")
Back to Journals » OncoTargets and Therapy » Volume 10
Adenocarcinoma of the lung with EGFR gene mutation and subsequent resistance mechanisms exploration: case report
Received 8 June 2017
Accepted for publication 11 August 2017
Published 12 September 2017 Volume 2017:10 Pages 4517—4525
DOI https://doi.org/10.2147/OTT.S143501
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Samir Farghaly
Li Xu,1,2 Qian Z Wang,1,2 Lin Wu1,2
1Department of the Second Chest Medicine, Hunan Cancer Hospital, Changsha, Hunan, People’s Republic of China; 2Department of the Second Chest Medicine, The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, Hunan, People’s Republic of China
Abstract: The treatment of lung cancer has made paradigm-shift advancements in the past decade with the development of therapies directed at specific genetic alterations, such as epidermal growth factor receptor (EGFR). Here, we present a rare case of lung adenocarcinoma harboring EGFR activating mutation and ALK overexpression. During the EGFR-tyrosine kinase inhibitors treatment, next-generation sequencing revealed phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway amplifications in tumor specimen and subsequent T790M mutation via plasma circulating tumor DNA. In conclusion, this case illustrates the existence of concomitant resistance mechanisms and demonstrates that circulating tumor DNA can reflect tumor heterogeneity.
Keywords: epidermal growth factor receptor, PI3K/Akt/mTOR pathway, T790M, next-generation sequencing, circulating tumor DNA
Introduction
Epidermal growth factor receptor (EGFR) mutations have been reported in ~50% of patients with lung adenocarcinomas. Clinical studies have demonstrated that non-small-cell lung cancer (NSCLC) patients with EGFR gene mutations may benefit from EGFR-tyrosine kinase inhibitors (EGFR-TKIs).1 Tissue biopsy is not only invasive but also limited in capturing spatial and temporal heterogeneity associated with tumors. Circulating tumor DNA (ctDNA) multiplex genotyping provides an alternative that can identify the heterogeneity across lesions. We report here a rare case of lung adenocarcinoma harboring EGFR mutation, which was followed by the exploration of the resistance mechanisms via next-generation sequencing (NGS).
Case report
In August 2014, a 40-year-old female, never-smoker, presented to our institution with left shoulder pain. The patient provided written informed consent for the publication of her case details. Computed tomography (CT) revealed a primary mass lesion in the left lower lobe, multiple bilateral pulmonary nodules, and enlargement of mediastinal lymph nodes. Her head magnetic resonance imaging scan revealed several small metastatic nodules, the biggest in cerebellum. The isotope bone scans revealed many metastatic mass lesions in the spine, scapula, rib, nasal, and hip bone. A left supraclavicular lymph node surgical biopsy was conducted (size 1.5×1.5 cm), and the pathological diagnosis was adenocarcinoma (cT4N3M1ab according to the 7th AJCC/UICC tumor staging system) (Figure 1A), with EGFR exon 19 deletion proven using amplication-refractory mutation system assay (Figure 1B) and ALK showing up as positive using Ventana immunohistochemistry (IHC) (Figure 1C). The metastatic lesions in the brain and bones are shown in Figure 1D and E. The main target lesion in the left lung prior to the treatment is shown in Figure 1F. The patient was administered gefitinib as first-line treatment at 250 mg qd in August 2014. Evaluation performed at 1 month revealed partial response across all lesions, with minimal disease burden at 7 months (Figure 1G). However, CT scan showed progression of the lung lesions after 8 months, because of several new lesions in both lungs (Figure 1H).
