oral volume of distribution of the unchanged drug is 150 L: this is indicative of extensive extravascular distribution due to the high lipophilicity of its base. There is no significant accumulation at the steady state.7
Clinical studies in early PD
A randomized, double-blind, placebo-controlled, multicenter, dose-finding trial was performed in 196 outpatients with idiopathic PD. A total of 172 subjects were randomized to receive 0.5 mg/kg SAF, 1.0 mg/kg SAF, or placebo as monotherapy or as adjunct therapy to a single DA. The primary objective was to determine the proportion of patients considered as responders, ie, obtaining ≥30% improvement at the Unified Parkinson’s Disease Rating Scale (UPDRS) part III compared to baseline. The primary efficacy variable showed a statistically significant difference between SAF 1.0 mg/kg and placebo (P=0.018) but not between SAF 0.5 mg/kg and placebo. An unexpected finding of this study was the superior benefit of SAF when added to a single DA, decreasing the UPDRS part III scores by an average of 27.8%.10
In a single-center, open, pilot trial, high doses of SAF (100 mg, 150 mg, and 200 mg daily) were tested and administrated to 13 parkinsonian patients as an add-on therapy at a stable dose of a DA. The initial dose of 100 mg/d was gradually increased in a 2-week interval to 150 and then 200 mg/d. A significant and progressive improvement of motor performance evaluated by UPDRS part III (4.2 points, P<0.001) for more than an 8-week period was observed.11
Study 01512 was a 24-week, randomized, double-blind, placebo-controlled, parallel group trial. Inclusion criteria were the diagnosis of idiopathic PD with <5 years of history, Hoehn and Yahr (H&Y) stages I–II, age between 30 years and 80 years, and a stable dose of a DA for at least 4 weeks before screening. Exclusion criteria were diagnosis or recent history of substance abuse, end-of-dose wearing off, on/off phenomena, disabling dyskinesias, psychosis or depression, severe postural hypotension, use of other PD medication with the exception of a single DA, or concomitant use of MAO-B inhibitor. Owing to the SAF selectivity for MAO-B, no restriction diet was asked of participants.
The study incorporated a run-in period, after which patients were randomized by a computerized system to receive 100 mg SAF, 200 mg SAF or placebo with a 1:1:1 ratio as add-on therapy to a single DA. A minimization procedure was used to ensure that the range of H&Y was equally distributed in all the three groups. A 2-week titration phase was scheduled to reach the target dose of 100 mg and 200 mg (divided into four doses of 50 mg SAF/placebo).
The dose of the DA had to remain stable for the duration of the study. A dose increase, or the addition of another PD medication, allowed for patients at maximum dosage had demonstrated a worsening of PD symptoms. All efficacy and safety measures needed to be performed before this intervention.
The primary efficacy measure was the change in UPDRS part III from baseline to endpoint (week 24). Secondary efficacy measures included responder analysis, ie, the proportion of patients showing improvement on the Clinical Global Impression – Change (CGI-C) at endpoint and the proportion of patients showing at least 30% improvement at UPDRS part III from baseline to endpoint, with no worsening at UPDRS part II and part IV total scores; the mean change was from baseline of UPDRS part II, CGI-C, and Clinical Global Impression – Severity of Illness (CGI-S). Tertiary efficacy variables were the Hamilton Rating Scale for Depression (HAM-D), UPDRS part I and part IV, and Mini Mental State Examination.
A total of 270 patients were randomized into the study, and the majority of them were able to reach and maintain the target dose of 100 mg/d or 200 mg/d. The primary endpoint showed no significant difference in the comparison between SAF 200 mg and placebo. The mean change in UPDRS part III from baseline to week 24 resulted in statistical significance in the group treated with 100 mg/d versus placebo with a value of −6.0 versus -3.6 (P=0.0419). The mean CGI-C score from the baseline score at week 24 for the SAF 200 mg and 100 mg groups was 3.1 and 3.4, respectively, indicating an improvement in the CGI score for both active treatments versus placebo (P=0.0325 and P=0.0293). The CGI-C score from the baseline score and the CGI-S mean change score at the endpoint were not statistically different between the groups. The mean change from baseline to week 24 in UPDRS part II showed an improvement in the activities of daily living (ADL) for the SAF 100 mg group (-2.2; P=0.0248) but not those for the SAF 200 mg group versus placebo. None of the tertiary efficacy measures indicated a significant benefit from the treatment with SAF.
