")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Adiponectin Protects Obese Rats from Aggravated Acute Lung Injury via Suppression of Endoplasmic Reticulum Stress
Authors Wei K, Luo J, Cao J, Peng L, Ren L, Zhang F
Received 25 August 2020
Accepted for publication 13 October 2020
Published 5 November 2020 Volume 2020:13 Pages 4179—4190
DOI https://doi.org/10.2147/DMSO.S278684
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Antonio Brunetti
Ke Wei,1 Jie Luo,1 Jun Cao,1 Lihua Peng,1 Li Ren,1 Fan Zhang2
1Department of Anesthesiology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China; 2Department of Anesthesiology, Jianyang People’s Hospital, Jianyang, Sichuan 641400, People’s Republic of China
Correspondence: Ke Wei
Department of Anesthesiology, The First Affiliated Hospital of Chongqing Medical University, 1# Youyi Road, Yuzhong District, Chongqing, People’s Republic of China
Tel +86 23 89011069
Fax +86 23 89011062
Email [email protected]
Introduction: Endoplasmic reticulum (ER) stress seems to mediate the obesity-induced susceptibility to acute lung injury (ALI). The present study was designed to evaluate the role of ER stress in adiponectin (APN)-induced lung protection in an obese rat model treated with lipopolysaccharide (LPS).
Methods: Four-week-old male Sprague-Dawley rats fed either a normal chow diet or a high-fat diet for 12 weeks were randomly assigned to one of the following groups: lean rats, diet-induced obesity rats, lean rats with ALI, obese rats with ALI, obese rats pretreated with 4-phenylbutyric acid (4-PBA) before ALI or obese rats pretreated with APN before ALI. At 24 h after instillation of LPS into the lungs, cell counts in the bronchoalveolar lavage fluid (BALF) were determined. Lung tissues were separated to assess the degree of inflammation, pulmonary oedema, epithelial apoptosis and the expression of ER stress marker proteins.
Results: The 78-kDa glucose-regulated protein (GRP78) and C/EBP homologous protein (CHOP) expression in the lung tissues of obese rats was upregulated before ALI, as well as the elevated apoptosis in epithelial cells. During ALI, the expression of ER stress marker proteins was similarly increased in both lean and obese rats, while significant downregulation of Mitofusin 2 (MFN2) was detected in obese epithelial cells. The lung tissues of obese rats showed higher concentrations of tumor necrosis factor-alpha (TNF-α), Interleukin 6 (IL-6) and IL-10, enhanced neutrophil counts and elevated wet/dry weight ratios. APN and 4-PBA decreased the degree of ER stress and suppressed LPS-induced lung inflammation, pulmonary oedema and epithelial apoptosis.
Conclusion: APN may exert protective effects against the exacerbated lung injuries in obese rats by attenuating ER stress, which operates as a key molecular pathway in the progression of ALI.
Keywords: adiponectin, endoplasmic reticulum stress, obesity, lung injury
Introduction
Acute lung injury (ALI), a life-threatening condition that causes substantial health problems worldwide, is an inflammatory lung process with a complex mechanism. In recent years, the impact of obesity on the incidence and outcome of ALI has received increasing attention due to the worldwide prevalence of overweight.1,2 Epidemiological studies suggest that obesity is an important predisposing factor in the pathogenesis of ALI.3,4 Increased saturated fatty acids may lead to widespread inflammation and cellular dysfunction.5,6 In the lung tissues, disturbance in the production of adipocytokines with pro-inflammatory or anti-inflammatory activities may prime the lungs of obese individuals for an acute inflammatory response.7,8
Adiponectin (APN) is a highly abundant adipocyte-derived adipocytokine with pleiotropic effects on various pulmonary cell types, including the vascular endothelium, alveolar macrophages and epithelia.9–11 The anti-inflammatory activity of APN in lung tissues is linked to attenuation of leukocyte-endothelium interactions.12 However, this APN-induced alleviation of lung inflammation was found to be impaired during obesity. Investigations have revealed that circulating APN levels are drastically decreased in obesity.12,13 Although the mechanism is unknown, APN deficiency during obesity is now believed to be one of the main causes of exacerbated pulmonary endothelial dysfunction and lung inflammation.14
The endoplasmic reticulum (ER) is the main site of the synthesis, maturation, and secretion of proteins.15–17 A wide variety of disturbances, such as an unbalanced redox state, disruption of calcium homeostasis, and viral and bacterial infections, lead to the accumulation of unfolded or misfolded proteins within the ER, known as ER stress. Under conditions related to obesity and type 2 diabetes, ER stress is activated in various tissues and is found to be responsible for most of the comorbidities associated with obesity.18–21 According to the limited studies, ER stress is correlated to impairment of lung endothelial cells, and inhibition of ER stress is effective in attenuating lung injury.22
While both APN deficiency and ER stress contribute to exacerbated lung injury, the interaction between these two obesity-related pathophysiological conditions remains unclear. A recent study indicated that APN can ameliorate the progression of chronic obstructive pulmonary disease by reducing ER stress-induced epithelial apoptosis.23 In this study, the relationship between ER stress and APN in LPS-induced ALI was assessed in an obese rat model. We hypothesized that the protective effects of ANP against ALI are exerted through preventing the activation of ER stress, which also primes obese rats for exacerbated lung injuries.
Materials and Methods
Experimental Animals and Diet
All procedures were approved by the Institutional Animal Care and Use Committee of Chongqing Medical University. The animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health. Four-week-old male Sprague-Dawley (SD) rats were supplied by the Animal Center of Chongqing Medical University. The rats were maintained on a 12 h/12 h artificial light-dark cycle and housed in individual cages. Rats were fed either a normal chow diet (13.5% calories from fat, 58% from carbohydrates, and 28.5% from protein) or a high-fat diet/Western diet with 45% calories from fat (ReadyDietech, Shenzhen, China) for 12 weeks.
Experimental Design
Rats were randomly assigned to one of the six groups (n=5): lean rats with a normal diet (Lean group), diet-induced obesity rats (DIO group), lean rats with ALI (Lean-ALI group), obese rats with ALI (DIO-ALI group), obese rats pretreated with 4-phenylbutyric acid (4-PBA) before ALI (4-PBA+DIO-ALI group) and obese rats pretreated with APN before ALI (APN+DIO-ALI group). ALI was induced by the instillation of one dose (100 µg) of lipopolysaccharide (LPS) into the posterior oropharyngeal space of the anaesthetized rats with the tongue gently extracted with tweezers to facilitate fluid entry into the lung. Obese rats treated with 4-PBA (Sigma-Aldrich, St. Louis, MO, USA) were intraperitoneally injected daily with 0.5 g/kg/d 4-PBA for 2 weeks until the induction of ALI. Obese rats allocated for APN supplementation were intravenously injected with 10 µg of globular APN (BioVision, Milpitas, CA, USA) twice a week from 2 weeks before the induction of ALI.
