")
Back to Journals » OncoTargets and Therapy » Volume 7
Additive effect by combination of Akt inhibitor, MK-2206, and PDGFR inhibitor, tyrphostin AG 1296, in suppressing anaplastic thyroid carcinoma cell viability and motility
Authors Che H, Guo H, Si X, You Q, Lou W
Received 9 November 2013
Accepted for publication 8 January 2014
Published 14 March 2014 Volume 2014:7 Pages 425—432
DOI https://doi.org/10.2147/OTT.S57324
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Huan-yong Che, Hang-yuan Guo, Xu-wei Si, Qiao-ying You, Wei-ying Lou
Shaoxing People's Hospital, Shaoxing Hospital of Zhejiang University, Shaoxing, Zhejiang, People's Republic of China
Abstract: The phosphatidylinositol-3-kinase/Akt pathway and receptor tyrosine kinases regulate many tumorigenesis related cellular processes including cell metabolism, cell survival, cell motility, and angiogenesis. Anaplastic thyroid carcinoma (ATC) is a rare type of thyroid cancer with no effective systemic therapy. It has been shown that Akt activation is associated with tumor progression in ATC. Here we observed the additive effect between an Akt inhibitor (MK-2206) and a novel platelet-derived growth factor receptor inhibitor (tyrphostin AG 1296) in ATC therapy. We found an additive effect between MK-2206 and tyrphostin AG 1296 in suppressing ATC cell viability. The combination of MK-2206 and tyrphostin AG 1296 induces additive apoptosis, additive suppression of the Akt signaling pathway, as well as additive inhibition of cell migration and invasion of ATC cells. Furthermore, the combination of MK-2206 and tyrphostin AG 1296 induced additive suppression of ATC tumor growth in vivo. In summary, our studies suggest that the combination of Akt and receptor tyrosine kinase inhibitors may be an efficient therapeutic strategy for ATC treatment, which might shed new light on ATC therapy.
Keywords: anaplastic thyroid carcinoma, Akt inhibitor, PDGFR inhibitor, synergy, additive effect, viability, motility
Introduction
Anaplastic thyroid cancer (ATC) is a rare, usually lethal malignancy in older adults, accounting for about 2% of thyroid cancers. It is a fast growing, poorly differentiated thyroid cancer starting from differentiated thyroid cancer or a benign tumor of the gland. This type of cancer grows very quickly, so it is more difficult to treat successfully. It remains one of the most deadly diseases. The mean survival time for ATC is usually less than 6 months from diagnosis.1–4 Unfortunately, this outcome is not fundamentally altered by available treatments, and there is no effective systemic therapy. So, it is urgent to find an effective therapeutic method or promising targets.
Due to the phosphatidylinositol-3-kinase (PI3K) mutant and constitutive activation in ATC, Akt is highly activated, which makes Akt a potential therapeutic target in ATC treatment.5–10 Upstream of PI3K are receptor tyrosine kinases (RTKs), which are activated by its ligands. Platelet-derived growth factor receptors (PDGFR) are cell surface tyrosine kinase receptors for the platelet-derived growth factor (PDGF) family. They are very important RTKs. Both Akt and the PDGF/PDGFR system play crucial roles in cell proliferation, differentiation, migration, invasion and tumorigenesis, and development and metastasis of ATC.5–15 Therefore, both Akt and PDGFR are promising targets in ATC therapeutics. Here we have characterized the potential synergistic or additive effect between an Akt inhibitor and a RTK inhibitor in ATC therapeutics. Our studies demonstrate that there is an additive effect between the Akt inhibitor, MK-2206, and a novel PDGFR inhibitor, tyrphostin AG 1296, in suppressing cancer cell viability and motility in vitro, as well as tumor growth in vivo.