For the measurements of plasma concentration of SAF and its metabolites, blood samples were taken at the baseline visit before the administration of first study medication, at 5 hours after the administration, and then at each visit. Samples were analyzed with liquid chromatography using tandem mass spectrometry. Plasma concentrations in both the SAF groups were consistent with the dose level and showed a linear dose exposure.
In 26% of the blood samples taken in the placebo group, the presence of SAF and its metabolites was detected, meaning that at some point during study visits, patients in the placebo group were exposed to SAF. An audit revealed that two of 51 bulk placebo bottles contained both placebo and SAF capsules.
The 015 study has demonstrated a significant improvement in the UPDRS part III, UPDRS part II, and CGI-C total score in the SAF 100 mg group; the percentage of responders was higher in both the SAF groups compared with placebo. The reason why the treatment with 200 mg failed to reach the significance is unclear. The percentage of patient discontinuation was higher in this group. The causes were adverse event (AE) and withdrawal of consent, and some of the patients who withdrew consent may have done so for lack of efficacy. This higher rate of discontinuation may have masked the clinical benefit. The UPDRS improvement detected in the placebo group may have contributed to the lack of efficacy reported in the 200 mg group, and the presence of SAF in the plasma sample of placebo group patients must be considered when analyzing the data from this study.12
A subset of 151 PD patients performed a computerized Cogtest battery as a part of this study: the test included auditory number sequencing test, spatial working memory test, strategic target detection test (STDT), tapping speed test, simple reaction time test, and choice reaction time test. In PD, executive functioning and working memory are most often impaired, but widespread frontal-lobe cognitive dysfunction with mild-to-moderate impairment in attention and memory and slight visuospatial alterations are present as well.13,14 Cognitive assessments of working memory, executive function, and simple motor speed were administered at baseline and again after 12 weeks and 24 weeks of treatment.
Improvement in cognition was seen in tests of executive function and working memory, deficits that are central in PD.15
Study 01716 was a 12-month, randomized, double-blind, placebo-controlled extension study of the 015 study. The 017 study recruited all subjects who had completed the core study or subjects who discontinued from the 015 study but who had completed efficacy evaluation at weeks 12 and 24. The primary efficacy endpoint was the time to intervention from baseline (ie, randomization in the 015 study) defined as an increased dose of DA; the addition of another DA, levodopa (L-dopa), or other PD treatment; or discontinuation due to the lack of efficacy. Secondary endpoints were change in the UPDRS part III scores; response rates to UPDRS part II, CGI-C, and CGI-S; proportion of patients requiring intervention; and H&Y scores. Tertiary endpoints were GRID-HAM-D, UPDRS part IV, UPDRS part I, and Mini Mental State Examination.
A total of 227 subjects were enrolled in the study, and 187 terminated the extension phase. Regarding the primary endpoint, the median time to intervention was 559 days in the combined SAF group and 466 days in the placebo group, with these values failing to represent statistical significance (P=0.3342). There were no statistically significant differences found for any variable, when compared with placebo, at the secondary and tertiary endpoints.