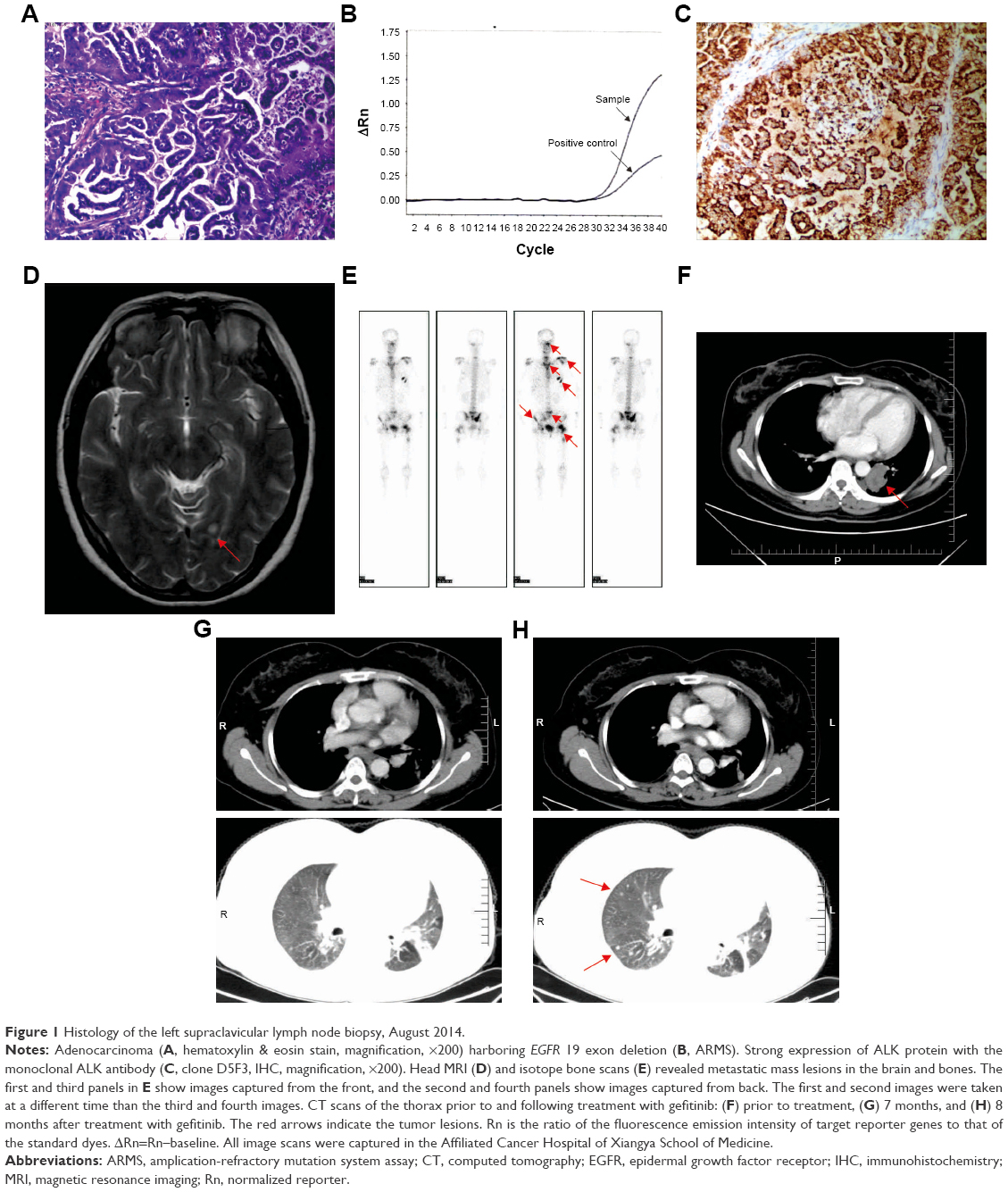 | Figure 1 Histology of the left supraclavicular lymph node biopsy, August 2014. |
Ventana IHC has been approved by the China Food and Drug Administration in identifying patients who are eligible for treatment with crizotinib. Based on ALK overexpression assessed using Ventana IHC, the patient was switched to a combinatorial treatment consisting of gefitinib and crizotinib, an ALK inhibitor, in May 2015. The patient was still kept on gefitinib given that the main target lesions were still in control, indicating their lasting response to gefitinib. During the treatment, the patient showed no major adverse events; however, CT scan still showed slow progression (Figure 2A and B). The patient remained on the combinatorial treatment for 10 months; in April 2016, the patient suffered left shoulder pain again and it was accompanied with shortness of breath. CT showed rapid progression, with several new lesions in both lungs. Repeat biopsy of the metastatic lymph node in R4 mediastinum coupled with NGS was performed to evaluate associated resistance mechanisms.
