")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Additive effects of cilnidipine, an L-/N-type calcium channel blocker, and an angiotensin II receptor blocker on reducing cardiorenal damage in Otsuka Long-Evans Tokushima Fatty rats with type 2 diabetes mellitus
Authors Mori Y, Aritomi S, Niinuma K, Nakamura T, Matsuura K, Yokoyama J, Utsunomiya K, Ishikawa T
Received 30 April 2013
Accepted for publication 27 February 2014
Published 17 June 2014 Volume 2014:8 Pages 799—810
DOI https://doi.org/10.2147/DDDT.S47441
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Yutaka Mori,1,2 Shizuka Aritomi,3 Kazumi Niinuma,3 Tarou Nakamura,3 Kenichi Matsuura,1 Junichi Yokoyama,1 Kazunori Utsunomiya1
1Division of Diabetes and Endocrinology, Department of Internal Medicine, The Jikei University School of Medicine, Minato-ku, Japan; 2Department of Clinical Research, National Hospital Organization, Utsunomiya National Hospital, Utsunomiya, Japan; 3Research Center, Ajinomoto Pharmaceuticals Co, Ltd, Kanagawa, Japan
Abstract: Cilnidipine (Cil), which is an L-/N-type calcium channel blocker (CCB), has been known to provide renal protection by decreasing the activity of the sympathetic nervous system (SNS) and the renin–angiotensin system. In this study, we compared the effects of the combination of Cil and amlodipine (Aml), which is an L-type CCB, with an angiotensin (Ang) II receptor blocker on diabetic cardiorenal damage in spontaneously type 2 diabetic rats. Seventeen-week-old Otsuka Long-Evans Tokushima Fatty rats were randomly assigned to receive Cil, Aml, valsartan (Val), Cil + Val, Aml + Val, or a vehicle (eight rats per group) for 22 weeks. Antihypertensive potencies were nearly equal among the CCB monotherapy groups and the combination therapy groups. The lowering of blood pressure by either treatment did not significantly affect the glycemic variables. However, exacerbations of renal and heart failure were significantly suppressed in rats administered Cil or Val, and additional suppression was observed in those administered Cil + Val. Although Val increased the renin–Ang system, Aml + Val treatment resulted in additional increases in these parameters, while Cil + Val did not show such effects. Furthermore, Cil increased the ratio of Ang-(1–7) to Ang-I, despite the fact that Val and Aml + Val decreased the Ang-(1–7) levels. These actions of Cil + Val might be due to their synergistic inhibitory effect on the activity of the SNS, and on aldosterone secretion through N-type calcium channel antagonism and Ang II receptor type 1 antagonism. Thus, Cil may inhibit the progression of cardiorenal disease in type 2 diabetes patients by acting as an N-type CCB and inhibiting the aldosterone secretion and SNS activation when these drugs were administered in combination with an Ang II receptor blocker.
Keywords: calcium channel blocker, combination therapy, angiotensin, type 2 diabetes mellitus
Introduction
The renin–angiotensin system (RAS) is a dual system that is composed of two arms: vasoconstriction, which involves the angiotensin (Ang)-converting enzyme (ACE)–Ang II–Ang II type 1 receptor (AT1-R); and vasodilation, which involves the ACE2–Ang-(1–7)–Mas receptor. The ACE2–Ang-(1–7)–Mas receptor arm primarily acts as a counterregulatory mechanism to the vasoconstrictor arm.1 The final functional effects of the RAS thus reflect the balance between these two arms.2–4 The circulating RAS plays a role in hemodynamic regulation through Ang II, which is a potent vasoconstrictor, and the counterregulatory peptide, Ang-(1–7), which is a vasodilator. Moreover, the ACE–Ang II–AT1-R arm primarily leads to the exacerbation of diabetic nephropathy, and the ACE2–Ang-(1–7)–Mas receptor arm mainly acts as a counterregulatory mechanism.5–7 Therefore, inhibition of the RAS with Ang II receptor blockers (ARBs) prevents the occurrence and progression of complications in patients with diabetes and diabetic nephropathy.8–10 However, ARBs increase plasma renin activity (PRA) and plasma Ang II levels by uncoupling this negative feedback loop through their inhibitory effects on the AT1-R.11 Experimental and clinical studies have shown that the combination treatment of ARB and an ACE inhibitor or a direct renin inhibitor (DRI) can prevent the increases in RAS activity that are induced by the ARB, and they can provide additive or synergistic effects by lowering blood pressure (BP) and preventing progressive organ damage.12–14 At the same time, recent data from the Ongoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial (ONTARGET)15 and Aliskiren Trial in Type 2 Diabetes Using Cardio–Renal Endpoints (ALTITUDE)16 studies suggest that kidney-related outcomes worsened with ACE inhibitor + ARB or DRI + ARB combinations. Consequently, these dual therapies are no longer recommended in patients with diabetes until further data become available from future studies.
Cilnidipine (Cil), which blocks both N- and L-type calcium channels,17 has a variety of unique actions that are mediated by its actions as an N-type calcium channel blocker (CCB), including the suppression of sympathetic nervous overactivity,18–20 cardiovascular system protection,21–24 and kidney protection.25–29 In addition, we have found that Cil prevents the increases of PRA and plasma Ang II levels that occur in spontaneously hypertensive rats30 and in a canine model of a chronic atrioventricular block that exhibits ventricular electrical remodeling,31 despite the fact that the L-type CCB amlodipine (Aml) causes an increase in PRA and plasma Ang II levels due to its antihypertensive actions.30,32 Moreover, Cil directly suppresses aldosterone secretion from adrenocortical cells through its N-type CCB actions.33 These results suggest that N-type calcium channels are important regulators of renin–Ang–aldosterone system (RAAS) activity. Moreover, Cil could suppress ARB-induced increases in RAS activity through its sympatholytic actions that are mediated by N-type calcium channel blockade.34 Additionally, a clinical trial that compared the CCBs Cil and Aml, in combination with a RAS inhibitor, showed that Cil is superior to Aml in preventing the progression of proteinuria25 and RAAS elevation35 in hypertensive patients. In addition, we previously showed that when each of these drugs was used in combination with valsartan (Val), Cil was more effective than Aml in preventing impaired kidney function and in preserving the glomerular slit membrane partly because of the suppression of ARB-induced RAS activation. These results showed that Cil suppresses ARB-induced increases in RAS activity and exerts organ-protective action when administered concomitantly with an ARB.36 However, very few studies have evaluated the effects of Cil in type 2 diabetes mellitus patients. Therefore, we compared the effects of Cil and the L-type CCB Aml on diabetic cardiorenal damage in spontaneously type 2 diabetic rats (Otsuka Long-Evans Tokushima Fatty [OLETF] rats) when these drugs were administered in combination with an ARB.
Materials and methods
Animals
Male OLETF rats (Otsuka Pharmaceutical Co, Ltd, Tokushima, Japan) were used at 17 weeks of age. Rats were kept at 22°C–25°C under a 12-hour light–dark cycle, and they had free access to chlorinated water and commercial chow (CRF-1; Oriental Yeast Co, Ltd, Tokyo, Japan). All animal experiments were performed in accordance with the guiding principles for the care and use of laboratory animals, which have been approved by the Japanese Pharmacological Society (Tokyo, Japan).