A post hoc analysis was then performed using stratification at two follow-up periods (0–240 days and 240–540 days) and then analyzing the two SAF groups separately for UPDRS part III and response rates. The SAF 100 mg group showed a significantly lower rate of intervention compared with DA monotherapy (25% vs 51%; P<0.05) as well as delay in median time to intervention versus DA by 9 days (P<0.05) in the 240–540 period. A significant improvement in the UPDRS part II and part III was also found, suggesting that SAF can be effective as an add-on therapy to DA.16
The MOTION17 study was a 6-month (24-week), randomized, double-blind, placebo-controlled international Phase III trial to evaluate the efficacy and safety of two fixed doses of SAF (50 mg/d and 100 mg/d). This trial enrolled patients with early idiopathic PD (<5 years of disease) treated with a single DA. A total of 679 patients were randomized to receive 50 mg, 100 mg SAF once daily or placebo. The study’s primary endpoint (change in UPDRS part III from baseline to week 24) was not met even though the analysis of the entire study population (n=679) showed a borderline statistically significant difference between the SAF 100 mg/d group and the placebo group (P=0.073). In the group of patients on monotherapy with a single DA (n=666 patients), SAF 100 mg/d significantly improved UPDRS part III (P=0.0396) and Parkinson’s disease questionnaire (PDQ)-39 compared to placebo. There was a borderline significant improvement in ADL for 100 mg/d measured by UPDRS part II and European quality of life.18
Clinical studies in advanced PD
Study 01619 was a Phase III, multicenter, randomized, double-blind, placebo-controlled, parallel group trial with four phases: a 10-day screening period, a 4-week L-dopa stabilization period, a 24-week treatment period, and an optional 1-week taper period. This trial recruited patients who were aged 30–80 years, were able to complete a diary, had been diagnosed with PD ≥3 years, had H&Y stage I–IV during off state, and had the presence of motor fluctuations with >1.5 hours off a day. Patients with disabling peak dose or biphasic dyskinesias as well as patients with the evidence of dementia, psychiatric complications, or severe concomitant disease were excluded. The primary efficacy measure was the change in mean daily on time with no or non-troublesome dyskinesias as detected by Hauser’s diaries. Secondary efficacy measures included total daily off time, UPDRS part III score in on time, UPDRS part II score during on time, CGI-C and CGI-S scores, dyskinesia rating scale (DRS) scores during on time, and percentage of change in the L-dopa dose. Tertiary efficacy variables were the GRID-HAM-D and PDQ-39 subscales scores. Clinic diaries were completed each day during L-dopa stabilization period and in 5 days preceding each study visit.
A total of 699 patients were randomized and equally divided into the three treatment groups; overall 88% of the enrolled subjects completed the study. At week 24, there were significant differences in the least squares mean change versus placebo in both the SAF 50 mg/d and SAF 100 mg/d groups. For off time, at week 24, least squares mean differences versus placebo were significantly higher in both the SAF 50 mg/d and SAF 100 mg/d groups. UPDRS part III (motor) scores were significantly improved in both the 50 mg/d and 100 mg/d groups compared to placebo. There were also significant improvements in CGI-C, CGI-S, and off time following the morning L-dopa dose in both the SAF groups compared with placebo. There were no significant between-group differences in DRS scores during on time. SAF 100 mg/d improved UPDRS part II (ADL) scores compared with placebo (P=0.006); however, this was not noted for the 50 mg/d group. There were also improvements in the PDQ-39 total score (P=0.0360) and subscale scores for emotional well-being (P=0.0116), communication (P=0.0361), and bodily discomfort (P=0.0159) for SAF 100 mg/d versus placebo. Changes in GRID-HAM-D scores from baseline to week 24 were numerically greater for both the SAF groups versus placebo.19
A total of 544 patients, who did not experience side effects after the completion of the core study, entered an 18-month, double-blind, placebo-controlled, parallel group extension study (Study 018).20 Subjects who had discontinued from Study 016 but had completed efficacy evaluations at weeks 12 and 24 were also enrolled in this trial. The primary endpoint was the mean change from baseline at Study 016 entrance in the DRS total score during on time. Secondary efficacy endpoints were the mean change from baseline to endpoint in diary on time without troublesome dyskinesia, diary responder rates, UPDRS part IV total score and scores of 32–35 (dyskinesia and dystonia) and 32–34, UPDRS part II scores, UPDRS part II response rates, change in L-dopa dose; UPDRS part III scores; CGI-C and CGI-S scores; and the change in individual diary categories. Tertiary efficacy variables were the GRID-HAM-D, PDQ-39 subscales scores, and H&Y. Change in DRS was not significantly different in the SAF versus placebo group; despite this, the SAF 50 mg/d and 100 mg/d groups were able to reduce the mean DRS scores by 31% and 27%, respectively, from baseline, compared with 3% observed in the placebo group. The mean total daily on time without troublesome dyskinesia significantly improved from baseline in Study 016 to week 78 in Study 018 in both the SAF 50 mg/d and 100 mg/d groups (P=0.0031 and P=0.0002, respectively).