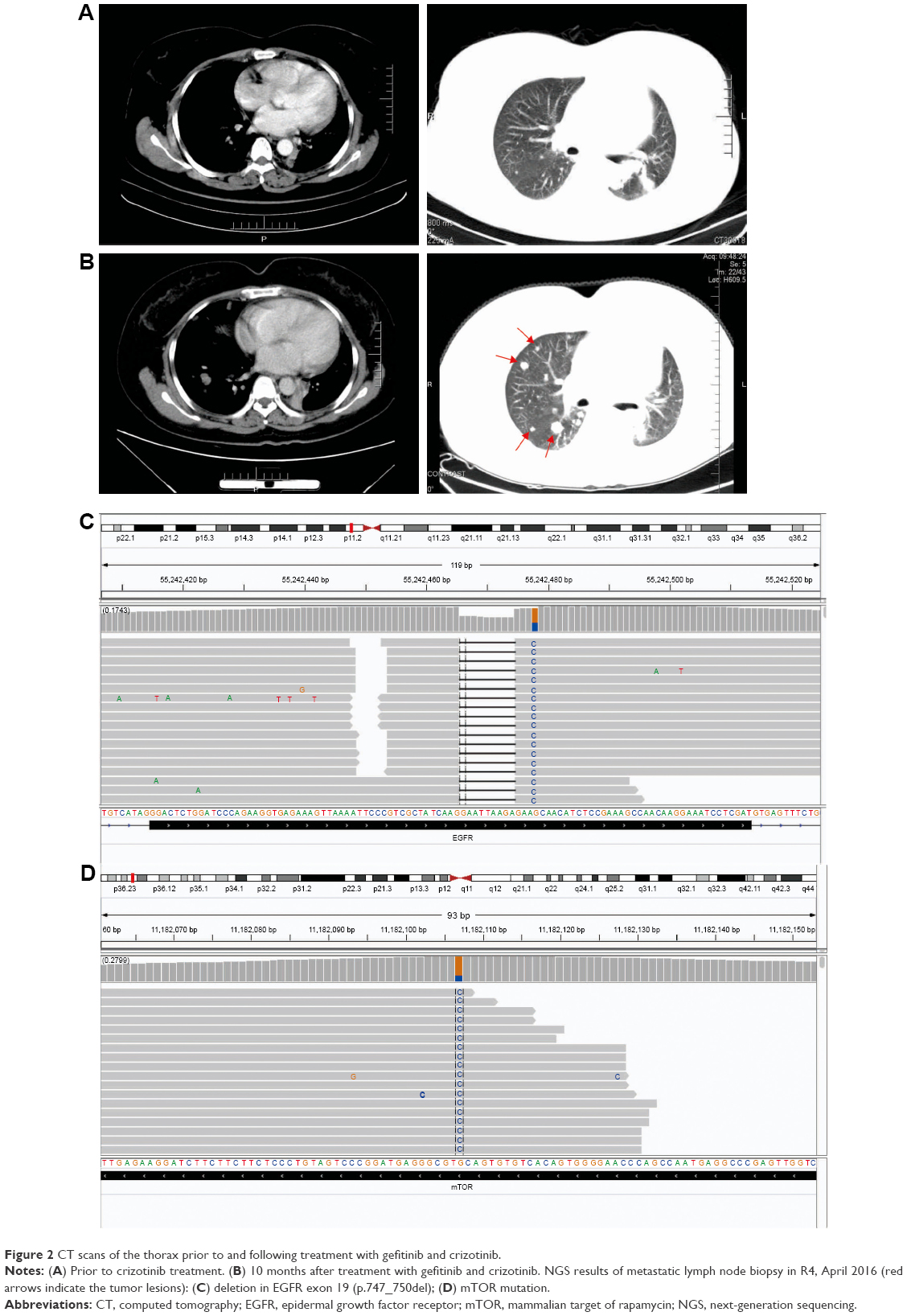 | Figure 2 CT scans of the thorax prior to and following treatment with gefitinib and crizotinib. |
NGS results showed EGFR exon 19 deletion (allele frequency [AF] 36.2%, Figure 2C) with PIK3CA/Akt/mammalian target of rapamycin (mTOR) pathway amplifications (PIK3CA, Akt1, mTOR copy number of 3.46, 3.51, 3.09, respectively) and mTOR mutation (AF 68.4%, Figure 2D). NGS did not reveal ALK fusion. Due to the presence of ALK overexpression at initial diagnosis, we assessed ALK status using Ventana IHC (Figure 3A) and fluorescence in situ hybridization (FISH) (Figure 3B), both of which were negative.
