")
Back to Journals » Journal of Inflammation Research » Volume 14
Acute P38-Mediated Enhancement of P2X3 Receptor Currents by TNF-α in Rat Dorsal Root Ganglion Neurons
Authors Jin Y, Wei S, Liu TT, Qiu CY, Hu WP
Received 22 April 2021
Accepted for publication 5 June 2021
Published 29 June 2021 Volume 2021:14 Pages 2841—2850
DOI https://doi.org/10.2147/JIR.S315774
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Ying Jin, Shuang Wei, Ting-Ting Liu, Chun-Yu Qiu, Wang-Ping Hu
Research Center of Basic Medical Sciences, School of Basic Medical Sciences, Hubei University of Science and Technology, Xianning, Hubei, 437100, People’s Republic of China
Correspondence: Wang-Ping Hu
Research Center of Basic Medical Sciences, School of Basic Medical Sciences, Hubei University of Science and Technology, 88 Xianning Road, Xianning, Hubei, 437100, People’s Republic of China
Email [email protected]
Purpose: Tumor necrosis factor-α (TNF-α) is a pro-inflammatory cytokine and involves in a variety of pain conditions. Some findings suggest that TNF-α may act directly on primary afferent neurons to induce acute pain hypersensitivity through non-transcriptional regulation. This study investigated whether TNF-α had an effect on functional activity of P2X3 receptors in primary sensory neurons. Herein, we report that a brief (5 min) application of TNF-α rapidly enhanced the electrophysiological activity of P2X3 receptors in rat dorsal root ganglia (DRG) neurons.
Methods: Electrophysiological recordings were carried out on rat DRG neurons, and nociceptive behavior was quantified in rats.
Results: A brief (5 min) exposure of TNF-α rapidly increased P2X3 receptor-mediated and α,β-methylene-ATP (α,β-meATP)-evoked inward currents in a dose-dependent manner. The potentiation of P2X3 receptor-mediated ATP currents by TNF-α was voltage-independent. TNF-α shifted the concentration–response curve for α,β-meATP upwards, with an increase of 31.57 ± 6.81% in the maximal current response to α,β-meATP. This acute potentiation of ATP currents by TNF-α was blocked by p38 mitogen-activated protein kinase (MAPK) inhibitor SB202190, but not by non-selective cyclooxygenase inhibitor indomethacin, suggesting involvement of p38 MAPK, but not cyclooxygenase. Moreover, intraplantar injection of TNF-α and α,β-meATP produced a synergistic effect on mechanical allodynia in rats. TNF-α-induced mechanical allodynia was also alleviated after local P2X3 receptors were blocked.
Conclusion: These results suggested that TNF-α rapidly sensitized P2X3 receptors in primary sensory neurons via a p38 MAPK dependent pathway, which revealed a novel peripheral mechanism underlying acute mechanical hypersensitivity by peripheral administration of TNF-α.