An ad hoc analysis performed on moderate-to-severe dyskinetic patients who entered Study 016 (n=242, 36%) showed a statistically significant reduction in the mean DRS total scores (P=0.0317). The exclusion of patients who experienced a reduction in their L-dopa daily dosage showed a continued benefit in the SAF 100 mg group, suggesting that the antidyskinetic effect was independent of the L-dopa dose reduction.
Significant improvements were also reported in off time; on time without dyskinesia; CGI-S; CGI-C (for SAF 50 mg); UPDRS part II, part III, and part IV total scores; PDQ-39; and GRID-HAM-D (SAF 100 mg/d only). Overall, the benefits observed during Study 016 in both the groups treated with SAF remained stable throughout the extension study; during the same period, the mean dose of L-dopa increased in the placebo and the SAF 50 mg group but decreased in the SAF 100 mg group from 579.6±310.0 mg to 556.0±381.9 mg.20
A post hoc analysis of Study 018 was recently performed with the aim to characterize the effects of SAF on dyskinesia. Patients enrolled were stratified on the basis of the absence or presence of dyskinesia (DRS =0 or DRS >0) at baseline and on whether or not the L-dopa dose had been changed over the entire study period of 24 months. The comparison between the SAF-treated patients and placebo group was performed using the Wilcoxon rank-sum test for independent samples. Changes in DRS scores were summarized as decreased (improvement of dyskinesia), unchanged (stable), and increased (worsening of dyskinesia). When analyzing the entire study population (DRS ≥0) who had not changed the L-dopa dosage in the 24-month period, SAF 100 mg was found to have significantly improved DRS scores compared to placebo with P=0.0488 (Figure 1). In the subgroup of patients exhibiting dyskinesia at baseline, improvement was also significant for SAF 100 mg both in subjects with or without L-dopa changes (P=0.0153). The analysis also showed a trend toward significance (P=0.0546) in dyskinetic patients who had not changed their L-dopa dose during the trial. These results confirm that the beneficial effects of SAF on dyskinesia in the 018 study were not entirely dependent on L-dopa dose reduction; in fact, a significant improvement was evidenced in the group of patients with stable L-dopa doses.21
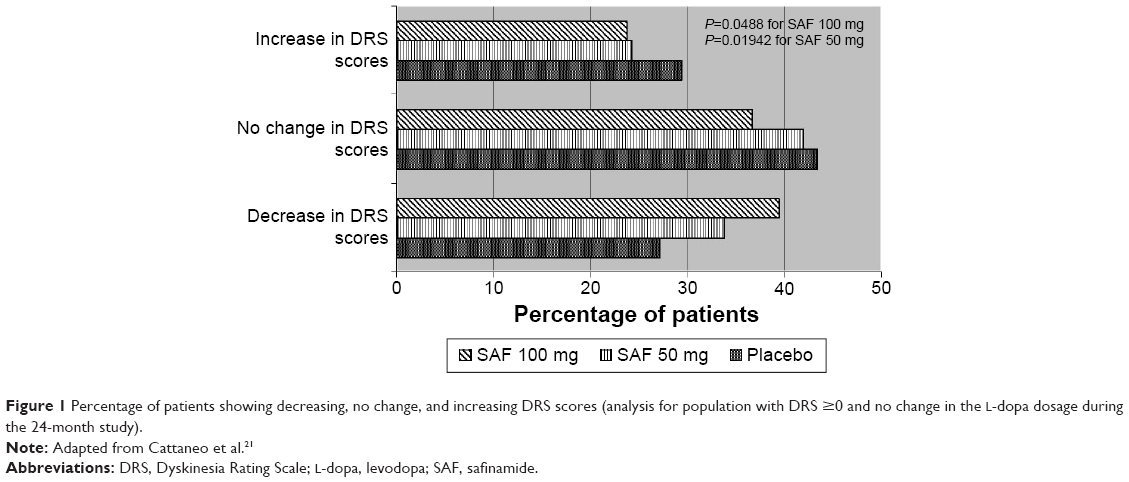 | Figure 1 Percentage of patients showing decreasing, no change, and increasing DRS scores (analysis for population with DRS ≥0 and no change in the L-dopa dosage during the 24-month study). |