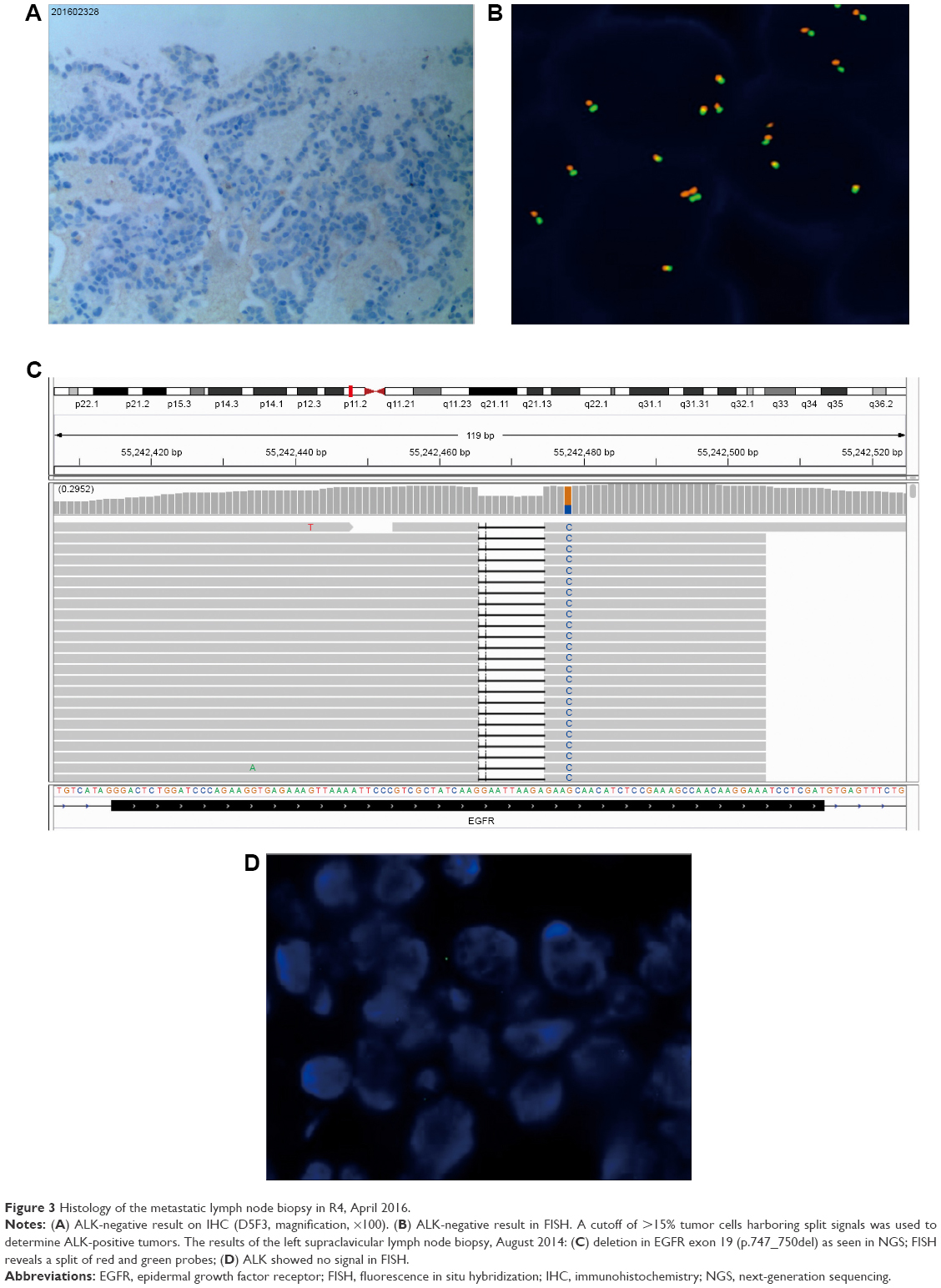 | Figure 3 Histology of the metastatic lymph node biopsy in R4, April 2016. |
We could not identify whether the PIK3CA/Akt1/mTOR pathway amplifications or mTOR mutation was the cause of resistance, so we selected the original biopsy specimen for NGS (the supraclavicular lymph node biopsy specimen in August 2014, when NGS was not popular). NGS showed that the tissue biopsy obtained at initial diagnosis only harbored EGFR mutation in exon 19 deletion (AF 34%, Figure 3C), and ALK FISH also showed no signal (Figure 3D).