Keywords: electrophysiology, dorsal root ganglion neuron, nociceptive response, P2X3 receptor, tumor necrosis factor-α
Introduction
During tissue damages and inflammation, a variety of mediators are released and contribute to peripheral sensitization by regulating activity of ion channels that mediate the transduction of pain signaling. Tumor necrosis factor-α (TNF-α), a pro-inflammatory cytokine, is synthesized and released by various cells, such as inflammatory, immune, glia, and neuronal cells.1,2 Intraplantar injection of TNF-α causes mechanical and thermal hyperalgesia in rats and mice.3–5 Elevated levels of endogenous TNF-α promote the development of pain and hyperalgesia.5,6 Neutralizing endogenous TNF-α with antibodies attenuates both thermal and mechanical hyperalgesia in neuropathic pain.7,8 Thus, TNF-α has been shown to play a critical role in the development and maintenance of pain.9 Peripheral treatment of TNF-α also causes acute thermal and mechanical hypersensitivity by rapidly enhancing the ongoing activity of nociceptive fibers.10–12 TNF-α can regulate pain signaling not only through genomic but also non-genomic mechanisms. TNF-α plays a long-lasting role in the expression of inflammatory mediators and signaling proteins. In addition, TNF-α has also a rapid regulatory effect on the functional activity of several ion channels. Studies have shown that TNF-α acutely regulates tetrodotoxin-resistant (TTX-R) sodium channels, L-type calcium channels, outward potassium channels, and transient potential receptor vanilloid (TRPV1) in primary sensory neurons and other cells.12–16 The acute regulatory effects of TNF-α on these ion channels may explain the rapid onset of hyperalgesia induced by TNF-α.17
ATP is also a major inflammatory mediator, and plays a prominent role in some pain states.18,19 ATP receptors or purinergic receptors, including P2X and P2Y, are expressed in primary sensory neurons.18 Among all purinergic receptors, P2X3 receptor subtypes, and P2X2/3 heteromer receptors are mainly distributed in small and medium-sized nociceptive sensory neurons.1,20,21 P2X3-containing receptors participate in the transmission of nociceptive signaling. For example, P2X3 knock-out mice display a reduced nociceptive behavior.22 Blockade of P2X3 by antagonist or antisense oligonucleotide can also attenuate pain.23,24 After inflammation and nerve injury, P2X3 receptor-mediated responses are more sensitive, including increased ATP-activated currents and ATP-induced nociceptive behaviors.25–28 However, the mechanisms responsible for the increase are not well understood.
Since both TNF-α and ATP are mediators released during inflammation, the purpose of this study was to investigate whether P2X3 receptor is also a target for rapid modulation of TNF-α. Herein, we show that a brief (5 min) pre-application of TNF-α to rat DRG neurons acutely increases P2X3 receptor-mediated ATP currents via a p38 mitogen-activated protein kinase (MAPK) dependent pathway, which may contribute to rapid onset of hyperalgesia induced by peripheral administration TNF-α.
Materials and Methods
Preparation of DRG Neurons
All studies were designed to minimize the sufferings of animals and approved by the animal research ethics committee of Hubei University of Science and Technology (No. 2020–08). Sprague-Dawley male rats (5–6 weeks old) were anesthetized and then killed. The DRGs were removed and chopped with thin spring scissors. The minced ganglia were transferred to a test tube containing Dulbecco’s modified Eagle’s medium (DMEM, Sigma) and incubated in a shaking for 25–30 min at 35°C. Incubation solution contained 1.0 mg/mL collagenase (type I-A, Sigma), 0.5 mg/mL trypsin (type II-S, Sigma), and 0.1 mg/mL DNase (type IV, Sigma). Trypsin digestion was terminated by adding 1.25 mg/mL Soybean trypsin inhibitor (type II-S, Sigma).
Electrophysiological Recordings
Electrophysiological experiments were carried out as described previously.29 Whole-cell patch clamp recordings were carried out at room temperature (22–25°C) using a MultiClamp-700B amplifier (Axon Instruments, CA, USA). The isolated DRG neurons were transferred to a 35 mm culture dish and kept in normal external solution for at least 60 min before electrophysiological recordings. The external solution contained the following (in mM): 150 NaCl, 5 KCl, 2 MgCl2, 2.5 CaCl2, 10 HEPES, 10 d-glucose. Its pH and osmolarity were adjusted to 7.4 with NaOH and 330 mOsm/L with sucrose, respectively. Recording pipettes were pulled using a Sutter P-97 puller (Sutter Instruments, CA, USA) and its resistance was in the range of 3–6MΩ. The micropipette solution contained (in mM): 140 KCl (or CsCl), 2 MgCl2, 11 EGTA, 10 HEPES, 4 ATP, and 0.3 Na2GTP. Its pH and osmolarity were adjusted to 7.2 with KOH and 310 mOsm/L with sucrose, respectively. In the present experiment, DRG neurons with a diameter of 15–35μm were used for electrophysiological recording. After whole-cell configuration established, 70–80% series resistance and membrane capacitance current were compensated. The recording currents were sampled at 10 kHz and filtered at 2 kHz. Data detection and analysis were performed using the pCLAMP 10 software (Axon Instruments, CA, USA).