Materials and methods
Materials
MK-2206 and tyrphostin AG 1296 were purchased from Selleck Chemicals LLC (Huston, TX, USA). Rabbit anti-phospho-Akt (Ser473, catalog number: 4060), anti-phospho-p70S6K (Thr389, catalog number: 9205), anti-phospho-S6 (Ser235/236, catalog number: 2211), anti-phospho-GSK-3β (Ser9, catalog number: 9336), anti-Akt (catalog number: 9272), anti-p70S6K (catalog number: 9202), anti-S6 (catalog number: 2217), anti-GSK-3β (catalog number: 9315), and secondary horseradish peroxidase-conjugated antibody (catalog number: 7074) were purchased from Cell Signaling Technology (Beverly, MA, USA). Other reagents and chemicals were purchased from Sigma Aldrich (St Louis, MO, USA).
Cells and cell culture
Human ATC cell lines (CAL62 and KAT4) were purchased from the American Type Culture Collection (Rockville, MD, USA). The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Life Technologies, Grand Island, NY, USA) with 10% fetal bovine serum, supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mM glutamine. All cells were cultured in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
Cell viability assay
ATC cells were seeded into 96-well white plates at a density of 5–10 × 103 cells per well in 100 μL of media. The compounds were added as a series of concentrations and incubated for 72 hours. A luminescence based commercial kit (CellTiter-Glo®; Promega, Madison, WI, USA) was used to test cell viability. Briefly, 30 μL of cell lysis/adenosine triphosphate detection reagent was added to each well, shaken for 10 minutes at room temperature, and the luminescence was measured with a plate reader (Molecular Devices, Sunnyvale, CA, USA). IC50 values were determined using Compusyn software (ComboSyn, Inc., Paramus, NJ, USA).
Cell migration assays
Transwell assay
The 24-transwell Boyden chamber (Costar, Bedford, MA, USA) with a polystyrene membrane (6.5 mm diameter, 10 μm thickness, and 8 μm pore size) was used. KAT4 cells were seeded in the upper compartment of the well in serum-free media (5 × 104 cells/well) with or without compounds. The lower compartment was supplied with 600 μL serum-free media supplemented with 20 μg/mL fibronectin. Cells were treated for 8 hours, then were fixed and stained with 0.1% crystal violet. The nonmigrating cells on the upper surface of the membrane were removed, and the migrated cells on the lower side were photographed with a microscope (Eclipse LV150L; Nikon Corporation, Tokyo, Japan) in five random fields. Then the migrated cells were lysed with 10% acetic acid, and colorimetric determination was made at 595 nm.
Wound healing assay
The KAT4 cells were seeded into fibronectin-coated 96-well plates (2 × 104 cells/well). After reaching confluence, the cell monolayers were scratched with a pipette tip. Fresh serum-free medium with or without compounds was added, and incubated for 8 hours. Then cells were photographed with a microscope in five random fields. The width of wounded cell monolayers was measured.
Cell invasion assay
The 24-transwell Boyden chamber was used for the cell invasion assay. The upper chamber was precoated with 1 mg/mL of matrigel (BD Biosciences, San Jose, CA, USA) for 4 hours at 37°C to form a basement membrane. Then the assay was performed and the result was analyzed as described above for the transwell assay.
Western blotting
KAT4 cells were lysed in 20 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid, pH 7.5, 150 mM sodium chloride, 1% NP-40, 10 mM tetrasodium pyrophosphate, 100 mM sodium fluoride, and 17.5 mM β-glycerophosphate buffer supplemented with complete mini protease inhibitor tablet. Samples separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis were transferred to nitrocellulose membranes, blocked with 5% bovine serum albumin (weight/volume [w/v]) at room temperature for 1 hour, and incubated with primary antibodies (1:1,000 dilution) at 4°C overnight. After incubation with secondary antibody (1:3,000 dilution) at room temperature for 1 hour, the membranes were developed with chemiluminescence ECL reagent (LumiGold™; SignaGen Laboratories, Rockville, MD, USA) and exposed to Hyperfilm MP (GE Healthcare, Buckinghamshire, UK).
Nuclei staining
KAT4 cells were seeded on fibronectin precoated glass coverslips, followed by treatment with compounds. Then cells were fixed with 4% paraformaldehyde for 30 minutes, permeabilized with 0.1% Triton X-100 for 20 minutes, and blocked with 5% normal serum for 30 minutes. For detection of cell nuclei, cells were incubated with Hoechst for 10 minutes. All images were obtained using Olympus BX51 microscope (Olympus America Inc., Melville, NY, USA).