Cell Counts in Bronchoalveolar Lavage Fluid (BALF)
At 24 h after LPS administration, the rats were euthanized, and the right main stem bronchi were clamped. Sterile saline (2.5 mL) was injected and aspirated through the trachea three times at 4°C. The BALF samples were centrifuged at 1500 rpm for 10 min to separate cells and supernatant. Total and differential cell counts in the BALF were determined by an automatic blood cell counter (Diatron, Budapest, Hungary).
Histologic Evaluation
The tissue samples from the apical lobe of the right lung were fixed in 10% formalin and embedded in paraffin wax. The paraffin-embedded tissue samples were sectioned into 5 µm thick pieces and stained with hematoxylin and eosin (H&E). Histologic alteration of the lung parenchyma was evaluated by assessing interstitial oedema, hemorrhage, and the infiltration or aggregation of neutrophils in the airspace or vessel walls.
Immunohistochemistry
The lung tissues were placed in 10% formalin at room temperature overnight for paraffin embedding and processed for immunohistochemical staining. Paraffin-embedded specimens were sectioned at 5 µm, deparaffinized and hydrated in PBS. Then, the sections were incubated in 3% H2O2 for 10 min and rinsed with PBS. A primary antibody against the 78-kDa glucose-regulated protein (GRP-78) (Abcam, Cambridge, UK) or Mitofusin 2 (MFN2) (Abcam, Cambridge, UK) was applied, followed by washing and incubation with a secondary antibody for 30 minutes. Sections were observed with an optical microscope.
Terminal Dump Nick-Labeling (TUNEL) Staining for Apoptosis
Cellular apoptosis was measured by TUNEL assay according to the manufacturer’s instructions (Roche Diagnostics, Basel, Switzerland). The lung tissues were placed in 10% formalin at room temperature overnight for paraffin embedding and then sectioned at 5 µm and processed for TUNEL staining. Five fields from each section were randomly selected and analysed with an optical microscope in a blinded manner, and the number of positive cells per section was counted. The apoptotic index (AI) was defined as the number of positive cells per 100 cells counted in the five fields from the same section.
Lung Wet/Dry (W/D) Weight Ratio
Tissue samples from the right inferior lobe of the lung were weighed and dried in an oven at 80°C for 72 h, and the sample was then weighed again to obtain the lung W/D.
Preparation of Lung Homogenates and Cytokines Assays
The intermediate lobes of the right lungs were harvested and homogenized in 1 mL of PBS with an Ultra-Turrax tissue homogenizer. The homogenates were then centrifuged at 2000 rpm for 10 min at 4°C. The supernatants were passed through a 0.45 µm filter, collected, and then stored at −80°C for cytokine production analysis. The levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-10 in the lung homogenate supernatants were determined using the enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Western Blotting
Western blot analysis of GRP78, C/EBP homologous protein (CHOP), Caspase-12 and MFN2 was performed. Proteins were extracted from the lung tissue samples with lysis buffer (Beyotime, Shanghai, China). The supernatants were collected after centrifugation (12,000 ×g for 15 minutes). Proteins were separated on 10–12% SDS-PAGE gels and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were blocked in 5% nonfat milk in Tris-buffered saline (TBS) for 1.5 h at 37°C and then incubated overnight at 4°C with primary antibodies against GRP78, CHOP, caspase12, MFN2, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1000) (Abcam, Cambridge, UK). After three washes, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Beyotime, Shanghai, China) for 1 h at room temperature. The relative intensities of the bands were semi-quantitatively analysed using Bio-Rad Quantity One software (version 4.4.0). All the results were normalized to levels of GAPDH and expressed as fold intensity compared with the Lean group.
Statistical Analysis
All data are expressed as the mean ± SD. Statistical analyses were performed using GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA, USA). Student’s t-test was used for two-group comparisons, whereas one-way ANOVA followed by Tukey’s post hoc analysis was used for multiple-group comparisons. P < 0.05 was used to indicate statistical significance.
Results
Weight Gain After High-Fat Diet Feeding
After 12 weeks, weight gain was observed in both groups of rats fed different diets. Rats receiving a high-fat diet are significantly heavier than those on a normal diet (Figure 1).
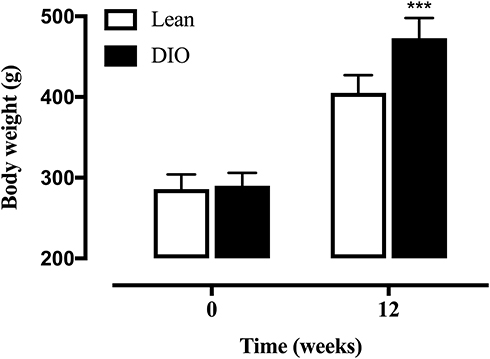 |
Figure 1 The body weight of SD rats fed either a normal or high-fat diet for 12 weeks. Data are presented as the mean ± SD. ***p < 0.001 vs Lean. |
Extents of ER Stress Before and After the LPS Administration
The basal expression levels of GRP78 and CHOP in the lung tissues of obese rats were elevated. The levels of these ER stress marker proteins were upregulated in both obese and lean rats during ALI (Figure 2A–D). The function of mitochondria-endoplasmic reticulum physical coupling, as measured by the expression of MFN2, was inhibited in the lung tissues of obese rats after LPS administration (Figure 2A and E). Increased GRP78 expression and reduced MFN2 expression during ALI were detected in positive epithelial cells by immunohistochemical staining (Figure 3A and B).