Drug Application
All drugs were obtained from Sigma (St. Louis, MO, USA). The working concentration of drugs was freshly prepared in normal external solution. Each working drug was stored in a series of independent reservoirs and applied by gravity. The distance was ∼30 μm between drug exit and recorded neurons.
Animal Behavioral Assay
Male rats were adapted to the environment for at least 30 min in a Plexiglas chamber before behavior experiments. Mechanical allodynia was measured by paw withdrawal threshold (PWT).30 All behavioral studies were carried out with double-blind experiment. After rats were encoded, TNF-α and/or α,β-meATP was injected into the rat's hind paws in separate groups. Another experimenter tested PWT of the ipsilateral hind plantar using a series of von Frey filaments (Stoelting, Wood Dale, IL) at 15 min after injection. In another behavioral experiment, the hind paws were pretreated with A-317491 or vehicle 10 min before TNF-α injection, and then PWT was measured at 0.5, 1, 2, 4, and 24 h after TNF-α injection.
Data Analysis
Data were expressed as mean ± SEM and statistically compared using the Student’s t-test or analysis of variance (ANOVA), followed by Bonferroni’s post hoc test. Nonlinear curve-fitting program ALLFIT was used for statistical analysis of concentration–response data.
Results
Acute Enhancement of P2X3 Receptor-Mediated Currents by TNF-α in Rat DRG Neurons
Inward currents (IATP) were evoked by α,β-methylene-ATP (α,β-meATP, 100 μM) or ATP (100 μM) in the majority of small- and medium-sized DRG cells (75.0%, 9/12; Figure 1A). IATP was blocked by 300 μM A-317491, a specific P2X3 receptor antagonist.24 And α,β-meATP is only an activator of P2X3 and P2X1 receptors.31 Therefore, we concluded that P2X3 receptors mediated the α,β-meATP induced currents.
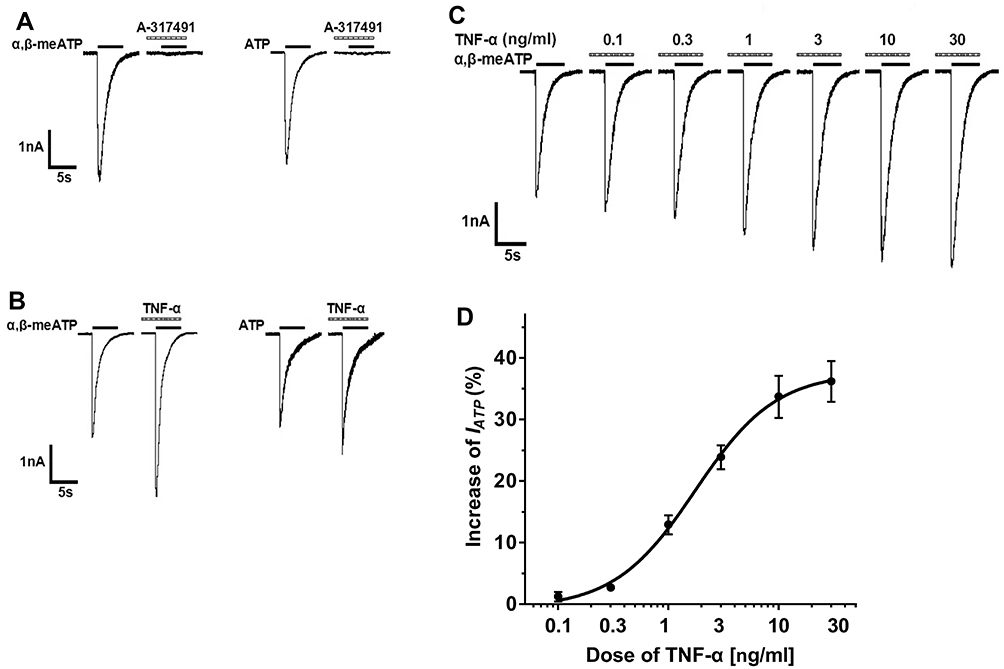 |
Figure 1 TNF-α rapidly increased P2X3 receptor-mediated ATP currents in DRG neurons. (A) In a representative DRG cell, 100 μM α,β-meATP and ATP induced similar inward currents. The currents could be blocked by 300 μM A-317491, a specific P2X3 receptor antagonist, indicating that they were P2X3 receptor-mediated ATP currents. Membrane potentials were clamped at −60 mV. (B) In a DRG neuron, a five minute application of TNF-α (10 ng/mL) increased α,β-meATP- and ATP-induced currents. (C) The sequential current traces illustrated that the amplitude of the currents induced by 100 μM α,β-meATP progressively increased after different doses of TNF-α was pre-treated to a representative DRG cell. (D) The graph shows the dose–response curve of TNF-α. The EC50 value of the curve was 1.75 ± 0.17 ng/mL. Each point represents the mean ± SEM of 7–11 cells. |