The SETTLE study22 was a 6-month (24-week), randomized, double-blind, placebo-controlled international Phase III trial. It enrolled 549 patients with mid-to-late-stage idiopathic PD (>3 years of disease) treated with optimized, stable doses of L-dopa and DA, catechol-O-methyltransferase inhibitor, anticholinergic, and/or amantadine. Patients experiencing a minimum of 1.5 hours off time during the day were randomized equally to the treatment with once-a-day SAF (50–100 mg) or placebo, as adjunctive treatment. The primary endpoint of the trial was the change in daily on time, as assessed by the patient-completed daily diary cards (18 hours/d). SAF 50–100 mg/d significantly improved on time (without worsening the troublesome dyskinesia), off time, UPDRS part III, CGI-S, CGI-C, PDQ-39, and off time following the first morning L-dopa dose (ie, latency to on) compared to placebo.
Safety evaluations in clinical studies
SAF’s safety was assessed through the recording of treatment emergent AEs (TEAEs), vital signs, laboratory tests, electrocardiogram (ECG), and physical and neurological examination in each of the published studies.
In the 015 study,12 after the first amendment, an accurate ocular examination was introduced. The AEs (<10% in each group) were mild or moderate in intensity. The most commonly reported were nausea, headache, abdominal pain, vomiting, pyrexia, cough, hypertension, blurred vision, gastritis, peripheral edema, nasopharyngitis, dizziness, back pain, and tremor. The incidence of severe AE was very low in the SAF 100 mg group (2.2%), but higher in the SAF 200 mg (10.15%) and placebo groups (6.7%). The four reported serious AEs (one accidental death, retinal vein occlusion, iron deficiency anemia, and gastroenteritis) were considered not related to the study drug. TEAEs accounted for the majority of premature discontinuation from the study together with the withdrawal of consent: six patients prematurely interrupted the study due to AEs (one in the 100 mg group, three in the 200 mg group, and two in the placebo group; Table 2).
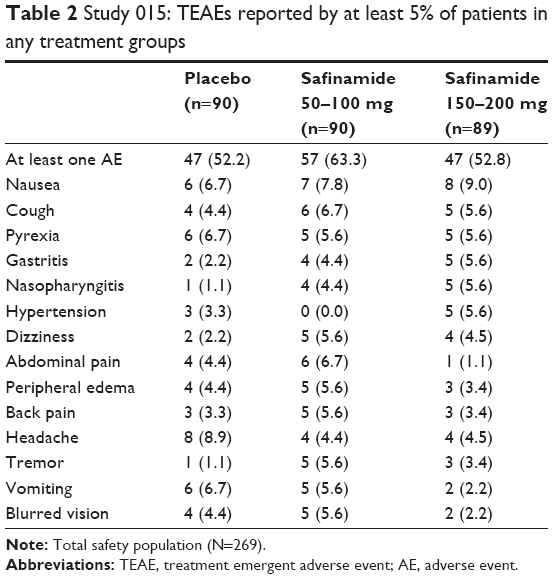 | Table 2 Study 015: TEAEs reported by at least 5% of patients in any treatment groups |
In the 01716 study, a total of 157 patients reported at least one AE, the majority of which were mild to moderate in severity. Two serious AEs were recorded during the study; both of them were considered not related to the study drug. There were no significant changes in vital signs, laboratory and ECG parameters, or the neuro-ophthalmologic examination.