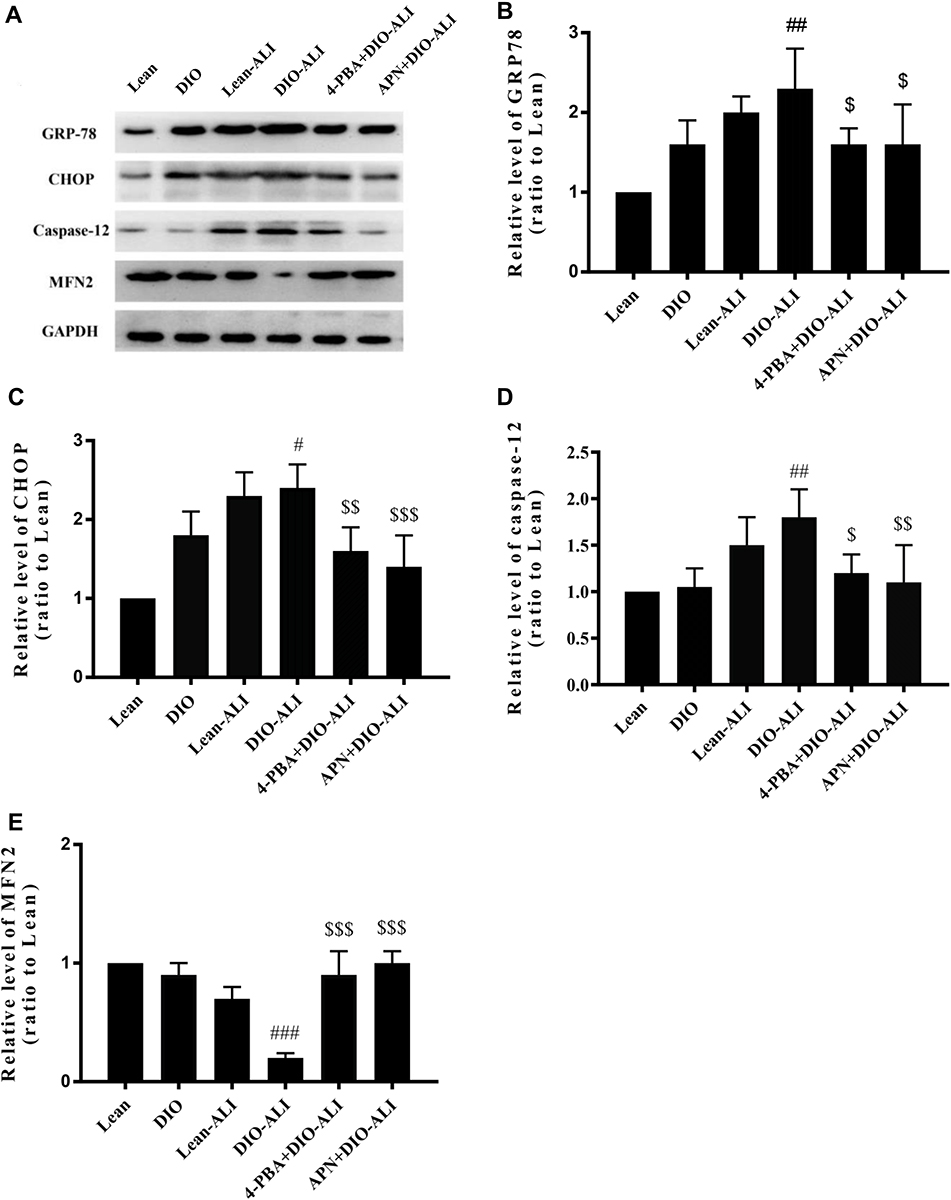 |
Figure 2 APN and 4-PBA alleviate the ER stress during ALI. (A) Western blot analysis for ER stress marker proteins and the mitochondria-ER physical coupling protein in lung tissues before and after LPS administration. Densitometry analysis of the expression of GRP78 (B), CHOP (C), Caspase 12 (D) and MFN2 (E). Data are presented as the mean ± SD. #, ##, ###p < 0.05, 0.01, 0.001 vs DIO; $, $$, $$$p < 0.05, 0.01, 0.001 vs DIO-ALI. |
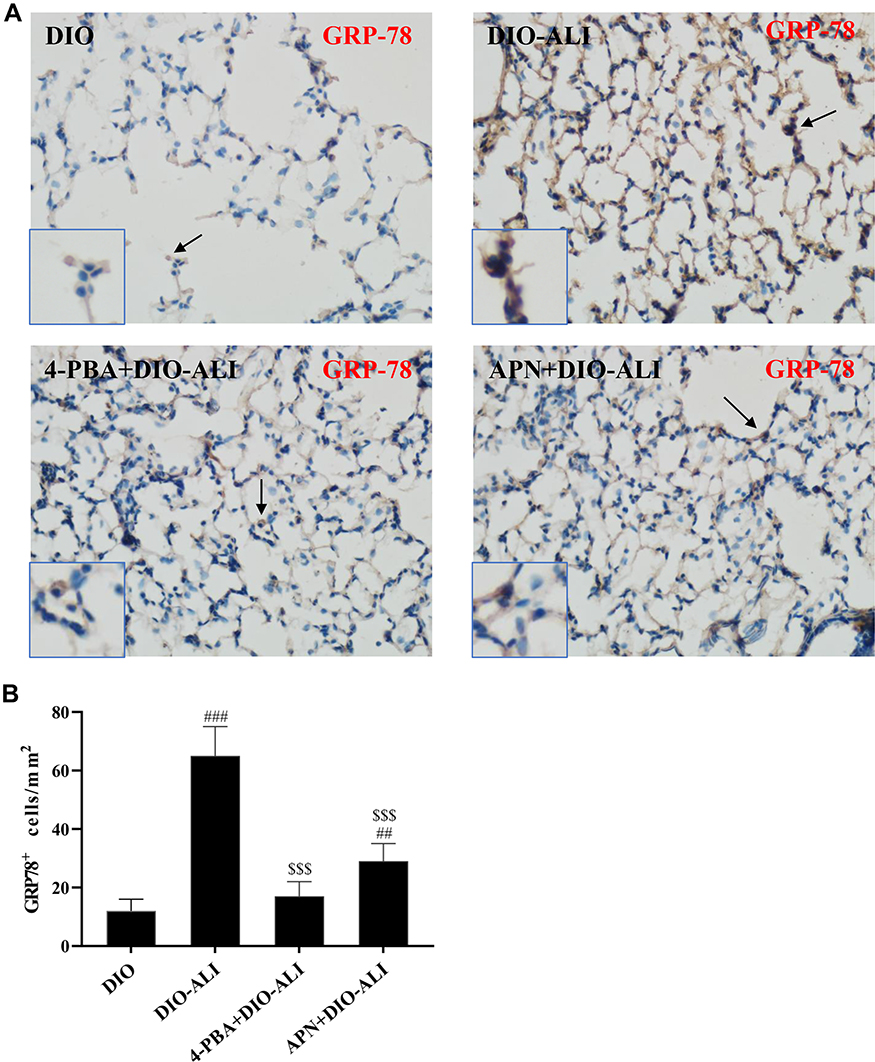 |
Figure 3 Continued. |
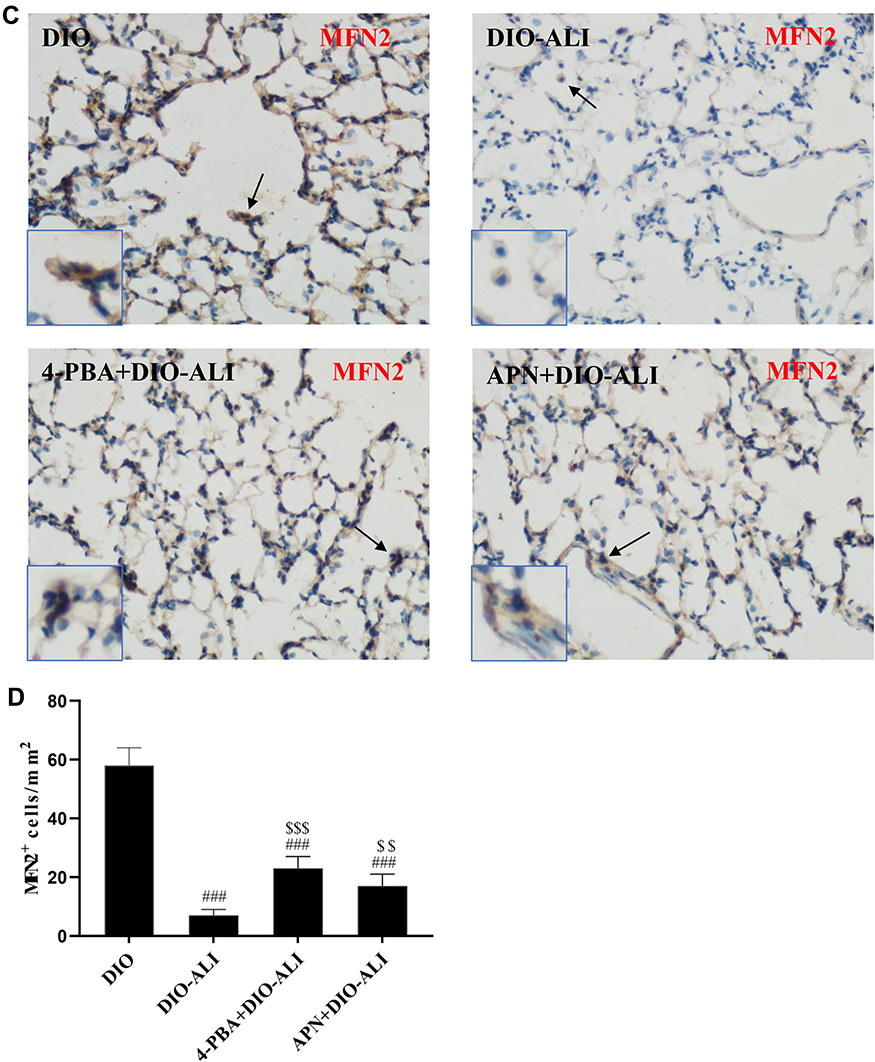 |
Figure 3 Immunohistochemical staining and quantification analysis of GRP78 (A, B) and MFN2 (C, D) in pulmonary epithelial cells (brown stain). Images were taken at 200× magnification. Data are presented as the mean ± SD. ##, ###p < 0.01, 0.001 vs DIO; $,$$,$$$,p < 0.01, 0.001 vs DIO-ALI. |
Effects of APN and 4-PBA on LPS-Induced ER Stress
Expression of the ER stress marker proteins GRP78, CHOP, and caspase-12 was significantly decreased in lung tissues from obese rats pretreated with APN or 4-PBA, and pretreatment also activated the expression of MFN2 (Figure 2B–E). Immunohistochemical staining to detect GRP78 and MFN2 expression in epithelial cells showed alterations in consistence with those shown by Western blotting (Figure 3A–D).
Impact of Obesity on Pulmonary Inflammation and Epithelial Apoptosis During ALI
Inflammatory responses to LPS in lung tissues were characterized by neutrophil cell counts in the BALF and the concentrations of inflammatory cytokines in lung homogenates. Determination of these parameters at 24 h after LPS administration showed increased concentrations of TNF-α and IL-6, IL-10 in lung homogenates from obese rats (Figure 4A). Elevated neutrophil counts in the BALF and lung W/D were observed in obese rats (Figure 4B and C). Histological examination showed aggravated structural damage, intra-alveolar oedema, hemorrhage and the infiltration of inflammatory cells in the lung tissues of the obese rats (Figure 4D). By TUNEL staining, a significantly higher rate of apoptosis in pulmonary epithelial cells was observed in obese rats compared to that in lean rats before the induction of ALI (Figure 5A and B).