In some DRG cells sensitive to α,β-meATP or ATP, the effects of TNF-α on the P2X3 receptor-mediated ATP currents were first evaluated. DRG cells were pre-incubated with TNF-α for 5 min prior to application of α,β-meATP or ATP. As shown in Figure 1B, the peak amplitudes of both α,β-meATP- and ATP-evoked currents were rapidly enhanced by a brief (5 min) application of TNF-α (10 ng/mL). In DRG cells pre-treated with TNF-α, the peak amplitudes of 100 μM α,β-meATP- and ATP-evoked currents increased from 2.64 nA to 4.13 nA and from 2.32 nA to 3.08 nA, separately (Figure 1B). The TNF-α-induced enhancement of IATP appeared 5 min after the onset of TNF-α application. Figure 1C and D show the effects of different doses of TNF-α pretreatment on the membrane currents evoked by 100 μM α,β-meATP. Figure 1C shows that the peak amplitudes of IATP increased with the increase of TNF-α pretreatment dose from 0.1 ng/mL to 30 ng/mL in a representative DRG cell. The dose–response curve in Figure 1D shows that EC50 (half-maximal effective dose) of TNF-α was 1.75 ± 0.17 ng/mL. The results suggested that TNF-α rapidly increased ATP currents mediated by P2X3 receptors in dose-dependent manner.
Concentration–Response and Current–Voltage Relationships for α,β-meATP in the Absence and Presence of TNF-α
Effects of TNF-α on the concentration–response curve for α,β-meATP was then investigated. Different concentration of α,β-meATP were used to induce IATP. Figure 2A shows that pretreatment of DRG cells with TNF-α (10 ng/mL) for 5 min enhanced the currents evoked by α,β-meATP at 3, 30, and 300 μM. Figure 2B shows that concentration–response curves for α,β-meATP in the absence and presence of TNF-α (10 ng/mL) pretreatment, which were fit with the Hill equation. We observed that the concentration–response curve for α,β-meATP was shifted upwards by TNF-α (10 ng/mL) pretreatment. First, maximal current response of both curves occurred at 300 μM α,β-meATP. However, the 300 μM α,β-meATP induced maximum current response increased 31.57 ± 6.81% in the presence of TNF-α (10 ng/mL). Second, the slope or Hill coefficient of curves in control and TNF-α treated cells were 0.93 ± 0.11 and 0.96 ± 0.14, respectively (P > 0.1, Bonferroni’s post hoc test). Third, TNF-α had no effect on EC50 of α,β-meATP for P2X3 receptors. EC50 of curves with and without TNF-α (10 ng/mL) pretreatment were 27.49 ± 3.05 μM and 29.45 ± 3.28 μM, respectively (P > 0.1, Bonferroni’s post hoc test). The results indicated that acute sensitization of P2X3 receptors by TNF-α was due to the increase of the α,β-meATP-induced maximum response, not the change of the apparent affinity of α,β-meATP for P2X3 receptors.