Drugs
The N-type and L-type CCB, Cil (Ajinomoto Co, Inc., Tokyo, Japan), the L-type CCB, Aml (Lek d. d., Kolodvorska, Slovenia), and Val (Livzon Group Changzhou Kony Pharmaceutical Co, Ltd, Changzhou, People’s Republic of China) were used as antihypertensive drugs. Each antihypertensive drug was initially dissolved in 0.5% hydroxypropyl methylcellulose (Sigma-Aldrich, St Louis, MO, USA).
Experimental procedures
Seventeen-week-old OLETF rats were divided into the following six groups: vehicle group (vehicle; number [n] =8); Cil group (2 mg/kg of Cil; n=8); Aml group (2 mg/kg of Aml; n=8); Val group (10 mg/kg of Val; n=8), Cil + Val group (combination of 2 mg/kg of Cil and 10 mg/kg of Val; n=8); and Aml + Val group (combination of 2 mg/kg of Aml and 10 mg/kg of Val; n=8). These groups were compared based on measurements of BP, body weight, urinary albumin excretion (UAE), and levels of glycohemoglobin (HbA1c), insulin, and blood glucose. The dosages were selected based on a preliminary examination. Rats were orally administered each drug for 22 weeks. Each drug was orally administered to the rats once a day every morning. Nineteen weeks after the start of drug administration, fasting venous blood samples were taken from the tail vein to assess blood glucose, insulin, HbA1c, and triglyceride levels. At the end of the study period, rats were anesthetized, blood was collected, and the heart and kidneys were quickly removed. Six Long-Evans Tokushima Otsuka (LETO) rats, which were treated identically to the vehicle group throughout the experimental period, served as the normal group.
BP measurements
Systolic BP was measured each week using the tail–cuff method and a Softron BP-98A BP meter (Softron Co, Ltd, Tokyo, Japan). Rats were introduced into a plastic wire holder and placed in a thermostatically warmed tube, which was maintained at 34°C–36°C during the measurements. The mean of three measurements was determined for each animal after environmental acclimation.
Measurements of biochemical parameters
HbA1c values were measured using ion-exchange high-performance liquid chromatography (HLC-723GHb VA1c2.2; TOSO, Japan). For the measurements of the other biochemical parameters, blood samples were centrifuged (3,000× g) for 15 minutes. Plasma insulin was measured using an insulin enzyme-linked immunosorbent assay (ELISA) kit (Morinaga, Takamatsu, Japan). Plasma glucose and triglyceride levels were determined using an autoanalyzer (Fuji Dri-Chem 5500; Fujifilm Holdings Corporation, Tokyo, Japan).
Measurement of plasma RAS activity
In order to measure the biochemical parameters, blood samples were centrifuged (1,000× g) for 15 minutes, and the plasma was stored at −80°C until analysis. PRA was determined using a radioimmunoassay system (Yamasa Shoyu Co, Ltd, Chiba, Japan), and Ang levels were purified by solid-phase extraction and measured using liquid chromatography–tandem mass spectrometry.
Measurement of noradrenaline (NA)
The noradrenaline (NA) concentration in the renal cortex was measured as follows. Briefly, pieces of the renal cortex were homogenized in a 3.3-fold volume of ice-cold saline containing 0.1 N HCl, and after centrifugation (12,000× g at 4°C) for 30 minutes, NA levels in the supernatant were measured using high-performance liquid chromatography.
Collection and analysis of urine
Rats were individually placed in metabolic cages, and urine samples were collected for 24 hours in order to determine UAE levels. Albumin concentrations were measured with a commercially available ELISA kit (Nephrat II; Exocell Inc., Philadelphia, PA, USA). Aldosterone levels were measured using an enzyme immunoassay system (Cayman Chemical Company, Ann Arbor, MI, USA).
Histological studies
The left kidney of each rat was fixed in fresh 10% formaldehyde and embedded in paraffin. Sections (3 μm thick) were stained with periodic acid–Schiff in order to assess glomerulosclerosis (GS). Fifty glomeruli in each specimen were examined. The severity of the lesions, according to the percentage of glomerular involvement, was observed under a light microscope (BX50; Olympus Corporation, Tokyo, Japan) with a blinded procedure and graded according to the following scale: 0, no lesions; 1+, less than 25%; 2+, 26%–50%; 3+, 51%–75%; and 4+, more than 76%.27 The myocardium was fixed in 10% formalin and stained with hematoxylin and eosin or Masson’s trichrome in order to determine the myocyte cross-sectional dimensions and interstitial fibrosis. Analyses of the slides were performed in a blinded fashion with a light microscope (BX50), and the relative volume occupied by each element of the ventricle (myocardial fibers and fibrous tissue) was measured using a special ocular apparatus (WinROOF version 3.5; Mitani, Fukui, Japan).
Quantitative reverse transcription–polymerase chain reaction
Total ribonucleic acid (RNA) was isolated from renal cortical tissues with the RNeasy Mini kit (Qiagen GmbH, Hilden, Germany). One microgram of total RNA was reverse-transcribed using SuperScript® III (Life Technologies, Carlsbad, CA, USA). Real-time quantitative reverse transcription–polymerase chain reaction was performed with a 7500 Real-Time PCR System with SYBR® Green Master Mix (Applied Biosystems; Life Technologies) and sequence-specific primers. Primer sequences were as follows: renin (forward primer, 5′-TCTCTCCCAGAGGGTGCTAA-3′; reverse primer, 5′-TTGCCCTGGTAATGTTGAGG-3′), (pro)renin receptor (forward primer, 5′-CATCCCTGGTGAGGAAGTCAAG-3′; reverse primer, 5′-CGCAAGGTTGTAGGGACTTTG-3′), ACE2 (forward primer, 5′-GTCGTGATGGGAACGGTAGT-3′; reverse primer, 5′-TTTCTCCTTTGCCAATGTCC-3′), ACE (forward primer, 5′-CGAATGTGCCATACATCAGG-3′; reverse primer, 5′-CCTGCTTCCTTGGATTGGTA-3′), and beta-actin (forward primer, 5′-AGATTACTGCCCTGGCTCCT-3′; reverse primer, 5′-CAGTGAGGCCAGGATAGAGC-3′). In addition, messenger RNA (mRNA) levels were normalized to beta-actin levels.
Statistical analysis
Data were expressed as means ± standard error of the mean. One-way analysis of variance, which was followed by Dunnett’s multiple-range test, was used to assess the differences between the vehicle group and the other OLETF rat groups. Parameters in the vehicle and normal groups were compared using unpaired t-tests. Differences were considered statistically significant when the probability values were less than 0.05.
Results
Systolic BP, body weight, and organ weight
Systolic BP, body weight, and organ weight of the vehicle group were significantly greater than those of the normal group (Table 1). Antihypertensive effects were compared among the OLETF rats of the six groups, and all of the antihypertensive drug treatment groups, except for the Aml group, showed significantly decreased systolic BPs compared to the vehicle group (Table 1).
