")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 13
ADAM17 Genetic Variants and the Response of TNF-α Inhibitor in Rheumatoid Arthritis Patients
Authors Kim HJ, Trinh NT, Choi Y, Kim W, Min KH, Kang SO, Kim JH , Kim HA, Jung JY, Choi IA , Lee KE
Received 17 October 2019
Accepted for publication 26 February 2020
Published 16 March 2020 Volume 2020:13 Pages 81—88
DOI https://doi.org/10.2147/PGPM.S235035
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Hyun Jeong Kim, 1,* Nga Thi Trinh, 1,* Yunjeong Choi, 1 Woorim Kim, 1 Kyung Hyun Min, 1 Sang Oh Kang, 1 Joo Hee Kim, 2 Hyoun-Ah Kim, 3 Ju-Yang Jung, 3 In Ah Choi, 4 Kyung Eun Lee 1
1College of Pharmacy, Chungbuk National University, Cheongju-si, Republic of Korea; 2College of Pharmacy, Ajou University, Suwon, Republic of Korea; 3Department of Rheumatology, Ajou University School of Medicine, Suwon, Republic of Korea; 4Division of Rheumatology, Department of Internal Medicine, Chungbuk National University Hospital, Cheongju, Republic of Korea
*These authors contributed equally to this work
Correspondence: Kyung Eun Lee
College of Pharmacy, Chungbuk National University, 660-1 Yeonje-ri, Osong-eup, Heungdeok-gu, Cheongju-si 28160, Republic of Korea
Tel +82 43 261 3590
Fax +82 43 268 2732
Email [email protected]
Purpose: TNF-α is a transmembrane protein which requires cleavage by ADAM17 in order to act systemically. The activation of ADAM17 to generate soluble TNF‑α results in an increased inflammatory activity. We hypothesized that variants spanning the ADAM17 gene contribute towards the observed variation in patient response defined by the number of changes in TNF‑α inhibitors.
Patients and Methods: Seven single-nucleotide polymorphisms (SNPs) of ADAM17 in 63 patients with rheumatoid arthritis who received TNF-α inhibitors were analyzed: rs57467365, rs62117540, rs117645314, rs6432013, rs532704607, rs117179141, and rs12692386. Univariate and multivariate regression analysis were employed to investigate the independent predictable factors for changes in TNF-α inhibitors.
Results: ADAM17 rs117645314 and rs117179141 showed significant association with the number of changes in TNF-α inhibitors. Patients with GA in rs117645314 and AT in rs117179141 had significantly higher chance of two or more changes of TNF-α inhibitors than those with wild homozygous alleles. Multivariate analysis showed that rs117179141 explained 5.7% of the 23.8% variability in TNF-α inhibitor response.
Conclusion: This study showed that the number of changes in TNF-α inhibitor is associated with ADAM17 SNPs.
Keywords: rheumatoid arthritis, TNF-α inhibitor, ADAM17, single-nucleotide polymorphism
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by chronic inflammation that mainly affects the joints. RA is an intricate, polygenic and heterogeneous disease characterized by complex interactions between genetic and environmental factors. The immune-mediated background is validated by the critical role of immune-suppressive therapies. Since the introduction of the first drugs of this class, the Tumor Necrosis Factor-α inhibitor (TNFi), the prognosis is much better than before. Recently, more immune mediators are under development, drugs targeting B cells, T cells or intracellular kinases.1,2 This choice of drugs is welcomed because none of them is effective in all patients. Typically, about 30% of the patients fail to respond to any of the drugs, and an additional 30% of patients show only a partial response. This variability in response drives rheumatologist to change from one drug to another, and by combining them with conventional antirheumatic drugs.3
Gene studies had been performed in order to investigate genetic biomarkers which could play an integral part in disease susceptibility and treatment. In regards to TNFi, most studies included small number of patients with inconsistent results.4–8 Previously investigated genes in associations with TNFi include tumor necrosis factor gene, PDE3A-SLCO1C1, protein tyrosine phosphatase receptor type C, fragment C gamma receptor, and mitogen-activated protein kinase 14.9–13 More than 40 candidate gene studies and 6 GWAS regarding the response to TNFi have been performed previously.14,15 However, much of the research conducted has been focused on candidate genes related to susceptibility of the disease that are not necessarily the same as those involved in the treatment response. Also, in many cases, results of different studies show contradictory or inconclusive conclusions making it difficult to apply the pharmacogenetics of TNFi into clinical practice.