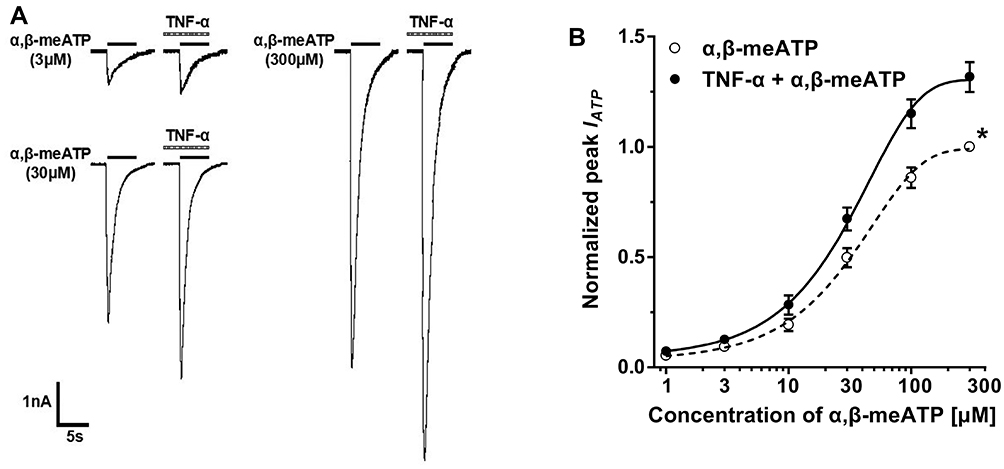 |
Figure 2 TNF-α shifted upwards the concentration–response curve for α,β-meATP. (A) Original currents show that TNF-α (10 ng/mL) pretreatment increased the currents induced by three different concentrations of α,β-meATP. (B) The graph shows concentration–response curves for α,β-meATP in the absence and presence of TNF-α (10 ng/mL). Both curves were fit by the Hill equation. Each point represents the mean ± S.E.M. of 7–10 neurons. All peak current values from the same cell were normalized to the current response induced by 300 μM α,β-meATP applied alone in the absence of TNF-α (marked with asterisk). The figure shows averaged data fitted with the Hill equation. The curves shown are a best fit of the data to the logistic equation I = Imax/[1 + (EC50/C)n], where C is the concentration of α,β-meATP, I is the normalized current response value, EC50 is the concentration of α,β-meATP for half-maximal current response, and n is the Hill coefficient. |
Membrane potential dependency of the effect of TNF-α on P2X3 receptor currents was then analyzed. We investigated the effects of TNF-α on α,β-meATP-evoked currents under different clamp potentials. Figure 3A shows that IATP was evoked by 100 μM α,β-meATP when the membrane potential was clamped at −80 mV, −40 mV, and +20 mV, respectively. The peak amplitudes of these IATP significantly increased after pretreatment of TNF-α (10 ng/mL) for 5 min. Figure 3B shows the current–voltage (I–V) curves for α,β-meATP in the absence and presence of TNF-α pretreatment. The reversal potential of each curve was close to 0 mV. However, TNF-α pretreatment increased the slope of I–V curve. There was no significant difference in the TNF-α-induced enhancement of IATP at all holding potentials from −80 to 20 mV (P > 0.1, Bonferroni’s post hoc test). The results indicated that acute enhancement of P2X3 receptor-mediated ATP currents by TNF-α was voltage-independent.