Based on NGS and subsequent FISH results, the overexpression detected using Ventana IHC at initial diagnosis was most likely to be a false positive. Therefore, chemotherapy consisting of pemetrexed and cisplatin was administered in May 2016. After 2 cycles, CT showed that the disease had progressed to liver and both lungs (Figure 4A). The patient was reluctant to undergo second-line chemotherapy. The patient was switched to everolimus, an mTOR inhibitor, in combination with gefitinib based on previous molecular testing results. Evaluation was performed after 1.5 months of treatment with everolimus, and CT showed stable disease (Figure 4B). Due to the advancements in liquid biopsy and the inability to obtain another tissue biopsy, we performed liquid biopsy coupled with capture-based targeted ultra-deep sequencing. ctDNA analysis showed EGFR 19 exon deletion (allele frequency 17.8%, Figure 4C) and EGFR20 T790M mutation (allele frequency 7.88%, Figure 4D). The patient was administered AZD 9291 at a dose of 80 mg/d and achieved partial response with significant reduction in the bilateral pulmonary and liver nodules after 1.4 months (Figure 4E) accompanied with the amelioration of left shoulder pain. Unfortunately, the patient experienced severe shortness of breath with a performance status of 3 after treatment with AZD9291 for 1.9 months. A CT scan showed dramatic bulky lung disease (Figure 4F), and the patient passed away on September 30, 2016; the time to progression or death from the initial administration of AZD9291 for the patient was 1.9 months. Overall survival was >24 months.
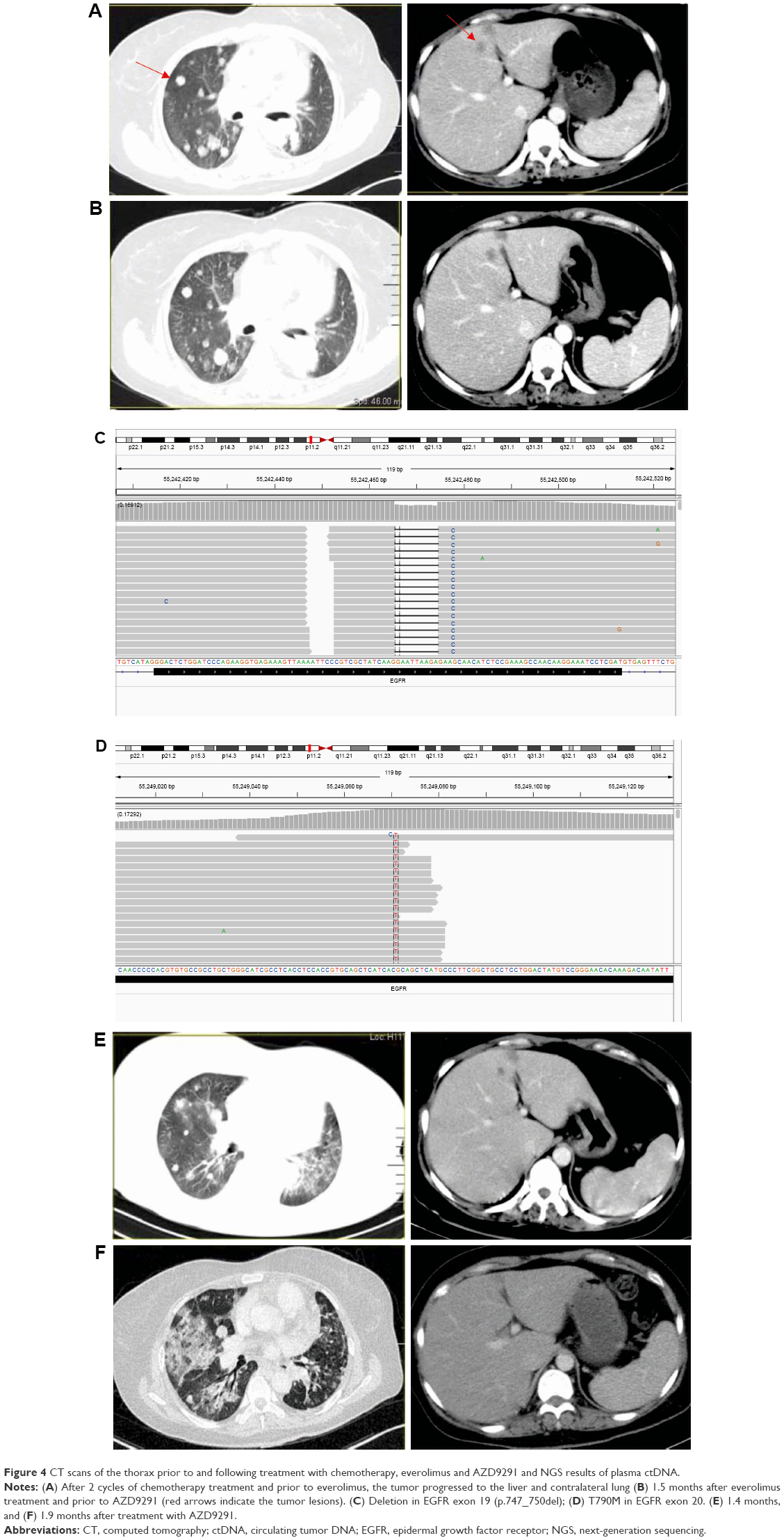 | Figure 4 CT scans of the thorax prior to and following treatment with chemotherapy, everolimus and AZD9291 and NGS results of plasma ctDNA. |
Discussion
Acquired resistance to EGFR inhibitors inevitably occurs in patients who initially respond to therapy, and they have a median progression-free survival of about 10 months.2 In the reported patient, after the treatment with gefitinib for 8 months, the primary lung lesion showed gradual progression. NGS plays an important role in detecting genetic status and identifying the heterogeneity of resistance mechanisms, which allows simultaneous upfront detection of clinically relevant point mutations, amplifications, deletions, and gene fusions in a one-stop technique which is highly efficient and tissue sparing.3,4 NGS detected the PIK3CA/Akt1/mTOR pathway and T790M mutation resistance mechanisms in the patient.
Activating mutations in EGFR result in activation of multiple downstream signal transduction pathways such as phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR pathway. PIK3CA encodes the p110α isoform of the catalytic subunit of PI3K. Activated PI3K phosphorylates Akt and leads to downstream activation of mTOR.5,6 PI3K/Akt/mTOR pathway which has been implicated in lung tumorigenesis, with mutations, amplifications, and epigenetic alterations reported at various points in the cascade and dysregulation of the pathway contributes to lung cancer progression and resistance to therapies that target EGFR-TKIs.7,8