Common AEs (≥5%) reported in the MOTION study were arthralgia, dizziness, somnolence, headache, nausea, nasopharyngitis, and back pain. Serious AEs were infrequent (<5%) and similar across treatments.
In the 01619 study, a total of 446 patients experienced TEAEs, the majority of which were rated as mild or moderate. There were no significant differences in the incidence of TEAEs (nervous system disorders and gastrointestinal disorders) among the treatment groups. Mild-to-moderate dyskinesias were more frequent in the SAF groups; severe dyskinesias were reported in 1.8%, 0.9%, and 2.3% of patients in the 100 mg/d, 50 mg/d, and placebo groups, respectively (Table 3). In the 018 extension study, the incidence of new/worsening dyskinesia AEs, as well as the incidence of the other dopaminergic side effects, was similar to placebo. New TEAEs were reported with a higher percentage in the placebo group (85.1%) than in the SAF 50 mg and 100 mg groups (76.7% and 78.3%, respectively).20 In the SETTLE22 study, the discontinuation rate and serious AEs were similar across treatments. The most frequent TEAEs (≥5% in one group or more) were back pain, dyskinesias, falls, headache, nausea, and urinary tract infections.
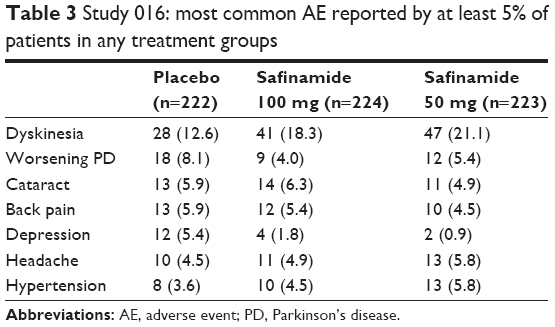 | Table 3 Study 016: most common AE reported by at least 5% of patients in any treatment groups |
Discussion
SAF showed to be efficacious in increasing on time in fluctuating PD patients of two large pivotal trials. The improvement was observed with SAF 50 mg/d and 100 mg/d, but only the higher dose was able to provide a further improvement, especially on ADL and quality of life (QoL) as measured by PDQ-39. Moreover, the signal noted in the ad hoc analysis of 016/018 studies about dyskinesias (improvement of dyskinesia in patients with DRS baseline value >4) was only related to 100 mg/d. The results of the recent post hoc analysis on Study 018 confirmed the long-term beneficial effects of SAF on dyskinesia.
SAF has both dopaminergic and non-dopaminergic mechanisms of action. While the antiparkinsonian effect can be ascribed in part to the inhibition of the MAO-B, which is complete at 50 mg, the enhanced benefit seen at the 100 mg dose is probably due to nondopaminergic mechanisms. The nondopaminergic properties could account for its potential neuroprotective properties observed in animal models. Indeed, it has demonstrated that increased glutamatergic innervation in the nigrostriatal pathway may contribute to the pathophysiology of PD.23 Glutamate may act as a neurotoxin leading to neurodegeneration in the nigrostriatal pathway, and SAF is able to modulate glutamate release.
In the advanced parkinsonian patients, SAF has been demonstrated to significantly improve on time with no or nontroublesome dyskinesias. As already mentioned, the ad hoc analysis performed on dyskinetic patients participating in the 016 study showed an improvement in DRS. This effect should be carefully studied: in fact, involuntary movements are perceived by patients and caregivers as one of the most disabling side effects of parkinsonian treatment.24 Interestingly, SAF induced an improvement of involuntary movements in 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine primate models of L-dopa-induced dyskinesias. This effect could be linked to the antiglutamatergic effect of SAF. Indeed, parkinsonian primates rendered hyperkinetic by L-dopa exhibit increased metabotropic glutamate receptor 5 density. Moreover, overactive glutamatergic transmission in the striatum has been found to be associated with L-dopa-induced dyskinesias.25,26
Furthermore, both short- (6 months) and long-term (18–24 months) treatments with SAF have demonstrated statistically significant improvement in the QoL, as assessed by the Parkinson’s disease quality of life (PDQ-39) and/or the European quality of life scales.