In an effort to discover genetic markers of TNFi response, we investigated a gene with a close relation to TNF-α pathway. TNF-α is a pro-inflammatory cytokine which undergoes proteolysis by TNF-α converting enzyme (TACE).16 A disintegrin and metalloproteinase 17 (ADAM17), also known as TACE, is a membrane bound enzyme that cleaves immune cell surface cytokine and cytokine receptor, such as TNFα and IL-6. Here, we examined the influence of ADAM17 genetic polymorphisms on the treatment response of TNFi.
Methods
Patients
The study population consisted of 63 patients with rheumatoid arthritis who were prescribed TNFi in Ajou University Hospital and Chungbuk National University Hospital between January 2008 and December 2018. Patients’ data were collected from electronic medical records from both hospitals. This study was approved by the ethics committees (Ajou University Hospital: AJIRB-BMR-OBS-17-153 and Chungbuk National University Hospital: 2017-06-011-004) and patients gave their written informed consent. The study was conducted according to the principles of the Declaration of Helsinki (2013).
Genotyping
Seven single-nucleotide polymorphisms (SNPs) of ADAM17 (rs57467365, rs62117540, rs117645314, rs6432013, rs532704607, rs117179141, rs12692386) were selected based on http://grch37.ensembl.org/index.html with minor allele frequency of greater than 10% in Japanese and Han Chinese in Beijing. Genotyping was conducted by SNaPshot assay in six SNPs and TaqMan Genotyping assay was used for rs12692386. Genomic DNA was purified from blood sample or buffy coat samples using the QIAamp DNA Blood Mini Kit (QIAGEN GmbH, Hilden, Germany) in accordance with the manufacturer’s protocol. All seven SNPs were in concordance with the Hardy–Weinberg equilibrium.
Statistical Analysis
We examined the number of changes in TNFi for the primary outcome reflecting the response of TNFi. The criteria for change in TNFi were lack of efficacy, presence of adverse events or noncompliance. We have divided the number of changes in TNFi into three groups as none, once, and two or more changes. The Pearson’s χ2-test or Fisher’s exact test was used for categorical and multinomial logistic regression was used to compare continuous variables. Stepwise linear regression analysis was used to assess multivariate relationships between demographic, clinical, and genetic variables and changes in TNFi. Factors having P-values < 0.05 from univariate analysis along with age and sex were included in the multivariate analysis. Variables were entered by stepwise selection for P < 0.05 and were removed for P > 0.1. P-values < 0.05 were considered statistically significant. Statistical Package for Social Sciences version 25.0 for Windows (SPSS, Chicago, IL, USA) was used for all analyses. The Haploview 4.2 software package was used to estimate pairwise linkage disequilibrium (LD), detect departure from the Hardy–Weinberg equilibrium, and construct haplotypes. The haplotype block was created based on the default algorithm of Burmester et al.17,18
Results
A total of 63 patients were included in the analysis and 17 patients showed one or more changes of therapy in TNFi. The median age of patients was 56 years, ranging from 19 to 78, and 77.8% were females. Most patients denied alcohol use or smoking (79.4% and 76.2%, respectively). Except three patients without the record of tuberculosis test, 20.7% of patient revealed pulmonary tuberculosis or positive interferon-gamma release assay (IGRA) or positive PPD test.
The number of changes in TNFi was associated with a significant increase in disease duration (p= 0.038). Nine patients had a history of hypertension, which showed statistical significance in relation to the number of changes in TNFi. (p= 0.042). Each component of disease activity score (DAS) as well as the total DAS28 score at baseline did not differ among the groups. Fifty-three patients (84.1%) were positive for rheumatoid factor (RF) and 45 patients (71.4%) were positive for anti-cyclic citrullinated peptide antibody (ACPA). Both tests of RF and ACPA were not statistically significant among the groups (Table 1).