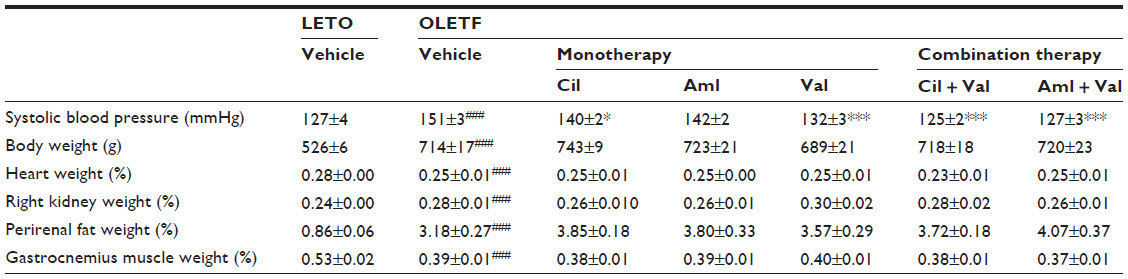 | Table 1 Blood pressure, body weight, and relative organ weight in LETO rats and OLETF rats at the end of the study period |
Biochemical parameters
The levels of all biochemical parameters, except for HbA1c, were significantly higher in the vehicle group than in the normal group (Table 2). HbA1c levels tended to be higher in the vehicle group than in the normal group (P=0.058; Table 2). Antihypertensive treatment had no effect on these parameters.
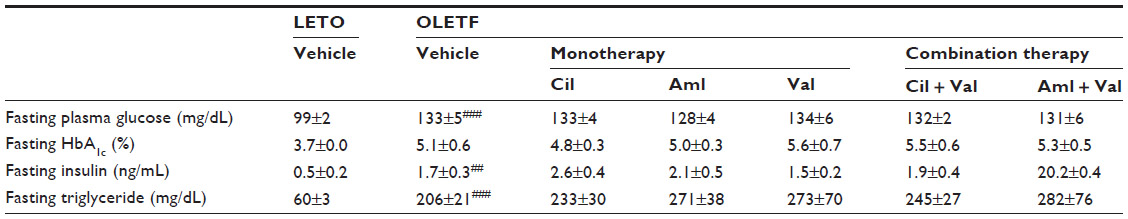 | Table 2 Biochemical measurements in LETO rats and OLETF rats at 19 weeks after the start of drug administration |
UAE and glomerulosclerosis score (GS)
The UAE levels of the vehicle group (244.3±85.4 mg/day) were significantly higher than those of the normal group (0.4±0.1 mg/day) (P<0.05; Figure 1A). All antihypertensive treatment groups exhibited a declining trend in UAE levels compared with those in the vehicle group, but only the Cil + Val group showed a significant reduction in UAE levels (66.4±13.8 mg/day) (P<0.05; Figure 1A). Additionally, the GS score of the vehicle group (1.45±0.05) was significantly higher than that of the normal group (0.50±0.03) (P<0.001; Figure 1B). The Cil, Val, and Cil + Val groups exhibited significantly lower GS scores (1.11±0.04, 1.12±0.06, and 1.10±0.03, respectively) than did the vehicle group (P<0.001 for each; Figure 1B).
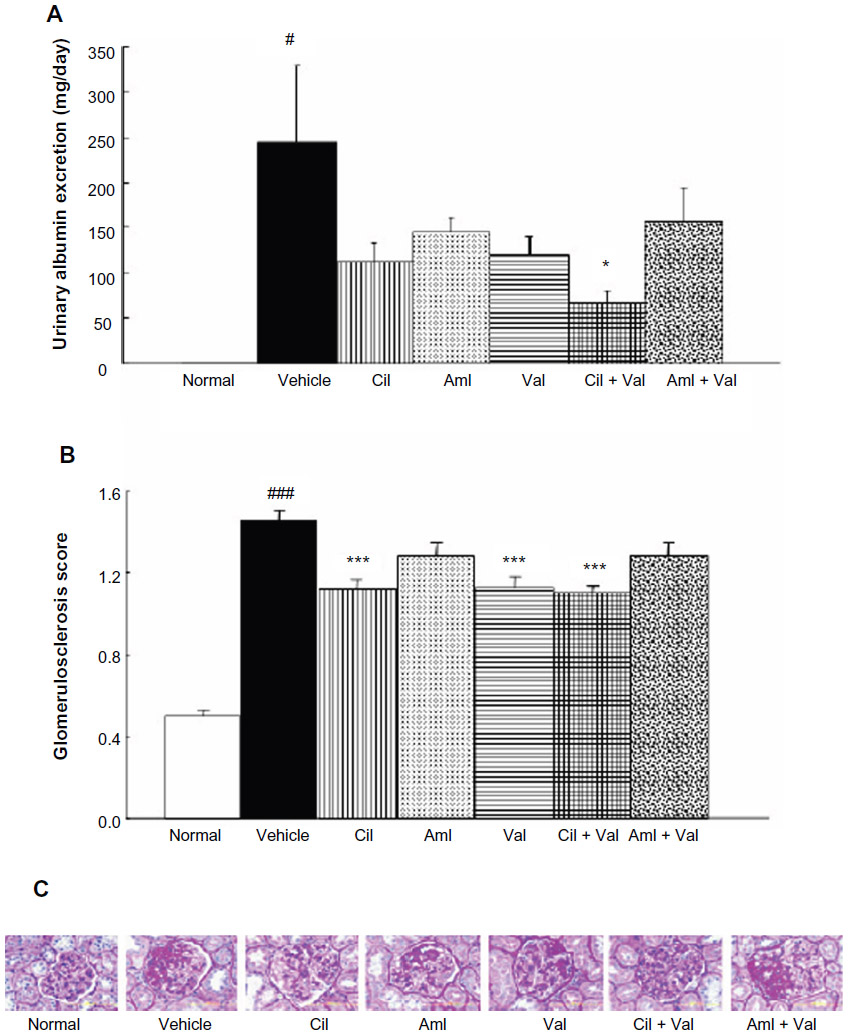 | Figure 1 Urinary albumin excretion levels. |
Cardiomyocyte size and cardiac fibrosis
Cardiomyocyte dimensions of the vehicle group (2,189.2±88.6 μm3) were significantly larger than those of the normal group (1,611.3±113.5 μm3) (P<0.01; Figure 2A). The Cil, Val, and Cil + Val groups had significantly smaller cardiomyocyte sizes (1,838.9±99.1 μm3; 1,867.2±55.7 μm3; and 1,707.6±66.4 μm3, respectively) than that of the vehicle group (P<0.05, P<0.05, and P<0.01, respectively; Figure 2A). In addition, only the Cil + Val group showed a significant reduction in cardiac fibrosis (4.93%±0.32%) compared to the vehicle group (6.49%±0.043%) (P<0.05; Figure 2B).