Flow cytometry analysis
KAT4 cells were seeded in 6-well plates (5 × 105 cells/well) with or without compound treatment. Then cells were harvested, fixed with 70% ethanol and stained with propidium iodide (5 mg/L) in the presence of RNase (1 g/L), 1 g/L sodium citrate, and 0.5% Triton X-100 (volume/volume [v/v]) in the dark for 30 minutes. Cells were collected for apoptosis analysis using FACSCalibur (BD Biosciences). The percentage of hypodiploidy was analyzed using ModFiT LT™ software (Verity Software House, Topsham, ME, USA).
Immunohistochemistry
Tumor tissues were fixed in phosphate-buffered formalin, embedded in paraffin, cut in 4 μm thickness, and applied to slides. The slides were deparaffinized in xylene using three changes for 5 minutes each, and hydrated gradually through graded alcohols: 100% ethanol twice for 10 minutes, 95% ethanol twice for 10 minutes, and then deionized water for 1 minute with stirring. For antigen unmasking, slides were placed in a container, covered with 10 mM sodium citrate buffer, pH 6.0, and heated in a convection steamer for 1 hour. The slides were washed in deionized water three times for 2 minutes and blocked with 5% normal goat blocking serum for 30 minutes. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using FragEL™ kit according to manufacturer’s instructions (Calbiochem, Darmstadt, Germany) for 1 hour, and incubated with an Alexa Fluor 488-conjugated secondary antibody for 30 minutes, and then incubated with Hoechst for 10 minutes. The slides were analyzed and photographed using an Olympus BX51 microscope (Olympus America Inc.).
Suppression of subcutaneous tumor growth in vivo
The nud/nud mice were purchase from Slack company in Shanghai (People’s Republic of China), and housed in a Biosafety level 2 lab. All care and treatment of experimental animals were in accordance with animal care and use committee guidelines. KAT4 cells were cultured in vitro in DMEM and then resuspended in phosphate-buffered saline (4×107 cells/mL). The cells were injected subcutaneously into the axillary regions of nud/nud mice (4×106 cells/100 μL/mouse). When the tumor volumes reached about 60 mm3, the mice were randomized to the control group or various treatment groups (n=6 per group), including MK-2206 (100 mg/kg), tyrphostin AG 1296 (100 mg/kg), a combination of MK-2206 (100 mg/kg) and tyrphostin AG 1296 (100 mg/kg), and vehicle (10% 1-methyl-2-pyrrolidinone and 90% polyethylene glycol 300). Tumors were measured every day for about 2 weeks with a microcaliper. The body weight was measured every day with a scale. The tumor volumes were calculated with the formula: mm3 = width × width × length × 0.5. After administration of the treatment for about 2 weeks, the mice were euthanized using carbon dioxide and tumors were harvested. Tumor cell apoptosis was analyzed using immunohistochemistry.
Statistics
The Student’s t-test and analysis of variance were performed using StatView (SAS Institute, Cary, NC, USA). P<0.05 was considered significantly different. The data shown are the mean values of triplicate measurements with error bars corresponding to standard deviation.
Results
MK-2206 synergizes with tyrphostin AG 1296 in suppressing ATC cell viability
PI3K/Akt and PDGFR signaling play crucial roles in promoting ATC cell survival. Upregulation of PI3K/Akt signaling is proven as an important mechanism of chemoresistance of cancer cells.5–18 We selected an Akt inhibitor, MK-2206, for in vitro anti-ATC studies. We examined the effect of MK-2206 on ATC cancer cell viability and found that MK-2206 significantly reduced the viability of the ATC cell lines, CAL62 and KAT4 (Figure 1A). We next used the Akt inhibitor, MK-2206, and a novel PDGFR inhibitor, tyrphostin AG 1296, for an in vitro synergy/additive study. To determine whether tyrphostin AG 1296 could potentiate MK-2206 effects, we treated KAT4 cells with MK-2206 and tyrphostin AG 1296 with a series of concentrations. The individual and combined survival curves and the isobologram showed that there was a significant synergism between MK-2206 and tyrphostin AG 129 (Figure 1B and C).