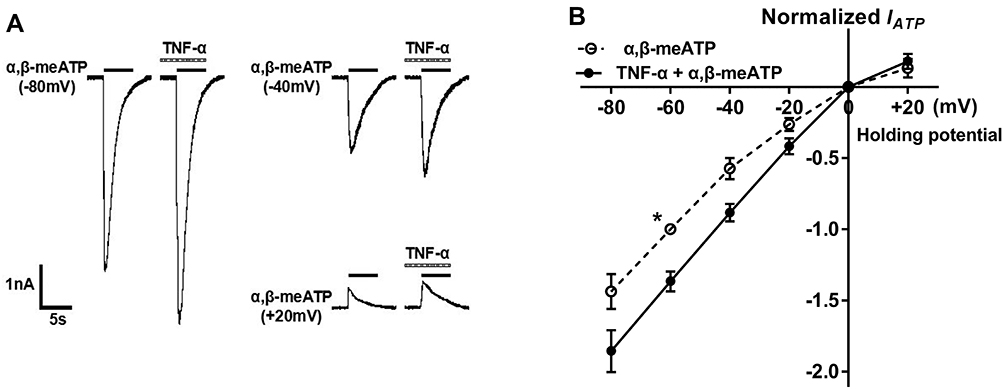 |
Figure 3 Current–voltage (I–V) relationships for α,β-meATP with or without the pre-application of TNF-α. (A) Original currents show that TNF-α (10 ng/mL) pretreatment increased the currents induced by 100 μM α,β-meATP at three different holding potentials. (B) The graph shows the effect of TNF-α (10 ng/mL) pretreatment on I–V curves for α,β-meATP. All current values were normalized to the current response induced by 100 μM α,β-meATP applied alone at −60 mV holding potential (marked with asterisk). Each point represents the mean ± SEM of 7–9 neurons. This experiment was carried out using recording pipettes filled with CsCl containing internal solution. |
Involvement of P38 MAPK, but Not COX, in the Acute Enhancement of P2X3 Receptor-Mediated ATP Currents by TNF-α
We further explored the pathway linking TNF-α to its effects on P2X3 receptor-mediated ATP currents. It has been shown that TNF-α can signal by activating p38 MAPK in DRG neurons and other cell types.12,32,33 TNF-α treatment also results in up-regulation of cyclooxygenase (COX) −2 expression in DRG neurons.34 We therefore observed whether p38 MAPK and COX-2 were involved in the acute enhancement of P2X3 receptor-mediated ATP currents by TNF-α. Application of TNF-α (10 ng/mL) alone increased the amplitude of IATP by 33.67 ± 3.43% (Figure 4A and B). However, the enhancement of IATP by TNF-α was only 6.54 ± 3.77% after SB202190, a fast-acting p38 MAPK inhibitor, was pre-treated to DRG cells (10 μM, for 3 min and then mixed with 10 ng/mL TNF-α for 5 min) (P < 0.01, Bonferroni’s post hoc test, compared with TNF-α pretreatment only, n = 7; Figure 4B), suggesting that the TNF-α-induced enhancement of IATP was substantially prevented by SB202190. Indomethacin, a potent COX-1 and COX-2 inhibitor, had no significant effect on the enhancement of IATP by TNF-α when it (30 μM) was treated to DRG cells similar to SB202190 administration (Figure 4A and B). The results suggested that p38 MAPK, but not COX, was involved in the acute enhancement of P2X3 receptor-mediated ATP currents by TNF-α.
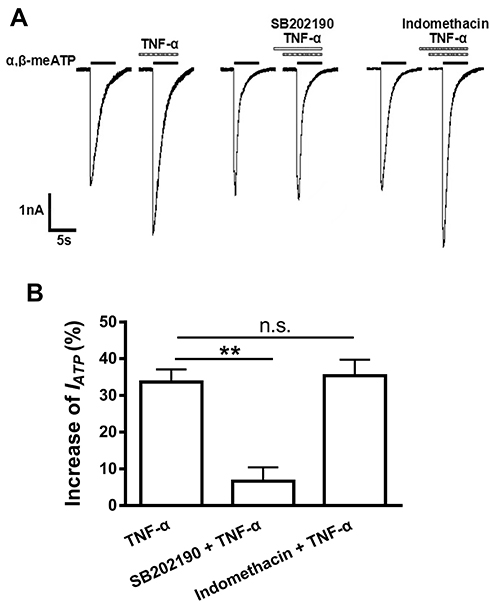 |