In the current case, rebiopsy of the lymph node at progressive disease after the commencement of gefitinib in combination with crizotinib revealed PI3K/Akt/mTOR amplifications and mutations, including the PIK3CA, Akt1, and mTOR amplification and mTOR mutation. The acquired development of resistance to EGFR-TKI through the copy number gains of PIK3CA/Akt/mTOR can serve as a potential resistance mechanism, but such mutations were not present prior to treatment. The patient’s condition was stable after the treatment with everolimus, revealing its effectiveness.
Liquid biopsy performed during the treatment with everolimus and gefitinib revealed EGFR T790M, one of the major mechanisms of resistance associated with first-generation EGFR-TKIs. AZD9291 (osimertinib), a potent drug for both EGFR-TKI sensitizing and T790M resistance mutations, displayed greater efficacy than platinum therapy plus pemetrexed in T790M-positive advanced NSCLC.9 In our case, the patient was switched to AZD9291 upon T790M detection, and significant response was achieved.
The most frequent mechanisms in addition to EGFR T790M are the activation of other receptor tyrosine kinases, including ALK (totally 29% in T790M− patients), RAS/MEK/ERK pathway (totally 17%), as well as PIK3CA/AKT/mTOR pathway (totally 20%). Genetic spectrums between EGFR T790M+ and T790M− groups showed that the EGFR T790M+ group did not possess any mutations in genes such as KRAS, ALK, and PIK3CA.10 There was another prospective study of 155 patients in which the most commonly observed mechanism of resistance was EGFR T790M, and tumor biopsy at the time of clinical acquired resistance to EGFR-TKIs displayed a spectrum of mechanisms, with rare overlap (4%) among mechanisms; the coexistence of T790M and PIK3CA mutations was not observed.11 Therefore, the case we have reported may be the first one with a dual resistance mechanism: PI3K/Akt/mTOR pathway amplifications and T790M mutation.
Several studies recently discovered that ALK rearrangement and EGFR mutation may coexist in the same tumor, with an incidence rate of 1.3% in patients with NSCLC in China.12 Crizotinib was administrated based on positive ALK expression revealed by Ventana IHC in the present case. However, no obvious clinical response was observed. Subsequent validations using samples obtained prior to and after treatment by various methods, including FISH, IHC, and NGS, suggested that the initial positive ALK expression result revealed by IHC may be a false positive. Validating IHC results by other methods is necessary.