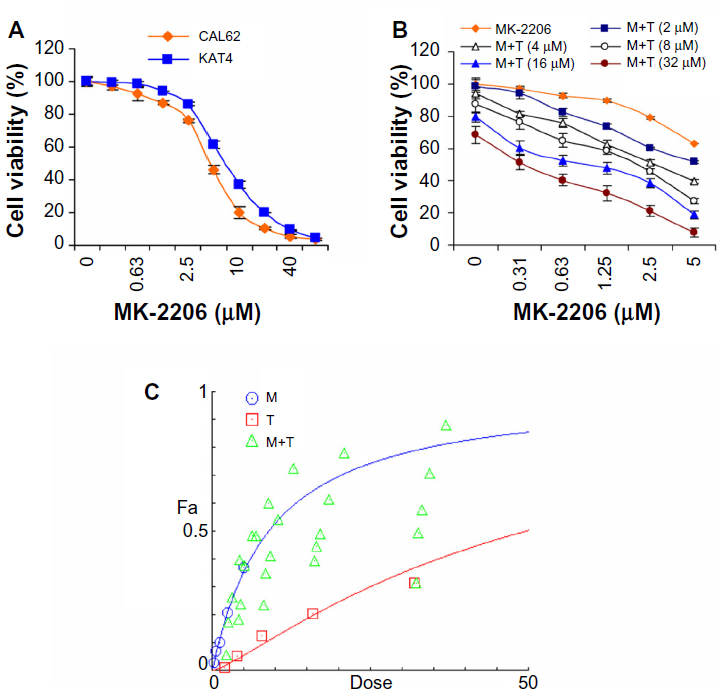 | Figure 1 MK-2206 synergizes with tyrphostin AG 1296 in suppressing anaplastic thyroid carcinoma cell viability. |
Combination of MK-2206 and tyrphostin AG 1296 induces an additive apoptosis in ATC cells
Since PI3K/Akt and PDGFR signaling play a critical role in the inhibition of apoptosis,7–13 we next examined MK-2206 induced apoptosis in ATC cells. We found MK-2206 caused a significant increase in subG1 deoxyribonucleic acid (DNA) content in KAT4 cells (P<0.01) (Figure 2A). In addition, after treatment with MK-2206, the nuclei of cancer cells exhibited a condensed and fragmented morphology, which is characteristic of apoptosis (Figure 2B), suggesting that the Akt inhibitor could induce dramatic apoptosis in ATC cells. We also found that the combination of MK-2206 and tyrphostin AG 1296 induced more pronounced cell apoptosis than either MK-2206 or tyrphostin AG 1296 alone. When these two compounds were combined, a more significant increase in subG1 DNA content was observed when compared to MK-2206 or tyrphostin AG 1296 alone (Figure 2A, P<0.01). Consistently, the combination of these two compounds induced a dramatic increase in condensed/fragmented nuclei number compared to MK-2206 or tyrphostin AG 1296 alone (Figure 2B and C, P<0.01).