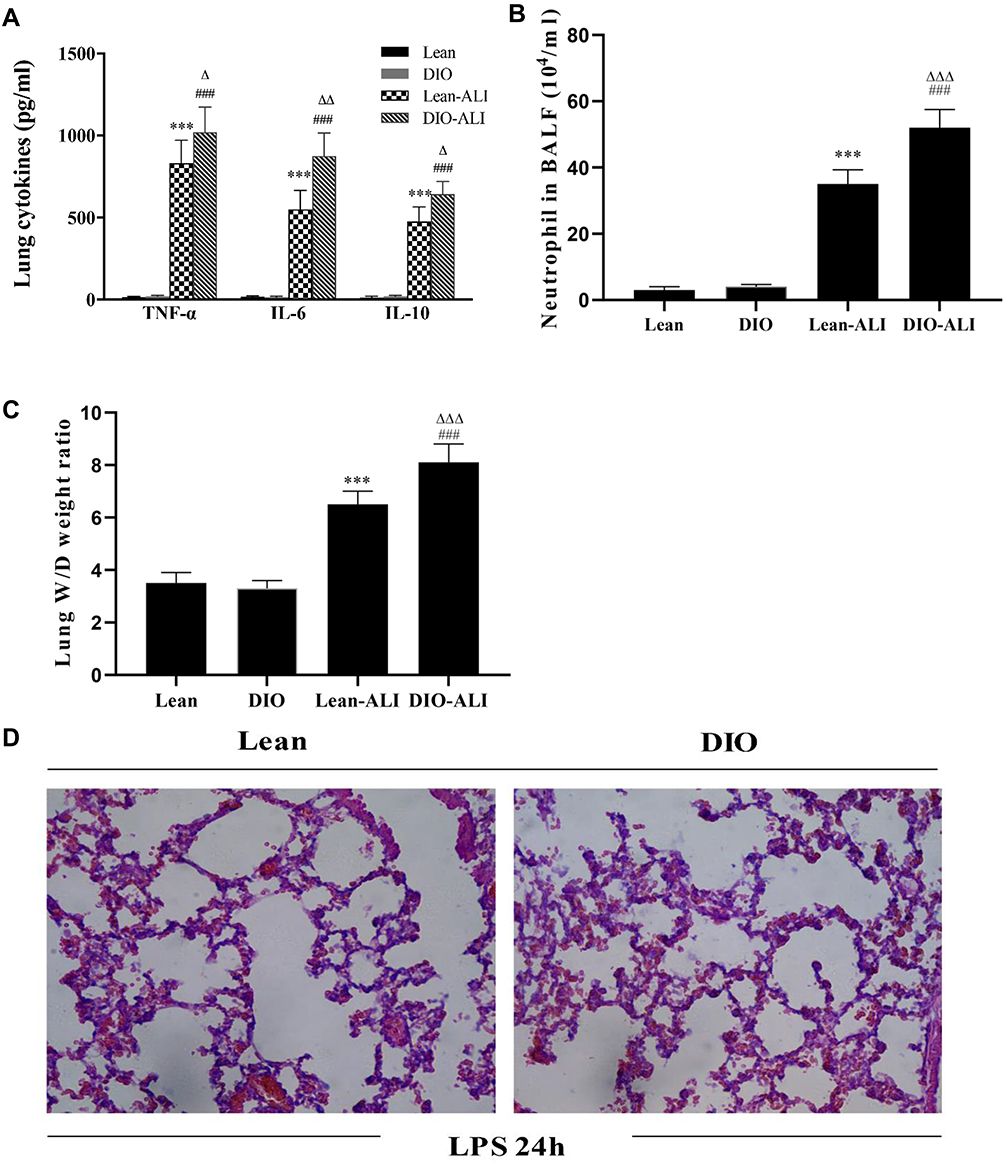 |
Figure 4 The impacts of obesity on the LPS-induced lung inflammatory response and tissue damage (n=5). (A) The concentrations of TNF-α, IL-6, and IL-10 in lung homogenate supernatants during ALI. (B) Neutrophil cell counts in the BALF of lean or obese rats at 24 hours after LPS administration. (C) Wet/dry weight ratio of LPS-injured lungs. (D) Representative H&E-stained lung sections from LPS-injured rats. Images were taken at 200× magnification. Data are presented as the mean ± SD. ***p < 0.001 vs Lean; ###p < 0.001 vs DIO; Δ, ΔΔ, ΔΔΔp <0.05, 0.01, 0.001 vs Lean-ALI. |
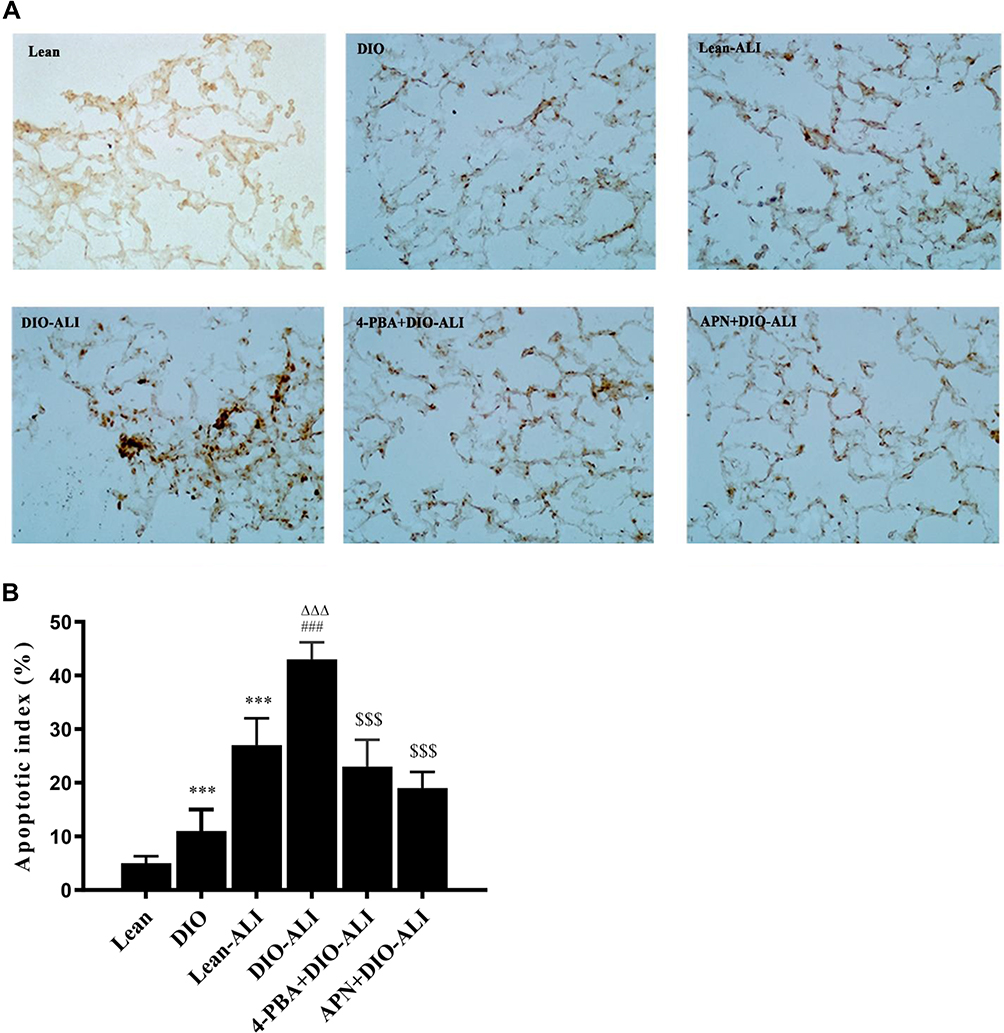 |
Figure 5 The effects of APN and 4-PBA on the apoptosis of pulmonary epithelial cells. (A) The apoptosis of epithelial cells was detected by TUNEL staining. TUNEL-positive cells observed under a light microscope were identified by brown-stained nuclei (images were taken at 200× magnification.). (B) The apoptotic index of the epithelial cells. A higher epithelial apoptosis ratio was found in the lung tissues of obese rats before ALI. Data are presented as the mean ± SD. ***p < 0.001 vs Lean; ###p < 0.001 vs DIO; ΔΔΔp <0.001 vs Lean-ALI; $$$p < 0.001 vs DIO-ALI. |
Effects of APN on LPS-Induced Lung Injury and Apoptosis
APN or 4-PBA pretreatment inhibited the LPS-induced accumulation of TNF-α, IL-6, IL-10 and neutrophils in lung tissues (Figure 6A and B) and attenuated pulmonary oedema. (Figure 6C). The rats in pretreatment groups also showed alleviated hemorrhage and infiltration of inflammatory cells in lung tissues (Figure 6D) and reduced epithelial apoptosis in TUNEL-stained lung sections (Figure 5A and B).