It is necessary to obtain genetic information from biopsy or liquid biopsy (ctDNA) prior to the treatment and during the disease course, as this can guide treatment options and help detect resistance to treatments. In order to deliver an appropriate first-line treatment regimen, detection of EGFR mutation and other gene alterations is recommended as routine genetic profiling for nonsquamous NSCLC. Only EGFR gene mutation detection is not enough prior to the treatment, because it is hard to find out details regarding newly acquired resistant mechanisms solely from posttreatment results due to the lack of comprehensive genetic information.
Tissue biopsies, the gold standards for mutation genotyping, often are not accessible, especially in advanced NSCLC patients who have progressed on prior treatments. Furthermore, tissue biopsy cannot reflect tumor heterogeneity due to its spatial and temporal snapshot nature. Use of ctDNA to detect activating mutations provides a promising diagnostic approach if tumor tissue is not accessible. Plasma ctDNA may overcome time and space heterogeneity, having the potential to reflect tumor heterogeneity. The ASSESS study suggested that detection of EGFR mutations in plasma was significantly more likely when patients had a higher metastatic tumor burden and distant metastases.13 In our case, the patient had multiple lesions in both lungs, liver, and bone, which may have different genetic alterations. Therefore, liquid biopsy is a better approach to overcome spatial heterogeneity. We recommend that ctDNA analysis should be performed at the same time as a tissue biopsy is ordered if available/feasible, as each provides complementary information.
To the best of our knowledge, this is the first report of the coexistence of PIK3CA/Akt1/mTOR pathway activation and T790M mutation in a patient with EGFR mutation. This unique case highlights the feasibility and necessity of using NGS and ctDNA multiplex genomic profiling in identifying molecular diagnosis of NSCLC or in the exploration of the underlying resistance mechanisms of targeted treatments. Further well-designed studies to assess its true effectiveness as well as criteria for eligible patient selection are warranted.
Acknowledgment
This work was supported by the Beijing Hope Run Special Fund of Cancer Foundation of China (grant number LC2016W09).
Disclosure
The authors report no conflicts of interest in this work.
References
Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. | ||
Paz-Ares L, Soulières D, Melezínek I, et al. Clinical outcomes in non-small-cell lung cancer patients with EGFR mutations: pooled analysis. J Cell Mol Med. 2010;14:51–69. | ||
Drilon A, Wang L, Arcila ME, et al. Broad, hybrid capture-based next-generation sequencing identifies actionable genomic alterations in “driver-negative” lung adenocarcinomas. Clin Cancer Res. 2015;21(16):3631–3639. | ||
Paweletz CP, Sacher AG, Raymond CK, et al. Bias-corrected targeted next-generation sequencing for rapid, multiplexed detection of actionable alterations in cell-free DNA from advanced lung cancer patients. Clin Cancer Res. 2016;22(4):915–922. | ||
Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–2899. | ||
Janmaat ML, Rodriguez JA, Gallegos-Ruiz M, Kruyt FAE, Giaccone G. Enhanced cytotoxicity induced by gefitinib and specific inhibitors of the Ras or phosphatidyl inositol-3 kinase pathways in non-small cell lung cancer cells. Int J Cancer. 2006;118:209–214. | ||
Ji M, Guan H, Gao C, Shi B, Hou P. Highly frequent promoter methylation and PIK3CA amplification in non-small cell lung cancer (NSCLC). BMC Cancer. 2011;11:147. | ||
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. | ||
Mok TS, Wu YL, Ahn MJ, et al. AURA3 investigators. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2016;376:629–640. | ||
Jin Y, Shao Y, Shi X, et al. Mutational profiling of non-small-cell lung cancer patients resistant to first-generation EGFR tyrosine kinase inhibitors using next generation sequencing. Oncotarget. 2016;7(38):61755–61763. | ||
Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. | ||
Yang JJ, Zhang XC, Su J, et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res. 2014;20:1383–1392. | ||
Normanno N, Brown H, Haddad V, et al. 580_PR: Clinical and demographic features that influence EGFR mutation detection in plasma from patients (pts) with aNSCLC: The ASSESS experience. J Thoracic Oncol. 2016;11:S151. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.