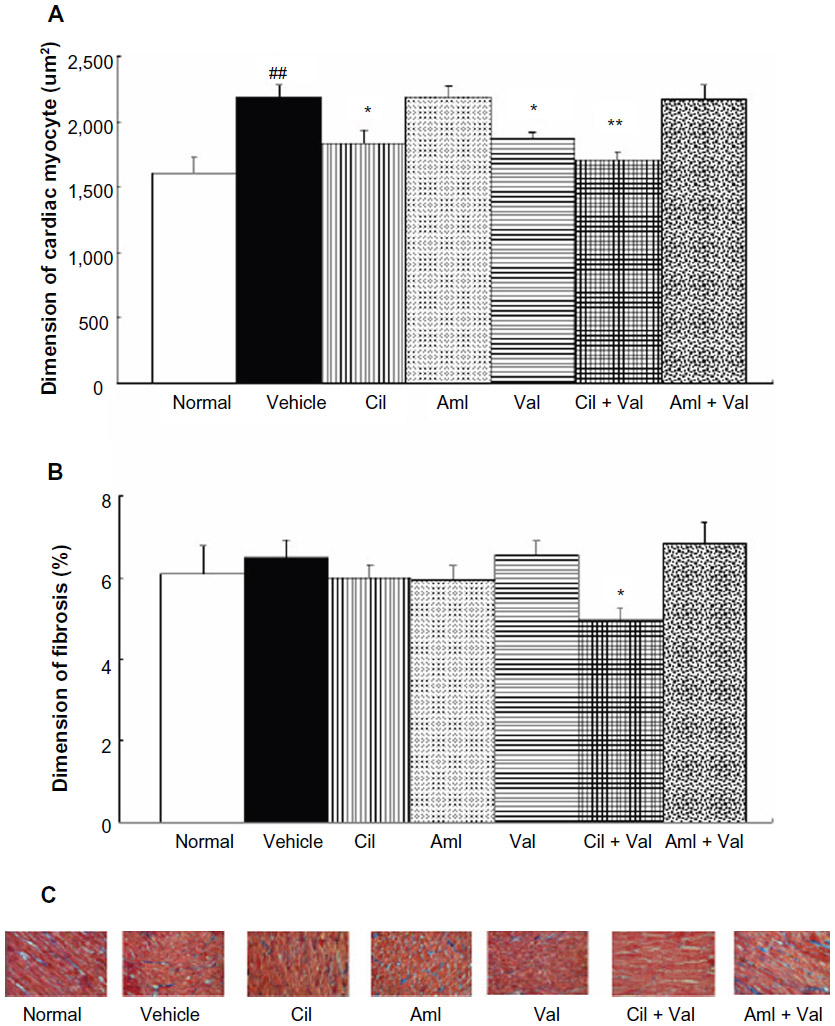 | Figure 2 Myocardial hypertrophy, fibrosis, and Masson’s trichrome-stained sections in OLETF rats treated with each drug for 22 weeks. |
Analysis of kidney cortex
The NA contents in the kidneys of the vehicle group (505.4±12.6 ng/kidney) were significantly greater than those of the normal group (401.4±12.8 ng/kidney) (P<0.05; Figure 3A), and the Cil + Val group showed significantly reduced NA contents (407.4±17.3 ng/kidney) compared with those of the vehicle group (P<0.05; Figure 3A). Renin expression levels in the kidney cortex of the vehicle group (14.1±1.5) were significantly lower than those of the normal group (21.3±2.6) (P<0.05; Figure 3B), and the Aml + Val group showed significantly increased levels of expression of renin (26.0±3.7) compared with those of the vehicle group (P<0.05; Figure 3B). For (pro)renin receptor expression, there were no significant distinctions between the normal and vehicle groups (63.2±3.7 and 70.2±4.6), but the Cil + Val group showed a significant reduction in the levels of expression of the (pro)renin receptor (43.9±1.7) compared to the vehicle group (P<0.05; Figure 3C).
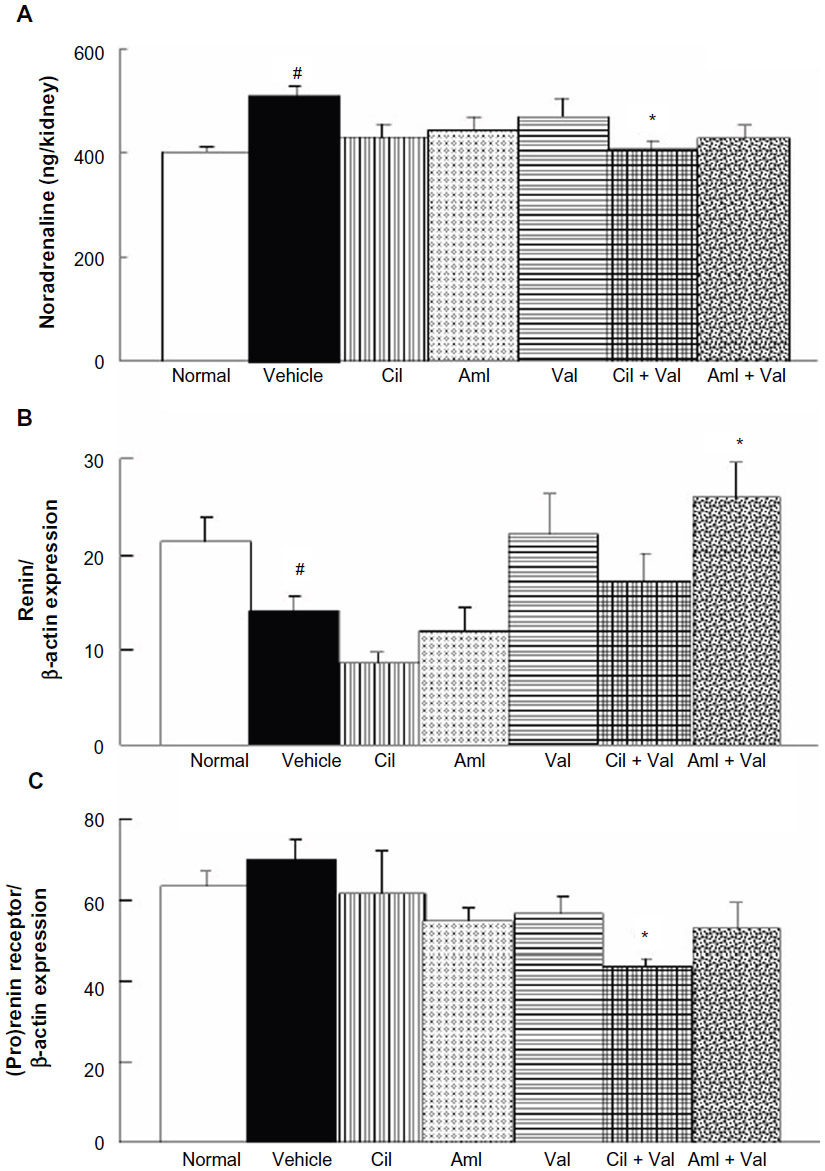 | Figure 3 Effects of the combined administration of Val + Aml and Val + Cil on noradrenalin levels and renin and (pro)renin receptor expression in the renal cortex. |
RAAS
As for PRA, only the Aml + Val group showed significantly higher PRA activity (19.8±4.0 mg/mL/hour) than the vehicle group (4.2±0.5 mg/mL/hour; P<0.001; Figure 4A), and no differences were observed between the vehicle group and the normal group (5.7±0.7 mg/mL/hour; Figure 4A). On the other hand, the plasma Ang I level of the vehicle group (189±10 pM) was significantly lower than that of the normal group (276±33 pM; P<0.001; Figure 4B). Further, the groups treated with Val showed a significantly higher level of plasma Ang I (Val: 418±61 pM, P<0.05; Cil + Val: 419±29 pM, P<0.05; Aml + Val: 777±128 pM, P<0.001) than did the vehicle group (Figure 4B). As is the case in PRA, only the Aml + Val group showed a significantly higher level of plasma Ang II (82.2±4.2 pM) than that of the vehicle group (35.4±12.5 pM, P<0.001; Figure 4C), and no differences were observed between the vehicle group and the normal group (48.2±1.2 pM; Figure 4C). Further, the plasma Ang-(1–7) level of the vehicle group (78.7±6.9 pM) was significantly lower than that of the normal group (125.8±13.5 pM, P<0.01; Figure 4D), and the combination therapy groups exhibited significantly higher level of plasma Ang-(1–7) (Cil + Val: 187.0±12.5 pM; P<0.01, Aml + Val: 174.8±26.7 pM; P<0.001) than the vehicle group did (Figure 4D).