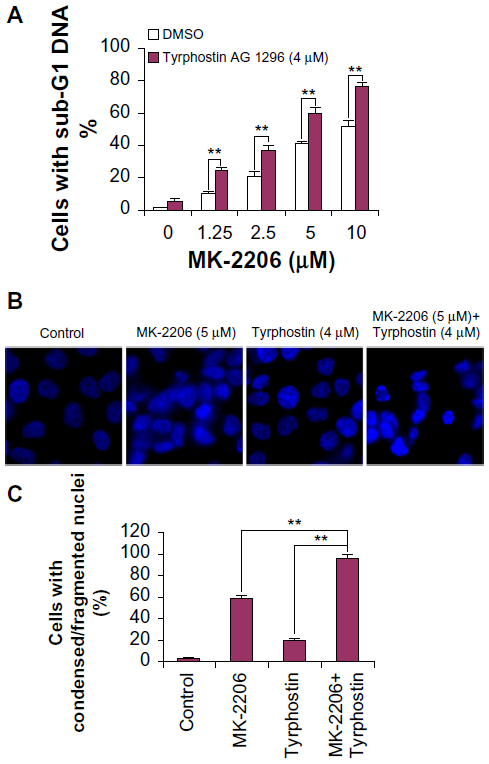 | Figure 2 Combination of MK-2206 and tyrphostin AG 1296 induces an additive apoptosis in anaplastic thyroid carcinoma cells. |
Combination of MK-2206 and tyrphostin AG 1296 induces an additive suppression of Akt signaling pathway in ATC cells
We next examined the effect of MK-2206 and tyrphostin AG 1296 on PI3K/Akt signaling in ATC cells. As shown in Figure 3, both MK-2206 and tyrphostin AG 1296 significantly suppressed the Akt signaling pathway including phosphorylation of Akt, p70S6, S6, and Gsk-3β in KAT4 cells, but no significant changes in total protein levels were observed. The addition of MK-2206 in conjunction with tyrphostin AG 1296 led to a stronger suppression of the Akt signaling pathway when compared to either MK-2206 or tyrphostin AG 1296 alone.
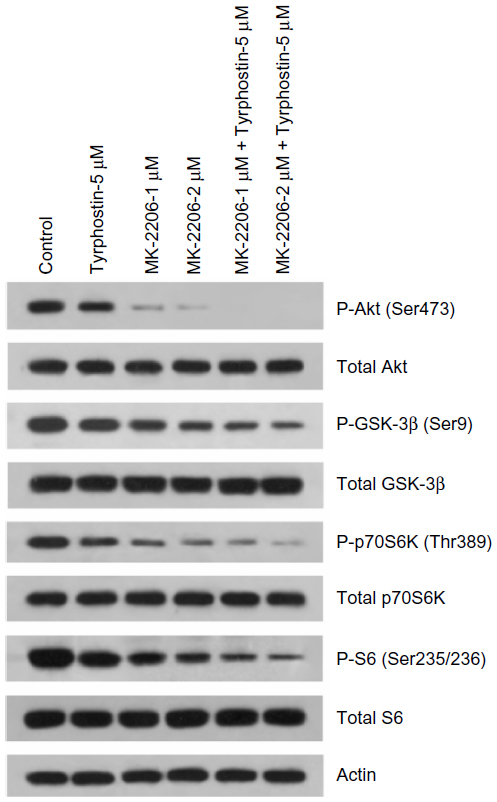 | Figure 3 Combination of MK-2206 and tyrphostin AG 1296 induces an additive suppression of Akt signaling pathway in anaplastic thyroid carcinoma cells. |
Combination of MK-2206 and tyrphostin AG 1296 induces an additive inhibition of cell migration and invasion in ATC cells
PI3K/Akt and PDGFR play crucial roles in cancer cell motility.19–21 We next investigated the effect of MK-2206 and tyrphostin AG 1296 on migration of ATC cells. As shown in Figure 4A and B, both MK-2206 and tyrphostin AG 1296 inhibited transwell migration of KAT4 cells. Similar inhibition of cell migration was also observed in a wound healing assay (Figure 4C and D). In addition, we found that the combination of MK-2206 and tyrphostin AG 1296 induced an additive inhibition of migration of KAT4 cells. Inhibition in the combination group is significantly higher than that of the MK-2206 or tyrphostin AG 1296 treatment groups (Figure 4B and D, P<0.01). Furthermore, both of the inhibitors efficiently suppressed cancer cell invasion through matrigel (Figure 4E and F). The combination of MK-2206 and tyrphostin AG 1296 also induced an additive inhibition of cell invasion. Inhibition in the combination group is significantly higher than that of the MK-2206 or tyrphostin AG 1296 treated groups (Figure 4F, P<0.01). Importantly, treatment with MK-2206, tyrphostin AG 1296, or both had no significant cytotoxic effects on the cells in parallel migration and invasion experiments using the same concentrations for the same treatment time (P>0.05) (Figure 4G), implying that the effect of MK-2206 or tyrphostin AG 1296 on cell motility was not a consequence of cellular toxicity.