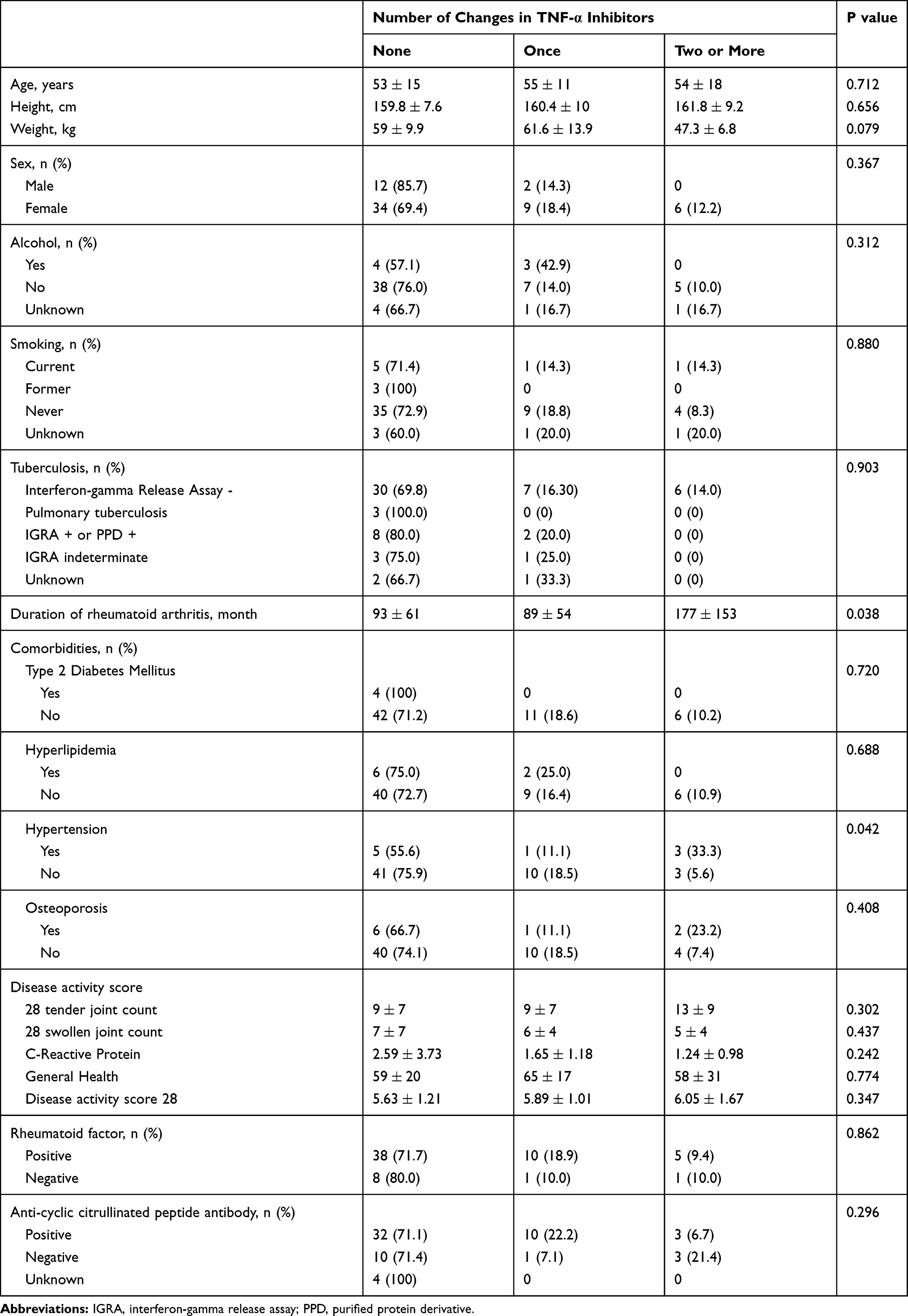 |
Table 1 Baseline Characteristics of Patients |
The most frequently prescribed TNFi was adalimumab (n=25), followed by golimumab (n=13). Non-TNF inhibitors, which were used prior to TNFi or concurrently, include conventional disease-modifying anti-rheumatic drugs such as methotrexate, leflunomide, sulfasalazine and hydroxychloroquine and newer agent such as tofacitinib. There were 18 patients who used hydroxychloroquine and these users were statistically more likely to fail the first TNFi (p=0.003). Steroids were used in all patients showing no statistical difference among the groups (Table 2).