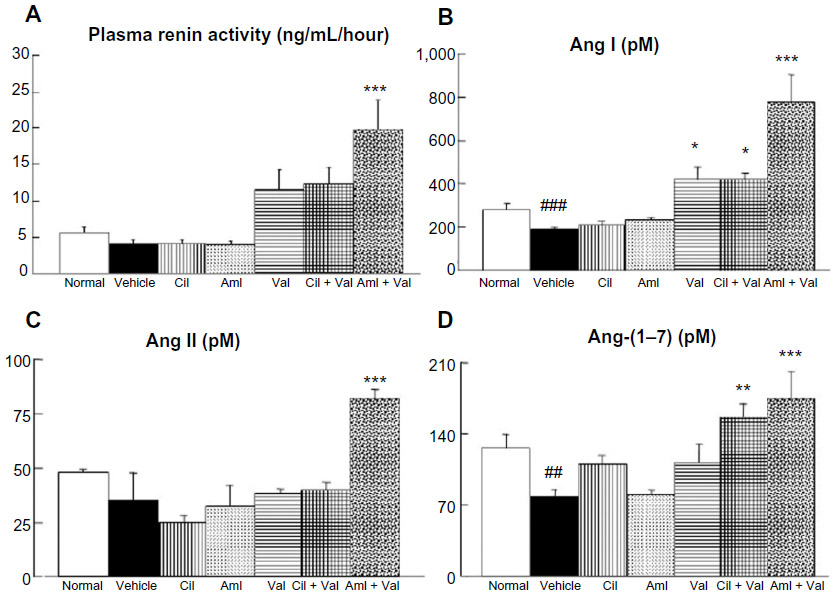 | Figure 4 Effects of the combined administration of Val + Aml and Val + Cil on plasma renin activity, Ang I, Ang II, and Ang-(1–7) levels. |
Furthermore, Ang ratios were determined, and the Ang-(1–7)/Ang I ratio was greater in the Cil group (0.54±0.04) than in the vehicle group (0.41±0.02; P<0.05; Figure 5A), whereas the Ang-(1–7)/Ang I ratios of the Val and the Aml + Val group (0.28±0.04 and 0.24±0.03) were lower than that of the vehicle group (P<0.05 and P<0.01, respectively; Figure 5A). On the other hand, no differences were observed in the Ang II/Ang I ratios among the groups (Figure 5B). In addition, there were no significant differences in the Ang-(1–7)/Ang II ratios and the ACE2/ACE ratios, but these ratios were generally similar (Figure 5C and D). In addition, only the Cil + Val group showed a significant reduction in aldosterone excretion (10.4±1.9 ng/day) compared to the vehicle group (22.8±4.4 ng/day; P<0.05; Figure 6).
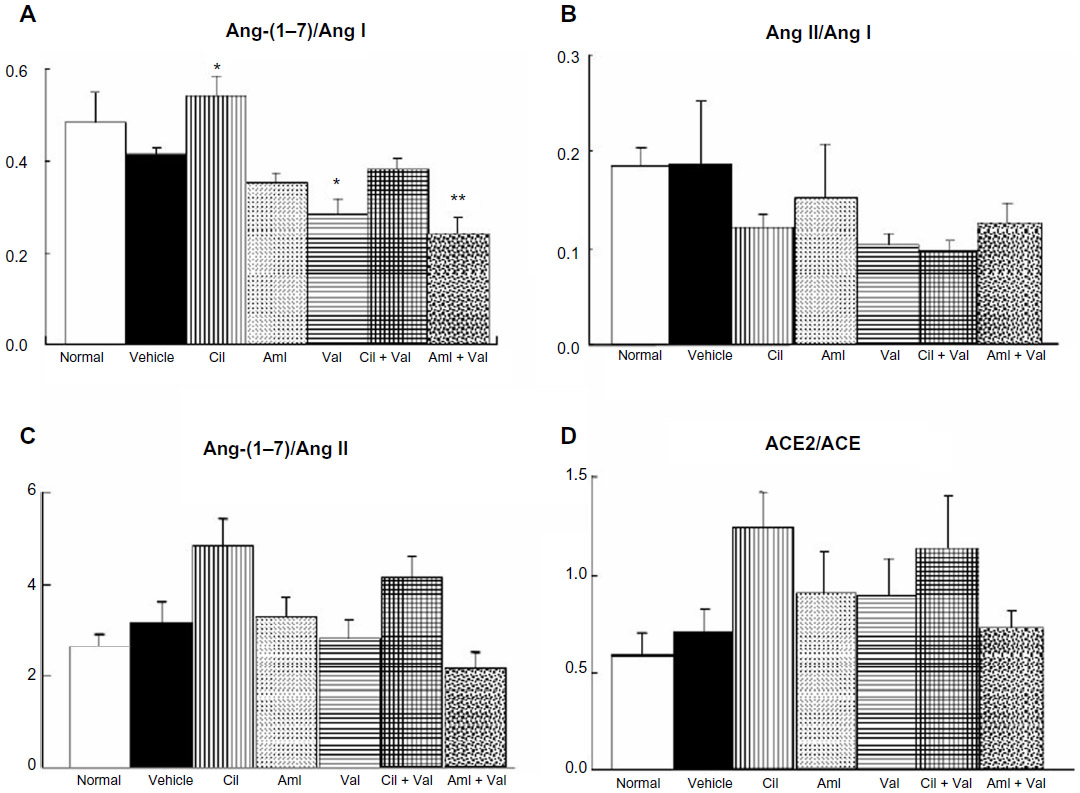 | Figure 5 The ratios of angiotensin peptides. |
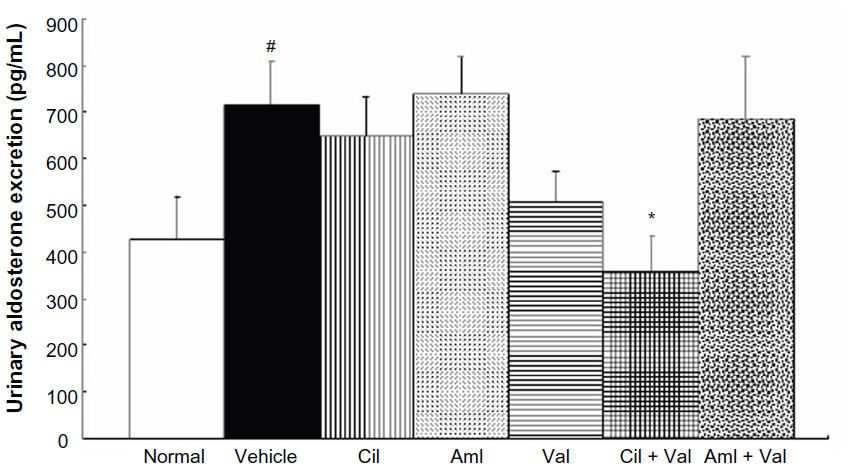 | Figure 6 Effects of the combined administration of Val + Aml and Val + Cil on the levels of aldosterone. |
Discussion
High BP has been well established as an important risk factor for type 2 diabetes.37,38 Recently, combination therapy with an ARB and diuretic drugs and/or CCBs has been recommended in order to treat hypertension and achieve strict depressor effects, thereby preserving organ function.39 In our study, the combination therapy groups (Val + Cil or Aml) showed strong BP-suppressive actions compared to the monotherapy groups. When comparing the Cil + Val and Aml + Val groups, only the combination of Cil + Val ameliorated the increases in UAE levels and cardiac fibrosis, despite the fact that these treatments had the same effects on hypertension. It has been reported that the development of diabetes is accompanied by an increase in albuminuria in OLETF rats and an increase in GS, mesangial expansion,40 and cardiac fibrosis.41 These data suggested that combination therapy with Cil and Val prevented the cardiorenal damage induced by the development of diabetes. With regard to UAE, our data support the additive effect of Cil and Val because treatment with Cil + Val resulted in a statistically significant reduction in UAE, although such effects were not observed with Cil or Val treatment alone (Figure 1A). The same holds true for the dimensions of cardiac myocytes (Figure 2A) and fibrosis (Figure 2B). As for GS, our data do not seem to show the additive or synergistic effects of Cil and Val, although the results of previous studies using Dahl–salt sensitive rats36 and SHR-STZ rats42 have shown that the effects of Cil and Val on GS were additive. One reason for this could be that in this study, the individual monotherapies may have generated the maximum potential therapeutic response, and Cil + Val might have failed to produce any synergistic effects because either both Cil and Val may act on GS through the same mechanism, or each of these may completely suppress GS progression at the start of treatment. Nevertheless, further studies are required to determine whether combination therapy using Cil with Val in rats with type 2 diabetes could produce a synergistic effect on GS.
In addition, the inhibition of the RAS is an important component in the treatment of patients with cardiovascular disease and/or kidney disease.43,44 Moreover, it has been suggested that the activation of the RAS that is induced by ARBs may cause aldosterone breakthrough,45 myocardial infarction,46 and/or renal injury.47 In this study, the groups that were administered Val (Val, Cil + Val, and Aml + Val groups) showed a tendency towards an increase in RAS, which was caused by Val’s AT1-R blocking actions. In addition, the Aml + Val group showed further RAS activation in comparison with the Val group through the AT1-R blocking actions and BP-lowering effects.34 Furthermore, it has already been reported that calcium inhibits renin gene expression in juxtaglomerular cells through the concerted actions of the inhibition of renin gene transcription and the destabilization of renin mRNA.48 In other words, Aml may accelerate renin gene transcription through the inhibition of the flow of calcium into juxtaglomerular cells. However, despite its CCB effects, the Cil + Val group did not exhibit further RAS activation, as reported in our previous study.35 In addition, we previously reported that Cil has suppressive effects on the Val-induced increases in RAS activity through its sympatholytic actions that are mediated by N-type calcium channel blockade.34 In the AVOID (Aliskiren in the Evaluation of Proteinuria in Diabetes) trial,49 the combination of a DRI with an ARB was found to have stronger organ-protective effects than ARB monotherapy, suggesting that inhibiting increases in ARB-induced RAS activity promotes the protection of organs and tissues. Taken together, these data suggest that the synergistic organ-protective effects of treatment with Cil and an ARB were attributable to the inhibitory effects of Cil on RAS activation.