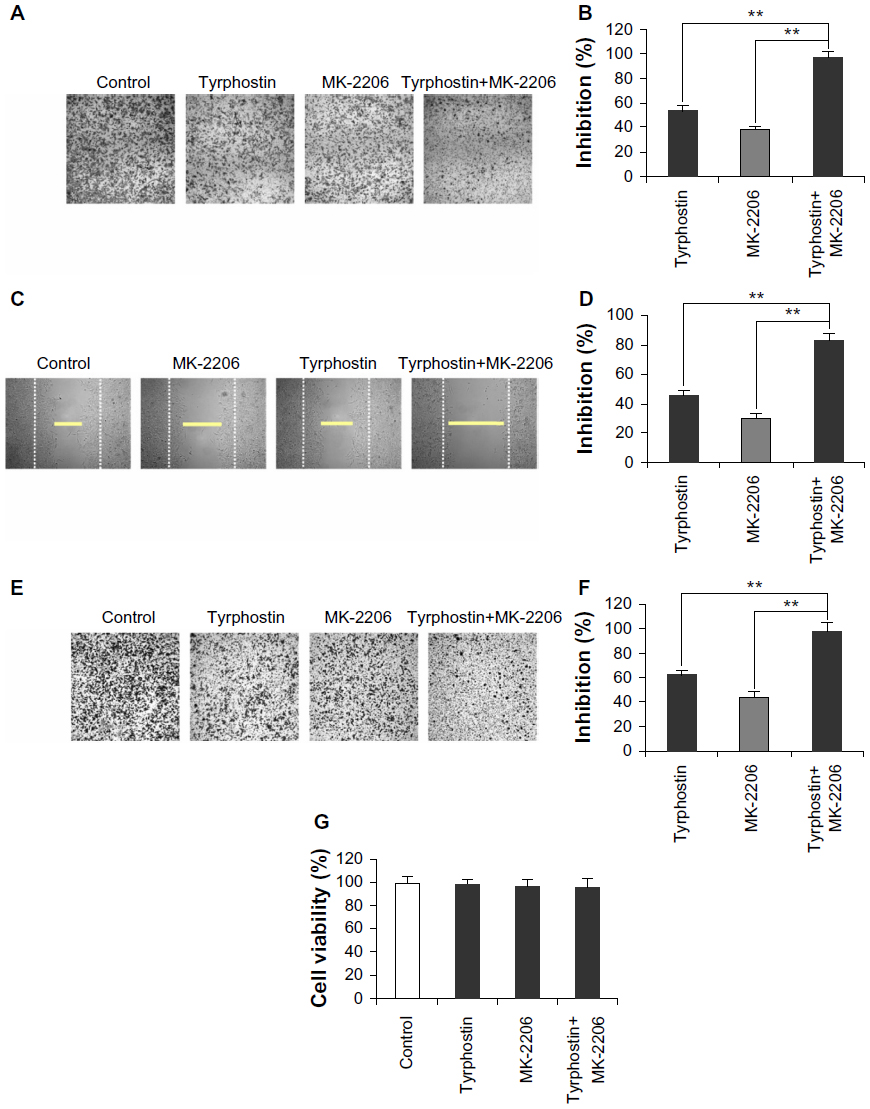 | Figure 4 Combination of MK-2206 and tyrphostin AG 1296 induces an additive inhibition of cell migration and invasion in anaplastic thyroid carcinoma cells. |
Combination of MK-2206 and tyrphostin AG 1296 induces additive suppression of ATC tumor growth in vivo
We next treated animals with MK-2206 (100 mg/kg), tyrphostin AG 1296 (100 mg/kg), or a combination of MK-2206 (100 mg/kg) and tyrphostin AG 1296 (100 mg/kg), and examined the effect of intraperitoneal administration of the compounds on the growth of KAT4 cells in nud/nud mice. Both MK-2206 and tyrphostin AG 1296 led to an intermediate level of growth suppression, whereas the combination showed strong additive effects (Figure 5A). Consistent with the in vitro data, the combination of MK-2206 and tyrphostin AG 1296 induced more significant apoptotic characteristics in tumors than either MK-2206 or tyrphostin AG 1296 alone as measured using TUNEL and nuclei staining assays (Figure 5B). In addition, administration of MK-2206, tyrphostin AG 1296, or both was well tolerated by healthy mice without significant signs of overt toxicity or loss of weight (P>0.05, Figure 5C). Taken together, these results provide evidence that the combination of MK-2206 and tyrphostin AG 1296 could effectively potentiate apoptosis and enhance suppression of both viability in vitro and tumor growth in vivo.