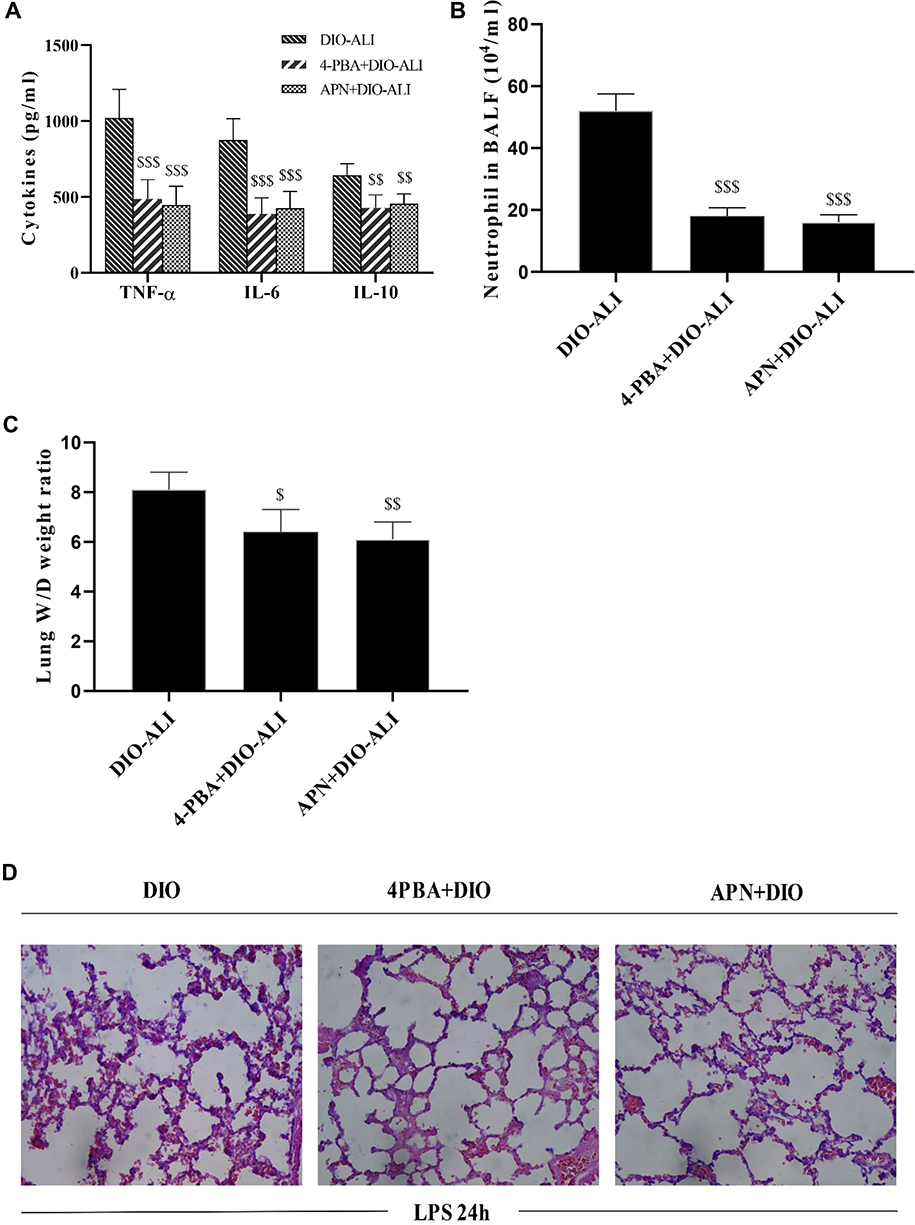 |
Figure 6 The effects of APN and 4-PBA on the LPS-induced lung inflammatory response and tissue damage in obese rats. (A) The concentrations of TNF-α, IL-6, and IL-10 in lung homogenate supernatants at 24 hours after LPS administration. (B) Neutrophil cell counts in the BALF at 24 hours after LPS administration. (C) Wet/dry weight ratio of LPS-injured lungs. (D) Representative H&E-stained lung sections from LPS-injured rats. Images were taken at 200× magnification. Data are presented as the mean ± SD. $$, $$$p < 0.05, 0.01, 0.001 vs DIO-ALI. |
Discussion
Several biochemical changes that predispose obese patients to ALI have been proposed in previous studies.1,8 Obesity-induced chronic inflammation is one of the most commonly accepted causes of this susceptibility. APN deficiency further predisposes obese patients to amplified inflammatory responses and lung injury upon a second insult (eg, sepsis). However, little is known about the exact mechanism by which obesity exacerbates lung injury or the pathway that mediates the lung-protective effects of APN. The results of this study confirmed the anti-inflammatory and anti-apoptotic activities of APN by regulation of ER stress. ER stress, which operates as a key promoter of lung inflammation and apoptosis, was similarly activated in both obese and lean rats during ALI. Our study also showed a significant reduction in MFN2 in obese rats.