As for aldosterone and NA, the Cil + Val group exhibited a significant reduction in aldosterone excretion. Previous studies have demonstrated that aldosterone production and NA secretion are mediated by N-type calcium channels; thus, Cil has suppressive effects on aldosterone secretion through L-/N-type CCB actions.30,33,35,50 Furthermore, it is well known that ARB has a suppressive effect on aldosterone secretion50 and sympathetic nerve activation51 because AT1-R stimulates aldosterone secretion52 and NA release.53 In this study, monotherapy using neither Cil nor Val successfully produced significant reductions in aldosterone and NA, but Cil + Val treatment suppressed the aldosterone and NA increase associated with worsening diabetes, and this suppression was due to the additive effect of Cil and Val. In this study, the Cil group exhibited a declining trend in the NA level, which is an index of sympathetic nerve activity, although this decline was not statistically significant. Many reports have shown that Cil inhibited the hyperactivity of sympathetic nerves, despite L-type CCB activating the sympathetic nerve.19,20,24 However, the dose (Cil group: Cil, 2 mg/kg) used in this study might be too low to influence the NA level significantly because the effects observed in the previous animal studies were obtained using doses of at least 10 mg per day.
ACE2 has been identified as a member of the RAS and a component of the ACE2–Ang-(1–7) –Mas counter-regulatory axis of the RAS, and it has recently been shown to have organ-protective effects. It has already been suggested that ACE2 may play a pivotal role in diabetes.7,54 In our study, Cil induced an increase in the levels of expression of ACE2 and in serum Ang-(1–7) levels. These results suggest that the L-/N-type CCB has the potential to change the balance between Ang-(1–7) and Ang II, and increase Ang-(1–7) levels in order to achieve cardioprotection and antihypertensive effects. However, ARBs increase PRA and plasma Ang II levels by uncoupling the negative feedback loop through their inhibitory effects on the AT1-R.11 For organ protection, combination therapy with Cil and an ARB may be better than treatment with an ARB alone because Cil converts the Ang II that is induced by ARB into Ang-(1–7) and exhibits an additive effect on renal and cardiac protection. This effect of Cil may be caused by its N-type calcium channel blocking actions through the close connection between ACE2–Ang-(1–7) and sympathetic nerve activity.55 In fact, it has been reported that the suppression of sympathetic nerve activity by the drug induces the expression of ACE2,56 and Cil is a unique CCB that has inhibitory actions on the sympathetic N-type calcium channels. Furthermore, the aldosterone-suppressive effects of Cil may increase ACE2 mRNA levels and decrease ACE mRNA levels because a previous study indicated that aldosterone reduced ACE2 mRNA levels and increased ACE mRNA levels in rat cardiomyocytes through the mineralocorticoid receptor.57 In fact, our previous study demonstrate that the Cil changed the balance between Ang-(1–7) and Ang II, and it increased the proportion of Ang-(1–7).58 The results of these reports suggest that the inhibitory effects of Cil on sympathetic nerve activity and aldosterone secretion induces ACE2 expression. Thus, further study is necessary in order to determine the mechanisms by which Cil increases ACE2 expression and plasma Ang-(1–7) levels.
As for a comparison of OLETF rats with LETO rats, the vehicle group showed lower levels of RAS than did the LETO group, although RAS levels have been reported to be upregulated in OLETF rats with type 2 diabetes.22,56 Fan et al showed that OLETF rats developed hypertension, and then they subsequently showed elevated RAS levels. This suggests that hypertension involves a compensatory decrease in the RAS level during early-stage disease. Nevertheless, further studies are required to elucidate why RAS levels in OLETF rats were lower than those in LETO rats.