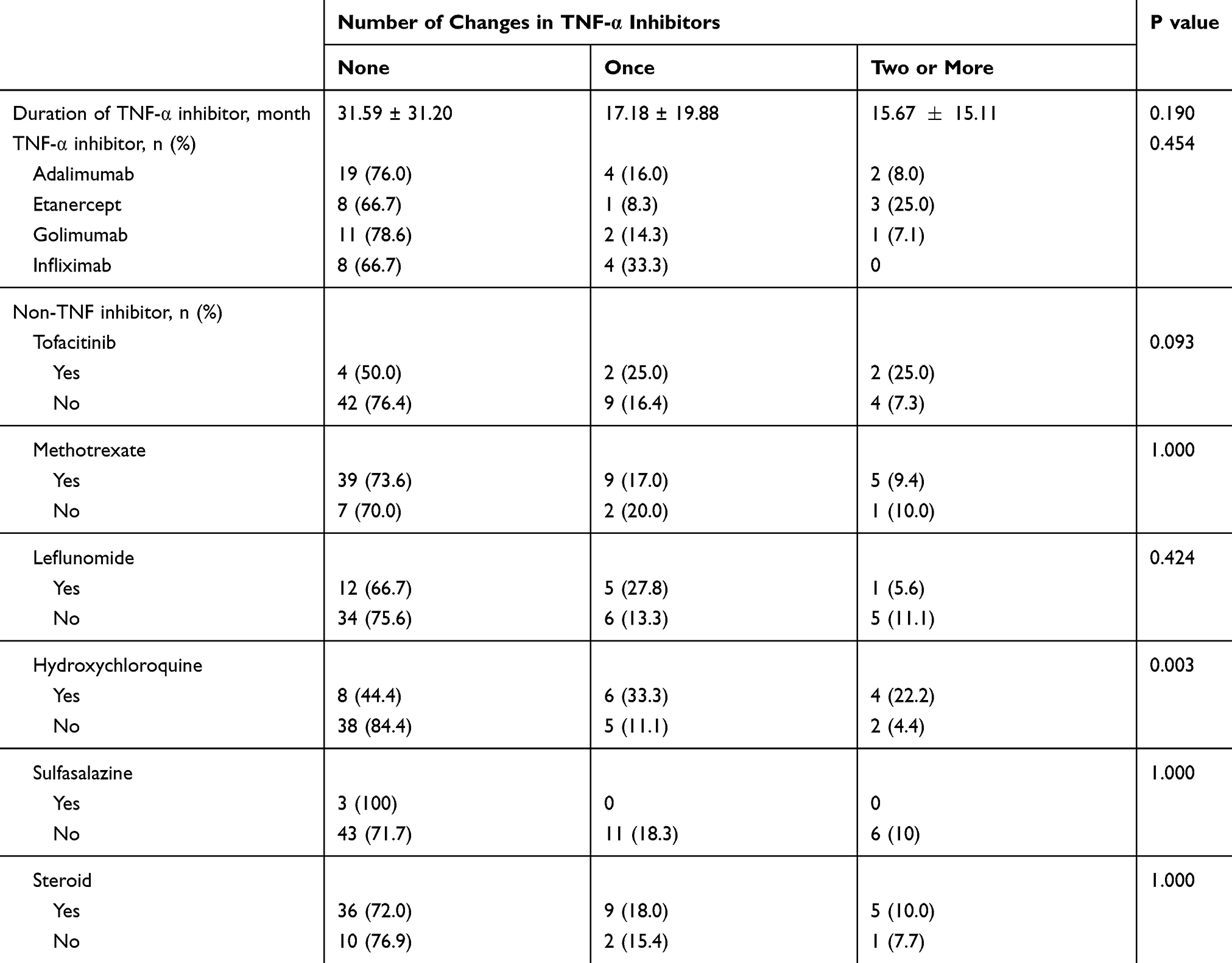 |
Table 2 TNF-α Inhibitors and Non-TNF-α Inhibitors Used for Study Subjects |
Of the 7 SNPs analyzed, no mutation was detected in rs532704607 SNP in our patients. For three SNPs, rs117645314, rs6432013, and rs117179141, homozygous variant allele carriers did not exist. Two SNPs showed statistical differences in number of changes in TNFi: rs117645314 and rs117179141 (Table 3). In rs117645314 SNP, GG-carriers had significantly lower chance of two or more changes in TNFi than those with GA genotypes (OR=0.12, 95% CI 0.02–0.78). For rs117179141 allelic variation, patients with AA genotype showed significantly lower chance of two or more changes in TNFi than those with AT-carriers (OR=0.12, 95% CI 0.02–0.78).