In the early PD patients, SAF showed an improvement in the UPDRS score when added to DAs in the 015 study. The results of the MOTION study, despite in line with what was observed in the 015 study, are controversial. The effect seen when SAF is added to a DA may be explained, thanks to the sum of the properties of both drugs.27 While DAs mainly act on D2/D3 receptor, SAF inhibits MAO-B activity, exerts glutamate release inhibition, and blocks DA reuptake. The combination of these effects leads to an optimization of endogenous DA action and in a full motor response compared to the one obtained with L-dopa.10 In the post hoc analyses performed for the 017 study, patients receiving SAF 100 mg/d experienced a significantly lower rate of intervention compared to placebo and a delay in median time to intervention. The addition of SAF to DAs may be an effective treatment strategy to improve motor function, prolonging the use of DAs and/or delaying the introduction of L-dopa.
Conclusion
SAF represents an important option for patients with PD already treated with L-dopa or other therapeutic combinations. Its dopaminergic and nondopaminergic properties introduce a novelty within the drugs for PD treatment. The effect on glutamate may improve dyskinesias and other nonmotor symptoms. The long-term, double-blind design studies performed to investigate SAF ensure a good safety profile and a long-term efficacy.
The lack of trials on de novo patients in monotherapy with SAF could be a limit for the early use of this compound. Moreover, despite the encouraging signals observed during the pivotal trials, a study to explore the effect on dyskinesia has not been conducted yet. The possible effect of SAF on nonmotor symptoms should also be explored.
SAF has been recently approved in Europe as an add-on therapy to L-dopa alone or in combination with other PD medication in mid-to-late-stage fluctuating PD patients.
Disclosure
The first author has been a consultant to TEVA, Novartis, GSK, Lundbeck, Merck Serono, MSD, UCB, Chiesi Pharma, IMPAX, Newron, Zambon, and Britannia. He has also received research funding from Novartis and GlaxoSmithKline as well as received honoraria for educational symposia and consultancy to GlaxoSmithKline. The second author reports no conflict of interest in this work.
References
Fariello RG. Safinamide. Neurotherapeutics. 2007;4:110–116. | ||
Chazot PL. Safinamide (Newron Pharmaceuticals). Curr Opin Investig Drugs. 2001;2:809–813. | ||
Moussa BH, Bakhle Y, Bakhle YS. Monoamine oxidase: isoforms and inhibitors in Parkinson’s disease and depressive illness. Br J Pharmacol. 2006;147:S287–S296. | ||
Youdim M, Gross A, Finberg J. Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br J Pharmacol. 2001;132:500–506. | ||
Mahmood I. Clinical pharmacokinetics and pharmacodynamics of selegiline. An update. Clin Pharmacokinet. 1997;33(2):91–102. | ||
Caccia C, Maj R, Calabresi M, et al. Safinamide: from molecular targets to a new anti-Parkinson drug. Neurology. 2006;67:S18–S23. | ||
Marzo A, Dal Bo L, Ceppi Monti N, et al. Pharmacokinetics and pharmacodynamics of safinamide, a neuroprotectant with antiparkinsonian and anticonvulsant activity. Pharmacol Res. 2004;50: 77–85. | ||
Cattaneo C, Caccia C, Marzo A, Maj R, Fariello RG. Pressor response to intravenous tyramine in healthy subjects after safinamide, a novel neuroprotectant with selective, reversible monoamine oxidase B inhibition. Clin Neuro Pharmacol. 2003;26:213–217. | ||
Marquet A, Kupas K, Johne A, et al. The effect of safinamide, a novel drug for Parkinson’s disease, on pressor response to oral tyramine: a randomized, double-blind, clinical trial. Clin Pharmacol Ther. 2012; 92(4):450–457. | ||