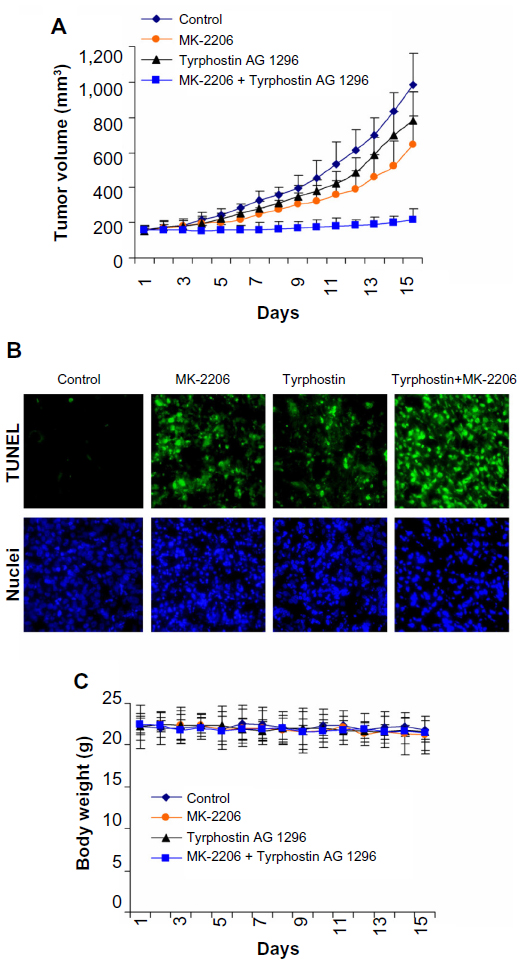 | Figure 5 Combination of MK-2206 and tyrphostin AG 1296 induces additive suppression of anaplastic thyroid carcinoma tumor growth in vivo. |
Discussion
ATC is an uncommon, typically lethal malignancy of older adults with a very low survival rate and without effective systemic therapy.1–4 Here we observed synergistic or additive effects between an Akt inhibitor and a novel PDGFR inhibitor in cell survival, apoptosis, cell signaling pathways, cell migration, and invasion in vitro, as well as tumor growth in vivo in ATC. We provide evidence that 1) MK-2206 synergizes with tyrphostin AG 1296 in suppressing cancer cell viability; 2) the combination of MK-2206 and tyrphostin AG 1296 induces additive apoptosis in ATC cancer cells; 3) the combination of MK-2206 and tyrphostin AG 1296 induces an additive suppression of Akt signaling pathways in ATC cells; 4) the combination of MK-2206 and tyrphostin AG 1296 induces an additive inhibition of cell migration and invasion in ATC cancer cells; and 5) the combination of MK-2206 and tyrphostin AG 1296 induces additive suppression of ATC tumor growth in vivo. The synergism/additive mode for MK-2206 and tyrphostin AG 1296 could be collaborative (eg, inhibition of PDGFR and Akt at the same time). Importantly, these effects are preferentially induced in cancer cells like ATC cells with high Akt activity, suggesting coinhibition of Akt and RTK signaling could be a promising strategy for human tumors characterized by elevated Akt activation levels, such as in ATC cells.10
In summary, we have documented an effective pharmacologic partnership between selective Akt inhibition and PDGFR antagonism that enhances cytotoxicity and reduces tumorigenesis. There are some reports about synergy between MK-2206 and RTK inhibitors.22,23 However, this is the first report about a synergistic/additive effect between MK-2206 and an PDGFR inhibitor, especially the novel inhibitor tyrphostin AG 1296. This is also a novel report about Akt targeting and a synergistic/additive effect in ATC. Our studies establish the feasibility of developing a synergistic Akt/RTK targeting strategy in ATC therapy, especially against Akt overexpressing ATC cells. This work opens avenues for Akt directed combination therapy, and it may be a promising starting point for further synergy strategy development in ATC therapeutics.