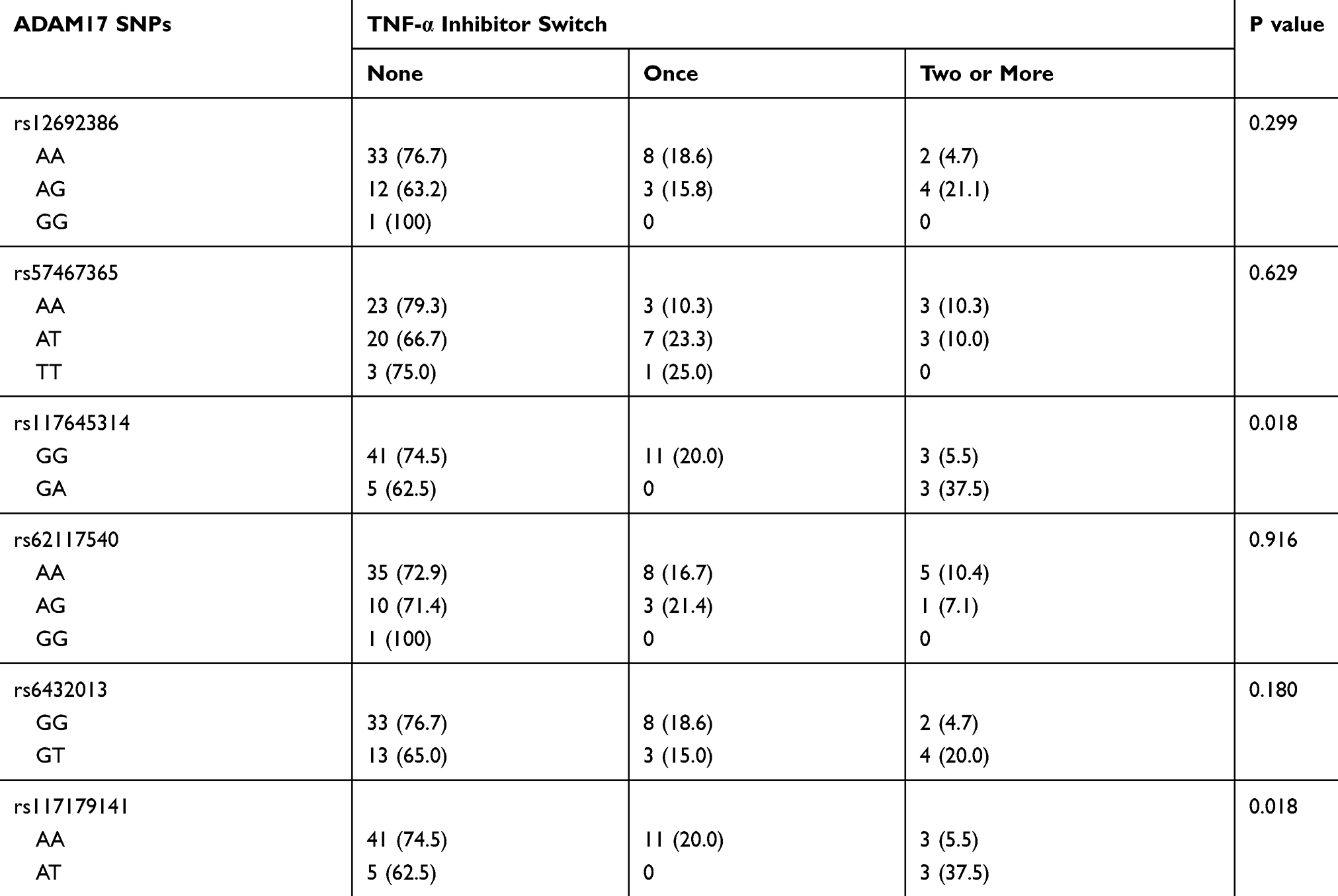 |
Table 3 Univariate Analysis of Genetic Polymorphisms of ADAM17 with Number of Changes in TNF-α Inhibitors |
Haplotype analyses were performed for 6 SNPs in ADAM17 gene with mutations in our patients. LD was observed across rs117645314, rs6432013 and rs117179141 (D’= 1 among all three SNPs, r2= 1 between rs117645314 and rs117179141, and r2=0.33 between rs6432013 and other two SNPs). There was a strong LD between rs6432013 and rs12692386 (D’= 0.79, r2= 0.63) (Figure 1).
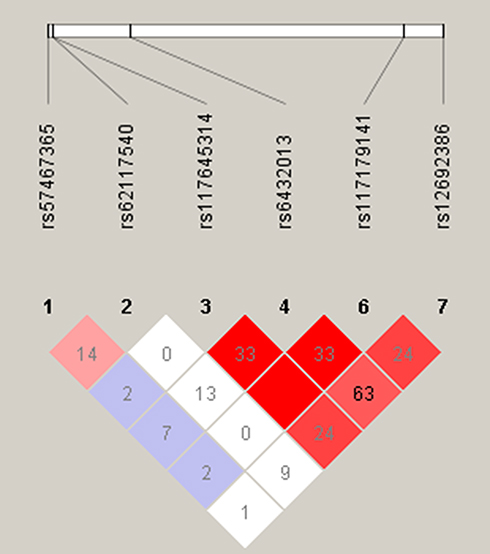 |
Figure 1 Relative positions and linkage disequilibrium (LD) estimates in ADAM17 polymorphisms in the analyzed population. Notes: Colored squares correspond to r2 values with numerical estimates given within the squares. The squares without a number correspond to r2 = 1. Shading represents the magnitude and significance of pairwise LD, with a red-to white gradient reflecting higher to lower LD values. |
Stepwise linear regression analysis was used to determine the independent effects of the possible genetic factors of TNFi response. Age and sex were fixed inclusion factors, and variables with p < 0.05 from univariate analysis, including two statistically significant SNPs, were included in our model. Multicollinearity was checked which showed variance inflation factor values from 1.00 to 1.13, indicating a low correlation of predictor variables. In the final model, hydroxychloroquine use accounted for 16.1% and rs117179141 accounted for 5.7% of the total 23.8% variability in observed number of changes in TNFi (Table 4).