Conclusion
In conclusion, the results of the present study showed that the L-/N-type CCB Cil has additive antihypertensive and UAE-lowering effects when administered in combination with an ARB, even in type 2 diabetic rats. Furthermore, combination therapy with Cil and Val significantly reduced NA secretion, suggesting that Cil suppressed the increases in PRA and Ang II that are associated with antihypertensive therapy by inhibiting sympathetic nervous system activity through its N-type calcium channel antagonism. Additionally, Cil increased Ang-(1–7) production by its inhibitory effects on sympathetic nerve activity and aldosterone secretion. Thus, combination therapy with Cil and an ARB may be more effective than treatment with an ARB alone because Cil converts the Ang II that is induced by ARB into Ang-(1–7) and exhibits additive protective effects on renal and cardiac organs.
Disclosure
The authors report no conflicts of interest in this work.
References
Santos RA, Ferreira AJ. Angiotensin-(1–7) and the renin-angiotensin system. Curr Opin Nephrol Hypertens. 2007;16(2):122–128. | |
Simões e Silva AC, Pinheiro SV, Pereira RM, Ferreira AJ, Santos RA. The therapeutic potential of angiotensin-(1–7) as a novel renin-angiotensin system mediator. Mini Rev Med Chem. 2006;6(5):603–609. | |
Matsui T, Tamaya K, Matsumoto K, Osajima Y, Uezono K, Kawasaki T. Plasma concentrations of angiotensin metabolites in young male normotensive and mild hypertensive subjects. Hypertens Res. 1999;22(4):273–277. | |
Nogueira AI, Souza Santos RA, Simões E, Silva AC, et al. The pregnancy-induced increase of plasma angiotensin-(1–7) is blunted in gestational diabetes. Regul Pept. 2007;141(1–3):55–60. | |
Oudit GY, Liu GC, Zhong J, et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes. 2010;59(2):529–538. | |
Burns WC, Velkoska E, Dean R, Burrell LM, Thomas MC. Angiotensin II mediates epithelial-to-mesenchymal transformation in tubular cells by ANG 1–7/MAS-1-dependent pathways. Am J Physiol Renal Physiol. 2010;299(3):F585–F593. | |
Bindom SM, Lazartigues E. The sweeter side of ACE2: physiological evidence for a role in diabetes. Mol Cell Endocrinol. 2009;302(2):193–202. | |
Ritz E, Schmieder RE, Pollock CA. Renal protection in diabetes: lessons from ONTARGET. Cardiovasc Diabetol. 2010;9:60. | |
Hoogwerf BJ. Renin-angiotensin system blockade and cardiovascular and renal protection. Am J Cardiol. 2010;105(Suppl 1):30A–35A. | |
Bichu P, Nistala R, Khan A, Sowers JR, Whaley-Connell A. Angiotensin receptor blockers for the reduction of proteinuria in diabetic patients with overt nephropathy: results from the AMADEO study. Vasc Health Risk Manag. 2009;5(1):129–140. | |
Levy BI. How to explain the differences between renin angiotensin system modulators. Am J Hypertens. 2005;18(9 Pt 2):134S–141S. | |
McMurray JJ, Ostergren J, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet. 2003;362(9386):767–771. | |
Forclaz A, Maillard M, Nussberger J, Brunner HR, Burnier M. Angiotensin II receptor blockade: is there truly a benefit of adding an ACE inhibitor? Hypertension. 2003;41(1):31–36. | |
Azizi M, Ménard J, Bissery A, et al. Pharmacologic demonstration of the synergistic effects of a combination of the renin inhibitor aliskiren and the AT1 receptor antagonist valsartan on the angiotensin II-renin feedback interruption. J Am Soc Nephrol. 2004;15(12):3126–3133. | |
Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008;372(9638):547–553. | |
Parving HH, Brenner BM, McMurray JJ, et al; ALTITUDE Investigators. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med. 2012;367(23):2204–2213. | |
Takahara A. Cilnidipine: a new generation Ca channel blocker with inhibitory action on sympathetic neurotransmitter release. Cardiovasc Ther. 2009;27(2):124–139. | |
Takahara A, Koganei H, Takeda T, Iwata S. Antisympathetic and hemodynamic property of a dual L/N-type Ca(2+) channel blocker cilnidipine in rats. Eur J Pharmacol. 2002;434(1–2):43–47. | |
Shiga T, Yamada Y, Matsuda N, et al. Influence of cilnidipine or nisoldipine on sympathetic activity in healthy male subjects. Heart Vessels. 2007;22(6):404–409. | |
Nagahama S, Norimatsu T, Maki T, Yasuda M, Tanaka S. The effect of combination therapy with an L/N-type Ca(2+) channel blocker, cilnidipine, and an angiotensin II receptor blocker on the blood pressure and heart rate in Japanese hypertensive patients: an observational study conducted in Japan. Hypertens Res. 2007;30(9):815–822. | |
Varagic J, Susic D, Frohlich ED. Cilnidipine improves spontaneously hypertensive rat coronary hemodynamics without altering cardiovascular mass and collagen. J Hypertens. 2002;20(2):317–322. | |
Sakata K, Yoshida H, Tamekiyo H, et al. Comparative effect of clinidipine and quinapril on left ventricular mass in mild essential hypertension. Drugs Exp Clin Res. 2003;29(3):117–123. | |
Takemori K, Ishida H, Dote K, Yamamoto K, Ito H. Prophylactic effects of an N- and L-type Ca2+ antagonist, cilnidipine, against cardiac hypertrophy and dysfunction in stroke-prone, spontaneously hypertensive rats. Can J Physiol Pharmacol. 2005;83(8–9):785–790. | |
Nagai H, Minatoguchi S, Chen XH, et al. Cilnidipine, an N+L-type dihydropyridine Ca channel blocker, suppresses the occurrence of ischemia/reperfusion arrhythmia in a rabbit model of myocardial infarction. Hypertens Res. 2005;28(4):361–368. | |
Fujita T, Ando K, Nishimura H, et al; Cilnidipine versus Amlodipine Randomised Trial for Evaluation in Renal Desease(CARTER) Study Investigators. Antiproteinuric effect of the calcium channel blocker cilnidipine added to renin-angiotensin inhibition in hypertensive patients with chronic renal disease. Kidney Int. 2007;72(12):1543–1549. | |
Zhou X, Ono H, Ono Y, Frohlich ED. N- and L-type calcium channel antagonist improves glomerular dynamics, reverse severe nephrosclerosis, and inhibits apoptosis and proliferation in an I-NAME/SHR model. J Hypertens. 2002;20(5):993–1000. | |
Tsuchihashi T, Ueno M, Tominaga M, et al. Anti-proteinuric effect of an N-type calcium channel blocker, cilnidipine. Clin Exp Hypertens. 2005;27(8):583–591. | |
Kojima S, Shida M, Yokoyama H. Comparison between cilnidipine and amlodipine besilate with respect to proteinuria in hypertensive patients with renal diseases. Hypertens Res. 2004;27(6):379–385. | |