Disclosure
The authors of this study declare no conflicts of interest in this work.
References
Are C, Shaha AR. Anaplastic thyroid carcinoma: biology, pathogenesis, prognostic factors, and treatment approaches. Ann Surg Oncol. 2006;13(4):453–464. | |
Giuffrida D, Gharib H. Anaplastic thyroid carcinoma: current diagnosis and treatment. Ann Oncol. 2000;11(9):1083–1089. | |
Ain KB. Anaplastic thyroid carcinoma: a therapeutic challenge. Semin Surg Oncol. 1999;16(1):64–69. | |
Wiseman SM, Masoudi H, Niblock P, et al. Anaplastic thyroid carcinoma: expression profile of targets for therapy offers new insights for disease treatment. Ann Surg Oncol. 2007;14(2):719–729. | |
Bruni P, Boccia A, Baldassarre G, et al. PTEN expression is reduced in a subset of sporadic thyroid carcinomas: evidence that PTEN-growth suppressing activity in thyroid cancer cells is mediated by p27kip1. Oncogene. 2000;19(28):3146–3155. | |
Weng LP, Gimm O, Kum JB, et al. Transient ectopic expression of PTEN in thyroid cancer cell lines induces cell cycle arrest and cell type-dependent cell death. Hum Mol Genet. 2001;10(3):251–258. | |
Dahia PL, Marsh DJ, Zheng Z, et al. Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res. 1997;57(21):4710–4713. | |
Gimm O, Perren A, Weng LP, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue and benign and malignant epithelial thyroid tumors. Am J Pathol. 2000;156(5):1693–1700. | |
Ringel MD, Hayre N, Saito J, et al. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001;61(16):6105–6111. | |
García-Rostán G, Costa AM, Pereira-Castro I, et al. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005;65(22):10199–10207. | |
Ha HT, Lee JS, Urba S, et al. A phase II study of imatinib in patients with advanced anaplastic thyroid cancer. Thyroid. 2010;20(9):975–980. | |
Zhang J, Wang P, Dykstra M, et al. Platelet-derived growth factor receptor-α promotes lymphatic metastases in papillary thyroid cancer. J Pathol. 2012;228(2):241–250. | |
Kim MJ, Kim SK, Park HJ, et al. PDGFRA promoter polymorphisms are associated with the risk of papillary thyroid cancer. Mol Med Rep. 2012;5(5):1267–1270. | |
Malkomes P, Oppermann E, Bechstein WO, Holzer K. Significantly high expression of platelet-derived growth factor (PDGF) in benign nodules of the thyroid: relevance in the development of goitre recurrence? Langenbecks Arch Surg. 2011;396(8):1165–1172. | |
Chen KT, Lin JD, Liou MJ, Weng HF, Chang CA, Chan EC. An aberrant autocrine activation of the platelet-derived growth factor alpha-receptor in follicular and papillary thyroid carcinoma cell lines. Cancer Lett. 2006;231(2):192–205. | |
Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. | |
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. | |
Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9(6):667–676. | |
Oka N, Nakahara S, Takenaka Y, et al. Galectin-3 inhibits tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by activating Akt in human bladder carcinoma cells. Cancer Res. 2005;65(17):7546–7553. | |
Moses SA, Ali MA, Zuohe S, et al. In vitro and in vivo activity of novel small-molecule inhibitors targeting the pleckstrin homology domain of protein kinase B/AKT. Cancer Res. 2009;69(12):5073–5081. | |
Kundra V, Escobedo JA, Kazlauskas A, et al. Regulation of chemotaxis by the platelet-derived growth factor receptor-beta. Nature. 1994;367(6462):474–476. | |
Hirai H, Sootome H, Nakatsuru Y, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9(7):1956–1967. | |
Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72(4):969–978. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.