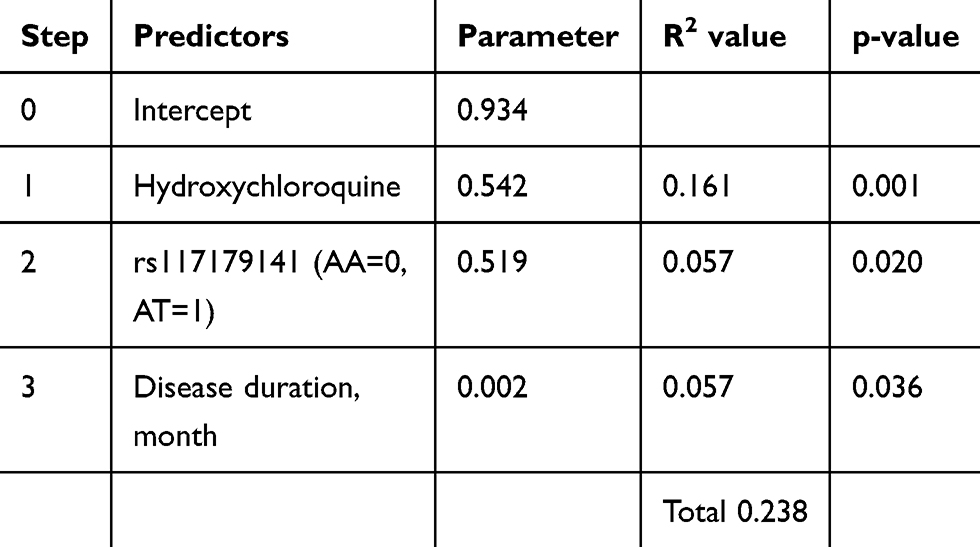 |
Table 4 Multivariable Regression Model Predicting Number of Changes in TNF-α Inhibitors |
Discussion
Despite the proven efficacy of anti-TNF drugs in the treatment of rheumatoid arthritis, a significant inadequate response rate is exhibited.19 Current clinical factors provide little power for predicting response; therefore, “trial and error” approach governs therapeutic decisions. In order to investigate clinically meaningful genetic polymorphism associated with TNFi, we examined ADAM17 SNPs. We found that rs117179141 and rs117645314 were statistically significant SNPs associated with the number of changes in TNFi.
The ADAM is a transmembrane protein, which is crucial for various biological processes, such as inflammation, migration and immunity. ADAM17, also recognized as TACE, is responsible for the shedding of many substrates including TNF-α and IL-6R which mediate various inflammation-promoting biological activities.20,21 TACE expressions were up-regulated in rheumatoid arthritis patients and TACE was shown to be an important regulator of the secretion of TNF-α in previous studies.22,23
Among the small number of studies examining the relationship between ADAM17 and pharmacotherapy, Umemura et al reported that suppression of ADAM17 can be a crucial therapeutic target in the treatment of abatacept in patients with rheumatoid arthritis.24 Another study showed that ADAM17 haplotype was associated with a clinical response to infliximab in Crohn’s disease patients.25 This haplotype included 11 SNPs spanning from the rs2001658 to the rs11684747, but none of the SNPs overlapped with our SNPs of interest. There was a study using a UK cohort which evaluated the association of TACE polymorphism with TNFi treatment response, but showed no significance in the 10 SNPs included.26 Seven SNPs in our study did not overlap with the UK study due to the ethnic difference although similar selection criteria of minor allele frequency was used.
Of the seven SNPs from our study, two SNPs, rs117645314 and rs117179141, were shown to be statistically significantly associated with TNFi response. Unfortunately, studies including these two SNPs are scarce, making it difficult to clarify the underlying mechanism accordingly. We can speculate about the possible influence of SNPs on the response of TNFi as an association with other regulatory SNPs in LD.27 One of the regulatory SNPs that may influence gene expression was found to be rs76947488 which was in strong LD with rs117179141. SNPs in strong LD with rs76947488 were rs72777025 in intergenic region and rs12692387 in regulatory region. But, rs117645314 was not in a strong LD with rs76947488 nor rs72777025, suggesting the putative functional role would be in rs117179141 and not rs117645314.28
In order to examine the effects of SNPs on ADAM17 gene expression, we examined the expression profiling using GTEx datasets.29 Unfortunately, two statistically significant SNPs (rs117179141 and rs117645314) were not found in GTEx datasets. We observed significant expression quantitative trait loci eQTL for linked SNPs of rs76947488, rs72777025 and rs12692387 in skeletal muscle tissue, which revealed significantly higher expressions with variant type alleles (p= 4.4 x 10−5, p= 1.8 x 10−11 and p= 1.6 x 10−17, respectively). Given these findings of statistically different gene expression profile of SNPs in LD with the significant SNPs of our interest, involvement of ADAM17 in the treatment response of TNFi warrants further studies in different patient groups and populations.