Konda T, Enomoto A, Matsushita J, Takahara A, Moriyama T. The N- and L-type calcium channel blocker cilnidipine suppresses renal injury in dahl rats fed a high-sucrose diet, an experimental model of metabolic syndrome. Nephron Physiol. 2005;101(1):p1–p13. | |
Konda T, Enomoto A, Aritomi S, et al. Different effects of L/N-type and L-type calcium channel blockers on the renin-angiotensin-aldosterone system in SHR/Izm. Am J Nephrol. 2009;30(2):155–161. | |
Takahara A, Nakamura Y, Wagatsuma H, et al. Long-term blockade of L/N-type Ca(2+) channels by cilnidipine ameliorates repolarization abnormality of the canine hypertrophied heart. Br J Pharmacol. 2009;158(5):1366–1374. | |
Fournier A, Oprisiu-Fournier R, Serot JM, et al. Prevention of dementia by antihypertensive drugs: how AT1-receptor-blockers and dihydropyridines better prevent dementia in hypertensive patients than thiazides and ACE-inhibitors. Expert Rev Neurother. 2009;9(9):1413–1431. | |
Aritomi S, Wagatsuma H, Numata T, et al. Expression of N-type calcium channels in human adrenocortical cells and their contribution to corticosteroid synthesis. Hypertens Res. 2011;34(2):193–201. | |
Aritomi S, Niinuma K, Ogawa T, Konda T, Nitta K. Effects of an N-type calcium antagonist on angiotensin II-renin feedback. Am J Nephrol. 2011;33(2):168–175. | |
Konoshita T, Makino Y, Kimura T, et al; Genomic Disease Outcome Consortium Study Investigators. A new-generation N/L-type calcium channel blocker leads to less activation of the renin-angiotensin system compared with conventional L type calcium channel blocker. J Hypertens. 2010;28(10):2156–2160. | |
Aritomi S, Koganei H, Wagatsuma H, et al. The N-type and L-type calcium channel blocker cilnidipine suppresses renal injury in Dahl rats fed a high-salt diet. Heart Vessels. 2010;25(6):549–555. | |
Gress TW, Nieto FJ, Shahar E, Wofford MR, Brancati FL. Hypertension and antihypertensive therapy as risk factors for type 2 diabetes mellitus. Atherosclerosis Risk in Communities Study. N Engl J Med. 2000;342(13):905–912. | |
Hsu CY, McCulloch CE, Darbinian J, Go AS, Iribarren C. Elevated blood pressure and risk of end-stage renal disease in subjects without baseline kidney disease. Arch Intern Med. 2005;165(8):923–928. | |
Japanese Society of Hypertension. Japanese Society of Hypertension guidelines for the management of hypertension (JSH 2004). Hypertens Res. 2006;29(Suppl):S1–S105. | |
Fukuzawa Y, Watanabe Y, Inaguma D, Hotta N. Evaluation of glomerular lesion and abnormal urinary findings in OLETF rats resulting from a long-term diabetic state. J Lab Clin Med. 1996;128(6):568–578. | |
Makino N, Sasaki M, Maeda T, Mimori K. Telomere biology in cardiovascular disease – role of insulin sensitivity in diabetic hearts. Exp Clin Cardiol. 2010;15(4):e128–e133. | |
Aritomi S, Niinuma K, Ogawa T, Konda T, Nitta K. Additive effects of cilnidipine and angiotensin II receptor blocker in preventing the progression of diabetic nephropathy in diabetic spontaneously hypertensive rats. Clin Exp Nephrol. 2013;17(1):41–50. | |
van den Born BJ, Koopmans RP, van Montfrans GA. The renin-angiotensin system in malignant hypertension revisited: plasma renin activity, microangiopathic hemolysis, and renal failure in malignant hypertension. Am J Hypertens. 2007;20(8):900–906. | |
Sechi LA, Novello M, Colussi G, et al. Relationship of plasma renin with a prothrombotic state in hypertension: relevance for organ damage. Am J Hypertens. 2008;21(12):1347–1353. | |
Rump LC. Secondary rise of albuminuria under AT1-receptor blockade – what is the potential role of aldosterone escape? Nephrol Dial Transplant. 2007;22(1):5–8. | |
Strauss MH, Hall AS. Angiotensin receptor blockers may increase risk of myocardial infarction: unraveling the ARB-MI paradox. Circulation. 2006;114(8):838–854. | |
Furumatsu Y, Nagasawa Y, Tomida K, et al. Effect of renin-angiotensin-aldosterone system triple blockade on non-diabetic renal disease: addition of an aldosterone blocker, spironolactone, to combination treatment with an angiotensin-converting enzyme inhibitor and angiotensin II receptor blocker. Hypertens Res. 2008;31(1):59–67. | |
Klar J, Sigl M, Obermayer B, Schweda F, Krämer BK, Kurtz A. Calcium inhibits renin gene expression by transcriptional and posttranscriptional mechanisms. Hypertension. 2005;46(6):1340–1346. | |
Riche DM, Minor DS, Holdiness AS, East HE. An issue of dependence: implications from the Aliskiren in the Evaluation of Proteinuria in Diabetes (AVOID) trial. J Clin Hypertens (Greenwich). 2009;11(2):89–93. | |
Kaneshiro Y, Ichihara A, Sakoda M, Kurauchi A, Takemitsu T, Itoh H. Cilnidipine and telmisartan similarly improves vascular damage in hypertensive patients. Clin Med Insights Cardiol. 2007;1:1–11. | |
Blankestijn PJ, Rupp H. Clinical profile of eprosartan: a different angiotensin II receptor blocker. Cardiovasc Hematol Agents Med Chem. 2008;6(4):253–257. | |
Aguilera G. Role of angiotensin II receptor subtypes on the regulation of aldosterone secretion in the adrenal glomerulosa zone in the rat. Mol Cell Endocrinol. 1992;90(1):53–60. | |
Reid AC, Mackins CJ, Seyedi N, Levi R, Silver RB. Coupling of angiotensin II AT1 receptors to neuronal NHE activity and carrier-mediated norepinephrine release in myocardial ischemia. Am J Physiol Heart Circ Physiol. 2004;286(4):H1448–H1454. | |
Batlle D, Jose Soler M, Ye M. ACE2 and diabetes: ACE of ACEs? Diabetes. 2010;59(12):2994–2996. | |
Zucker IH, Schultz HD, Patel KP, Wang W, Gao L. Regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure. Am J Physiol Heart Circ Physiol. 2009;297(5):H1557–H1566. | |
Kar S, Gao L, Zucker IH. Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing-induced heart failure. J Appl Physiol (1985). 2010;108(4):923–932. | |
Yamamuro M, Yoshimura M, Nakayama M, et al. Aldosterone, but not angiotensin II, reduces angiotensin converting enzyme 2 gene expression levels in cultured neonatal rat cardiomyocytes. Circ J. 2008;72(8):1346–1350. | |
Aritomi S, Niinuma K, Kawakami M, et al. Cilnidipine, an L-/N-type calcium channel blocker, changes the circulating angiotensin–(1–7)/angiotensin II ratio. J Hypertens. In press. | |
Fan YY, Kobori H, Nakano D, et al. Aberrant activation of the intrarenal renin-angiotensin system in the developing kidneys of type 2 diabetic rats. Horm Metab Res. 2013;45(5):338–343. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.