APN is synthesized as a monomer that is composed of four regions: an amino-terminal peptide, a short hypervariable region, a collagen-like domain and a carboxy-terminal globular domain.24 In serum, the APN monomeric form is present as a full-length form or as a globular form of the protein. APN acts through two major functionally distinct receptors, AdipoR1 and AdipoR2. AdipoR1 is a high affinity receptor for globular APN (gAPN) and is expressed ubiquitously, but most abundantly in skeletal muscle. AdipoR2 mainly recognizes full-length APN (fAPN) and is predominantly expressed in the liver. It was found that gAPN has much higher bioactivity than fAPN and may be the final active ligand of APN at its target cells.25,26 In a recent study, Li and colleagues reported that a single in vivo dose of gANP may exert potent protective functions against lung ischemia and reperfusion injury in rats with type 2 diabetes mellitus.27
Previous studies have attributed the effects of APN against lung injuries to its anti-inflammatory and anti-oxygenation properties in pulmonary vascular endothelial cells.9,14,28,29 Later studies on the pathogenesis of chronic obstructive pulmonary disease and lung cancer demonstrated multiple protective effects of APN on epithelia cells.23,30,31 In this study, gAPN pretreatment led to reduced ER stress and epithelial apoptosis and the decreased release of the pro-inflammatory mediators during ALI. Our results confirmed the inhibitory effect of APN on ER stress, which further prevented the progression of lung inflammation and epithelial apoptosis. The molecular pathway connecting APN and ER stress remains unknown. Liu et al observed in adipose tissues that APN alleviates ER stress-induced apoptosis via activating the AdipoR1/AMP-activated protein kinase (AMPK) signal pathway.32
ER stress, caused by the accumulation of unfolded or misfolded proteins in the ER, is an adaptive mechanism by which cells react to perturbations in the ER, but sustained ER stress may induce injury. Pathways mediated by three protein sensors are involved in ER stress and may induce cell apoptosis (ie, inositol-requiring enzyme 1a, protein kinase RNA-like endoplasmic reticulum kinase, and activating transcription factor).33 GRP78, which binds these three ER stress sensors under normal conditions, interacts in a more stably manner with misfolded or unfolded proteins during enhanced ER stress. Therefore, the upregulation of GRP78 is commonly used as an ER stress marker.34 CHOP is another marker for assessment of ER stress, as it is induced in response to ER stress as an apoptotic transcriptional factor.35 In this study, the expression of GRP78 and CHOP was significantly upregulated by LPS, while no difference in the level of these marker proteins between the obese and lean rats was found. In addition, reduction of the ER stress dramatically alleviated lung inflammation. Our results support the role of ER stress as a key signaling pathway in regulating the ALI rather than as a specific pathophysiological process that primes the lungs for injury in obese rats.
ER stress-induced apoptosis in endothelial cells may worsen pulmonary edema during septic shock, which indicates a novel target for lung protection by restricting the activation of ER stress.36,37 In the present study, enhanced epithelial apoptosis was observed in obese rats under normal conditions. During ALI, a higher level of apoptosis is kept in obese rats, accompanied by exacerbation of pulmonary inflammation. The results suggest a role of obesity in promoting epithelial damage.
MFN2 is a transmembrane GTPase on the outer mitochondrial membrane that serves to fuse mitochondria. It provides tethering force between the ER and mitochondria to ensure their proximity and communication.38,39 The expression of MFN2 is affected by the interactions between the two organelles and plays a key role in regulating mitochondrial dynamics. Previous studies demonstrated that MFN2 is a substrate of Jun N-terminal kinase (JNK), which is activated in obesity and ALI.40–42 JNK Phosphorylation of MFN2 in response to cellular stress results in ubiquitin-mediated proteasomal degradation of MFN2.40 In the present study, LPS induced a remarkable downregulation of MFN2 in epithelial cells, especially in those of obese rats. The inflammation-induced reduction in MFN2 may be correlated to the alterations in mitochondrial homeostasis.43 Our study is not sufficient to prove an association between reduced MFN2 and mitochondrial dysfunction, and the extended researches are required to confirm if mitochondria-related pathways are involved in mediating the epithelial damage.
Reduced MFN2 has been considered the upstream of altered mitochondrial dynamics and ER stress. Several studies have reported that obesity-induced changes in MFN2 expression result in mitochondrial dysfunction and activation of ER stress.44,45 A recent study on airway inflammation reported that ER stress may also lead to a reduction in MFN2, indicating the existence of reverse signal transduction in the progression of cellular inflammatory response.46 Similarly, our results demonstrate a role of ER stress as upstream of MFN2. The expression of MFN2 restored to normal levels when rats were pretreated with an ER stress inhibitor.
Conclusion
Taken together, our results suggest that APN may exert protective effects against exacerbated lung injuries in obese rats by attenuating ER stress, which is highly elevated by LPS and operates as a key molecular pathway in the progression of ALI.
Acknowledgments
The present work was supported by grants from Chongqing Science & Technology Bureau (Project no. CSTC2019jscx-msxmX0214), Chongqing Health Commission (Project no. 2017HBRC001) and National Key Clinical Specialty building Project
Disclosure
The authors declare that they have no potential conflicts of interest for this work.
References
1. McCallister JW, Adkins EJ, O’Brien JM. Obesity and acute lung injury. Clin Chest Med. 2009;30(3):495–508. doi:10.1016/j.ccm.2009.05.008
2. Stapleton RD, Suratt BT. Obesity and nutrition in acute respiratory distress syndrome. Clin Chest Med. 2014;35(4):655–671. doi:10.1016/j.ccm.2014.08.005
3. Gong MN, Bajwa EK, Thompson BT, Christiani DC. Body mass index is associated with the development of acute respiratory distress syndrome. Thorax. 2010;65(1):44–50. doi:10.1136/thx.2009.117572
4. Lederer DJ, Kawut SM, Wickersham N, et al. Obesity and primary graft dysfunction after lung transplantation: the lung transplant outcomes group obesity study. Am J Respir Crit Care Med. 2011;184(9):1055–1061. doi:10.1164/rccm.201104-0728OC
5. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi:10.1172/JCI200319246
6. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867.
7. Konter J, Baez E, Summer RS. Obesity: “priming” the lung for injury. Pulm Pharmacol Ther. 2013;26(4):427–429. doi:10.1016/j.pupt.2012.03.003
8. Wang C. Obesity, inflammation, and lung injury (OILI): the good. Mediators Inflamm. 2014;2014:978463. doi:10.1155/2014/978463
9. Konter JM, Parker JL, Baez E, et al. Adiponectin attenuates lipopolysaccharide-induced acute lung injury through suppression of endothelial cell activation. J Immunol. 2012;188(2):854–863. doi:10.4049/jimmunol.1100426
10. Summer R, Fiack CA, Ikeda Y, et al. Adiponectin deficiency: a model of pulmonary hypertension associated with pulmonary vascular disease. Am J Physiol Lung Cell Mol Physiol. 2009;297(3):L432–438.