The limitation of this study is the small sample size due to the limited number of rheumatoid arthritis patients in epidemic aspect. Thus, the effect of clinical and genetic variables on the treatment response according to each TNF-alpha inhibitor could not be revealed. The limited number of patients included and the absence of replicative cohort warrants future investigation with larger sample size in longer study period.
Conclusion
There is an evidence that ADAM17 affects clinical phenotype. However, as most SNP frequencies are relatively low and clinical variations associated with these SNPs are not severe, the impact of a single SNP on TNFi response may be limited. Further studies of the mechanisms underlying the observed association of SNPs with clinical outcomes could provide additional understandings that may assist the results of this study leading to new possibilities for drug treatment.
Abbreviations
RA, rheumatoid arthritis; TNFi, tumor necrosis factor-α inhibitor; TACE, TNF-α converting enzyme; ADAM17, A disintegrin and metalloproteinase 17; SNPs, single-nucleotide polymorphisms; DAS, disease activity score; RF, rheumatoid factor; ACPA, anti-cyclic citrullinated peptide antibody; LD, linkage disequilibrium.
Funding
This work was supported by the Basic Science Research Program through National Research Foundation (NRF) funded by the Korea government (MSIP; Ministry of Science, ICT & Future Planning) (NRF-2017R1C1B5016202) and the Medical Research Center Program (2017R1A5A2015541) of the NRF funded by the Korean government (MSIP). The funding sources did not have a role in the design, conduct, and analysis of the study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Burmester GR, Pope JE. Novel treatment strategies in rheumatoid arthritis. Lancet (London, England). 2017;389(10086):2338–2348. doi:10.1016/S0140-6736(17)31491-5
2. Smolen JS, Landewe R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977. doi:10.1136/annrheumdis-2016-210715
3. Lopez-rodriguez R, Perez-Pampin E, Marquez A, et al. Validation study of genetic biomarkers of response to TNF inhibitors in rheumatoid arthritis. PLoS One. 2018;13(5):e0196793. doi:10.1371/journal.pone.0196793
4. Castaneda S, Lopez-mejias R, Gonzalez-gay MA. Gene polymorphisms and therapy in rheumatoid arthritis. Expert Opin Drug Metab Toxicol. 2016;12(3):225–229. doi:10.1517/17425255.2016.1141405
5. Davila L, Ranganathan P. Pharmacogenetics: implications for therapy in rheumatic diseases. Nat Rev Rheumatol. 2011;7(9):537–550. doi:10.1038/nrrheum.2011.117
6. Kang CP, Lee KW, Yoo DH, Kang C, Bae SC. The influence of a polymorphism at position −857 of the tumour necrosis factor alpha gene on clinical response to etanercept therapy in rheumatoid arthritis. Rheumatology (Oxford). 2005;44(4):547–552. doi:10.1093/rheumatology/keh550
7. Lee YH, Ji JD, Bae S-C, Song GG. Associations between Tumor Necrosis Factor-α (TNF-α) −308 and −238 G/A polymorphisms and shared epitope status and responsiveness to TNF-α blockers in rheumatoid arthritis: a metaanalysis update. J Rheumatol. 2010;37(4):740–746. doi:10.3899/jrheum.090707
8. Pavy S, Toonen EJM, Miceli-richard C, et al. Tumour necrosis factor α −308G→A polymorphism is not associated with response to TNFα blockers in Caucasian patients with rheumatoid arthritis: systematic review and meta-analysis. Ann Rheum Dis. 2010;69(6):1022–1028. doi:10.1136/ard.2009.117622
9. Seitz M, Wirthmuller U, Moller B, Villiger PM. The −308 tumour necrosis factor-alpha gene polymorphism predicts therapeutic response to TNFalpha-blockers in rheumatoid arthritis and spondyloarthritis patients. Rheumatology (Oxford). 2007;46(1):93–96. doi:10.1093/rheumatology/kel175
10. Acosta-colman I, Palau N, Tornero J, et al. GWAS replication study confirms the association of PDE3A-SLCO1C1 with anti-TNF therapy response in rheumatoid arthritis. Pharmacogenomics. 2013;14(7):727–734. doi:10.2217/pgs.13.60