Stocchi F, Arnold G, Onofrj M, et al. Improvement in motor function in early Parkinson disease by safinamide. Neurology. 2004;63:746–748. | ||
Stocchi F, Vacca L, Grassini P, et al. Symptoms relief in Parkinson’s disease by safinamide. Biochemical and clinical evidence beyond MAO-B inhibition. Neurology. 2006;67:S24–S29. | ||
Stocchi F, Borgohain R, Onofrj M, et al; Study 015 Investigators. A randomized, double-blind, placebo controlled trial of safinamide as add on therapy in early Parkinson’s disease patients. Mov Disord. 2012; 27:106–112. | ||
Elgh E, Domellöf M, Linder J, Edström M, Stenlund H, Forsgren L. Cognitive function in early Parkinson’s disease: a population-based study. Eur J Neurol. 2009;16:1278–1284. | ||
Verbaan D, Marinus J, Visser M, et al. Cognitive impairment in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2007;78:1182–1187. | ||
Sharma T, Anand R, Stocchi F, Borgohain R, Rossetti R; 015 Study Group. Cognitive effects of Safinamide in early Parkinson’s disease patients. International Congress of Parkinson’s Disease and Movement Disorders; June 3–7, 2007; Istanbul, Turkey. | ||
Schapira AHV, Stocchi F, Borgohain R, et al; Study 017 Investigators. Long-term efficacy and safety of safinamide as add-on therapy in early Parkinson’ disease. Eur J Neurol. 2013;20:271–280. | ||
Barone P, Fernandez H, Ferreira JJ, et al. Safinamide as an add-on therapy to a stable dose of a single dopamine agonist: results from a randomized, placebo-controlled, 24-week multicenter trial in early idiopathic Parkinson disease patients (MOTION Study). 65th Annual Meeting of the American Academy of Neurology (AAN); March 16–23, 2013; San Diego Convention Center, San Diego, CA, USA. | ||
Clinical Study Report Newron Pharmaceutical S.p.A, Zambon S.p.A. Safinamide Protocol n. 27918. October 25, 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002396/WC500184967.pdf. | ||
Borgohain R, Szasz J, Stanzione P, et al; Study 016 Investigators. Randomized trial of safinamide add-on to Levodopa in Parkinson’s disease with motor fluctuations. Mov Disord. 2014;29(2):229–237. | ||
Borgohain R, Szasz J, Stanzione P, et al; Study 018 Investigators. Two year, randomized, controlled study of Safinamide as add-on to levodopa in mid to late Parkinson’s disease. Mov Disord. 2014;29(10): 1273–1280. | ||
Cattaneo C, La Ferla R, Bonizzoni E, et al. Long-term effects of safinamide on dyskinesia in mid-to late-stage Parkinson’s disease: a post-hoc analysis. J Parkinsons Dis. 2015;5:475–481. | ||
Schapira A, Fox S, Hauser R, et al. Safinamide add on to L-dopa: at randomized, placebo controlled 24 weeks global trial in patients with Parkinson’s disease and motor fluctuations. 65th Annual Meeting of the American Academy of Neurology (AAN); March 16–23, 2013; San Diego Convention Center, San Diego, CA, USA. | ||
Blandini F. An update on the potential role of excitotoxicity in the pathogenesis of Parkinson disease. Funct Neurol. 2010;25:65–71. | ||
Chapuis S, Ouchchane L, Metz O, et al. Impact of the motor complications of Parkinson’s disease on the quality of life. Mov Disord. 2005; 20:224–230. | ||
Gregoire L, Roach A, Di Paolo T. Safinamide reduces levodopa-induced dyskinesias in MPTP-lesioned primates while prolonging anti-parkinsonian efficacy. Mov Disord. 2010;25(suppl 2):S308. | ||
Verhagen Metman L, Del Dotto P, Blanchet PJ, van den Munckhof P, Chase TN. Blockade of glutamatergic transmission as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Amino Acids. 1998;14(14):75–82. | ||
Anand R, Onofrj M, Schapira AH, et al. Safinamide potentiates the effects of DA-agonists in early stage Parkinson’s disease (PD) patients. Mov Disord. 2007;22(16):S99. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.