11. Teoh H, Quan A, Bang KW, et al. Adiponectin deficiency promotes endothelial activation and profoundly exacerbates sepsis-related mortality. Am J Physiol Endocrinol Metab. 2008;295(3):E658–664. doi:10.1152/ajpendo.90384.2008
12. Arita Y, Kihara S, Ouchi N, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257(1):79–83. doi:10.1006/bbrc.1999.0255
13. Matsuzawa Y, Funahashi T, Kihara S, Shimomura I. Adiponectin and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2004;24(1):29–33. doi:10.1161/01.ATV.0000099786.99623.EF
14. Shah D, Romero F, Duong M, et al. Obesity-induced adipokine imbalance impairs mouse pulmonary vascular endothelial function and primes the lung for injury. Sci Rep. 2015;5(1):11362. doi:10.1038/srep11362
15. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115(10):2656–2664. doi:10.1172/JCI26373
16. Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3(1):399–425. doi:10.1146/annurev.pathmechdis.3.121806.151434
17. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7(12):1013–1030.
18. Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18(1):59–68. doi:10.1016/j.molmed.2011.07.010
19. Puri P, Mirshahi F, Cheung O, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134(2):568–576. doi:10.1053/j.gastro.2007.10.039
20. Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16(4):396–399. doi:10.1038/nm0410-396
21. Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi:10.1126/science.1103160
22. Shah D, Romero F, Guo Z, et al. Obesity-induced endoplasmic reticulum stress causes lung endothelial dysfunction and promotes acute lung injury. Am J Respir Cell Mol Biol. 2017;57(2):204–215. doi:10.1165/rcmb.2016-0310OC
23. Zhao YL, Li F, Liu YW, et al. Adiponectin attenuates endoplasmic reticulum stress and alveolar epithelial apoptosis in COPD rats. Eur Rev Med Pharmacol Sci. 2017;21(21):4999–5007.
24. Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26(3):439–451. doi:10.1210/er.2005-0005
25. Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7(8):941–946. doi:10.1038/90984
26. Tao L, Gao E, Jiao X, et al. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation. 2007;115(11):1408–1416. doi:10.1161/CIRCULATIONAHA.106.666941
27. Li D, Song LL, Wang J, Meng C, Cui XG. Adiponectin protects against lung ischemia-reperfusion injury in rats with type 2 diabetes mellitus. Mol Med Rep. 2018;17(5):7191–7201.
28. Xu L, Bao HG, Si YN, et al. Effects of adiponectin on acute lung injury in cecal ligation and puncture-induced sepsis rats. J Surg Res. 2013;183(2):752–759. doi:10.1016/j.jss.2013.01.055
29. Cai L, Yi F, Dai Z, et al. Loss of caveolin-1 and adiponectin induces severe inflammatory lung injury following LPS challenge through excessive oxidative/nitrative stress. Am J Physiol Lung Cell Mol Physiol. 2014;306(6):L566–573. doi:10.1152/ajplung.00182.2013
30. Cui E, Guo H, Shen M, et al. Adiponectin inhibits migration and invasion by reversing epithelial-mesenchymal transition in nonsmall cell lung carcinoma. Oncol Rep. 2018;40(3):1330–1338.
31. Nigro E, Scudiero O, Sarnataro D, et al. Adiponectin affects lung epithelial A549 cell viability counteracting TNFalpha and IL-1ss toxicity through AdipoR1. Int J Biochem Cell Biol. 2013;45(6):1145–1153. doi:10.1016/j.biocel.2013.03.003
32. Liu Z, Gan L, Wu T, et al. Adiponectin reduces ER stress-induced apoptosis through PPARalpha transcriptional regulation of ATF2 in mouse adipose. Cell Death Dis. 2016;7(11):e2487. doi:10.1038/cddis.2016.388
33. Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13(10):1211–1233. doi:10.1101/gad.13.10.1211
34. Kim HJ, Jeong JS, Kim SR, Park SY, Chae HJ, Lee YC. Inhibition of endoplasmic reticulum stress alleviates lipopolysaccharide-induced lung inflammation through modulation of NF-kappaB/HIF-1alpha signaling pathway. Sci Rep. 2013;3(1):1142. doi:10.1038/srep01142
35. Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12(7):982–995.
36. Endo M, Oyadomari S, Suga M, Mori M, Gotoh T. The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J Biochem. 2005;138(4):501–507. doi:10.1093/jb/mvi143
37. Zeng M, Sang W, Chen S, et al. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol Lett. 2017;271:26–37. doi:10.1016/j.toxlet.2017.02.023
38. van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 2014;1843(10):2253–2262. doi:10.1016/j.bbamcr.2014.03.009
39. Filadi R, Theurey P, Pizzo P. The endoplasmic reticulum-mitochondria coupling in health and disease: molecules, functions and significance. Cell Calcium. 2017;62:1–15. doi:10.1016/j.ceca.2017.01.003
40. Leboucher GP, Tsai YC, Yang M, et al. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell. 2012;47(4):547–557. doi:10.1016/j.molcel.2012.05.041
41. Pal M, Febbraio MA, Lancaster GI. The roles of c-Jun NH2-terminal kinases (JNKs) in obesity and insulin resistance. J Physiol. 2016;594(2):267–279. doi:10.1113/JP271457
42. Zheng Y, Zhang M, Zhao Y, Chen J, Li B, Cai W. JNK inhibitor SP600125 protects against lipopolysaccharide-induced acute lung injury via upregulation of claudin-4. Exp Ther Med. 2014;8(1):153–158. doi:10.3892/etm.2014.1684
43. Delmotte P, Sieck GC. Endoplasmic reticulum stress and mitochondrial function in airway smooth muscle. Front Cell Dev Biol. 2019;7:374. doi:10.3389/fcell.2019.00374
44. Diaz B, Fuentes-Mera L, Tovar A, et al. Saturated lipids decrease mitofusin 2 leading to endoplasmic reticulum stress activation and insulin resistance in hypothalamic cells. Brain Res. 2015;1627:80–89. doi:10.1016/j.brainres.2015.09.014
45. Arruda AP, Pers BM, Parlakgul G, Guney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20(12):1427–1435. doi:10.1038/nm.3735
46. Yap J, Chen X, Delmotte P, Sieck GC. TNFalpha selectively activates the IRE1alpha/XBP1 endoplasmic reticulum stress pathway in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2020;318(3):L483–L493. doi:10.1152/ajplung.00212.2019
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.