11. Ferreiro-iglesias A, Montes A, Perez-pampin E, et al. Replication of PTPRC as genetic biomarker of response to TNF inhibitors in patients with rheumatoid arthritis. Pharmacogenomics J. 2016;16(2):137–140. doi:10.1038/tpj.2015.29
12. Iotchkova V, Huang J, Morris JA, et al. Discovery and refinement of genetic loci associated with cardiometabolic risk using dense imputation maps. Nat Genet. 2016;48(11):1303–1312. doi:10.1038/ng.3668
13. Coulthard LR, Taylor JC, Eyre S, et al. Genetic variants within the MAP kinase signalling network and anti-TNF treatment response in rheumatoid arthritis patients. Ann Rheum Dis. 2011;70(1):98–103. doi:10.1136/ard.2010.133249
14. Bek S, Bojesen AB, Nielsen JV, et al. Systematic review and meta-analysis: pharmacogenetics of anti-TNF treatment response in rheumatoid arthritis. Pharmacogenomics J. 2017;17(5):403–411. doi:10.1038/tpj.2017.26
15. Massey J, Plant D, Hyrich K, et al. Genome-wide association study of response to tumour necrosis factor inhibitor therapy in rheumatoid arthritis. Pharmacogenomics J. 2018;18(5):657–664. doi:10.1038/s41397-018-0040-6
16. Deora A, Hegde S, Lee J, et al. Transmembrane TNF-dependent uptake of anti-TNF antibodies. mAbs. 2017;9(4):680–695. doi:10.1080/19420862.2017.1304869
17. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi:10.1093/bioinformatics/bth457
18. Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science (New York, NY). 2002;296(5576):2225–2229. doi:10.1126/science.1069424
19. Hyrich KL, Watson KD, Silman AJ, Symmons DP. Predictors of response to anti-TNF-alpha therapy among patients with rheumatoid arthritis: results from the british society for rheumatology biologics register. Rheumatology (Oxford). 2006;45(12):1558–1565. doi:10.1093/rheumatology/kel149
20. Menghini R, Fiorentino L, Casagrande V, Lauro R, Federici M. The role of ADAM17 in metabolic inflammation. Atherosclerosis. 2013;228(1):12–17. doi:10.1016/j.atherosclerosis.2013.01.024
21. Lisi S, D’amore M, Sisto M. ADAM17 at the interface between inflammation and autoimmunity. Immunol Lett. 2014;162(1Pt A):159–169. doi:10.1016/j.imlet.2014.08.008
22. Ishii S, Isozaki T, Furuya H, et al. ADAM-17 is expressed on rheumatoid arthritis fibroblast-like synoviocytes and regulates proinflammatory mediator expression and monocyte adhesion. Arthritis Res Ther. 2018;20(1):159. doi:10.1186/s13075-018-1657-1
23. Ohta S, Harigai M, Tanaka M, et al. Tumor necrosis factor-alpha (TNF-alpha) converting enzyme contributes to production of TNF-alpha in synovial tissues from patients with rheumatoid arthritis. J Rheumatol. 2001;28(8):1756–1763.
24. Umemura M, Isozaki T, Ishii S, et al. Reduction of serum ADAM17 level accompanied with decreased cytokines after abatacept therapy in patients with rheumatoid arthritis. Int J Biomed Sci. 2014;10(4):229–235.
25. Dideberg V, Theatre E, Farnir F, et al. The TNF/ADAM 17 system: implication of an ADAM 17 haplotype in the clinical response to infliximab in Crohn’s disease. Pharmacogenet Genomics. 2006;16(10):727–734. doi:10.1097/01.fpc.0000230117.26581.a4
26. Potter C, Gibbons LJ, Bowes JD, et al. Polymorphisms spanning the TNFR2 and TACE genes do not contribute towards variable anti-TNF treatment response. Pharmacogenet Genomics. 2010;20(5):338–341. doi:10.1097/FPC.0b013e32833878d7
27. Deng N, Zhou H, Fan H, Yuan Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget. 2017;8:66. doi:10.18632/oncotarget.v8i66
28. Hunt SE, McLaren W, Gil L, et al. Ensembl variation resources. Database. 2018;2018. doi:10.1093/database/bay119
29. Carithers LJ, Ardlie K, Barcus M, et al. A novel approach to high-quality postmortem tissue procurement: the GTEx project. Biopreserv Biobank. 2015;13(5):311–319. doi:10.1089/bio.2015.0032
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.