Figure 4 p38MAPK, but not COX, was involved in the potentiation of P2X3 receptor-mediated ATP currents by TNF-α. Representative current traces in (A) and the bar graph in (B) showed that the effects of TNF-α alone, p38 inhibitor SB202190 plus TNF-α, and non-selective COX inhibitor indomethacin plus TNF-α on α,β-meATP (100 μM) -evoked currents. IATP was significantly enhanced by TNF-α (10 ng/mL, 5 min) pre-treatment alone or combined treatment of indomethacin (30 μM, 3+5=8 min) plus TNF-α (10 ng/mL, 5 min). However, the enhancement of IATP did not occur when DRG cells were treated with SB202190 (10 μM, 3+5=8 min) plus TNF-α (10 ng/mL, 5 min). Statistical tests were performed using one-way ANOVA followed by post hoc Bonferroni’s test, and significance is shown **P < 0.01. n.s. Not significant. n = 7 in each column. |
Participation of P2X3 Receptors in TNF-α-Induced Mechanical Allodynia
We finally determined whether the acute sensitization of P2X3 receptors by TNF-α could be observed behaviorally, as shown by above electrophysiological results in vitro. Considering that both ATP and TNF-α can induce mechanical allodynia, we first investigated whether combined application of α,β-meATP and TNF-α had a synergistic effect on mechanical allodynia. To easily distinguish the synergistic effect and individual effect of either α,β-meATP or TNF-α, low dose of α,β-meATP (1 nmol in 50 μL) and TNF-α (0.1 ng in 50 μL) were selected for application. Intraplantar injection of α,β-meATP or TNF-α alone reduced the paw withdrawal threshold (PWT) by 24.11 ± 6.47% and 28.53 ± 6.14%, respectively (Figure 5A). However, the combined injection of α,β-meATP and TNF-α produced a decrease of 71.78 ± 4.89% in the PWT, which was significantly different from adding the predicted effect (24.11% + 28.53% = 52.64%) produced by separate treatment of α,β-meATP and TNF-α (P < 0.05; Figure 5A). The results suggested that there was a synergistic effect on mechanical allodynia when α,β-meATP and TNF-α were applied in combination.
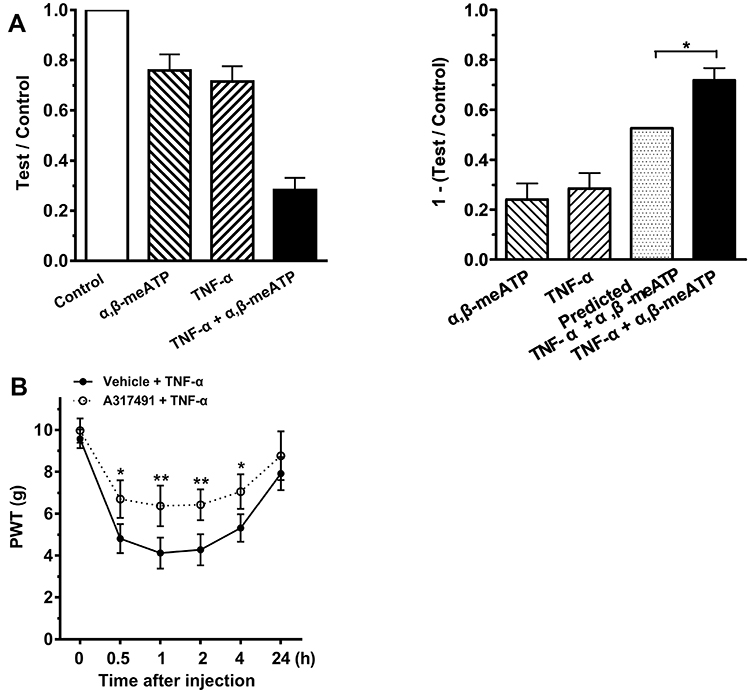 |
Figure 5 P2X3 receptors participated in TNF-α-induced mechanical allodynia. (A) Intraplantar individual injection of α,β-meATP (1 nmol in 50 μL) and TNF-α (0.1 ng in 50 μL) produced a moderate decrease in the mechanical threshold (1-test/control). However, a co-injection of α,β-meATP and TNF-α (TNF-α + α,β-meATP) produced a much large decrease in the threshold than adding the threshold reduction caused by TNF-α and α,β-meATP alone (predicted TNF-α + α,β-meATP). Paw withdrawal threshold (PWT) was tested at 15 min after intraplantar injection. PWT was normalized to the baseline values before every injection. n = 10 rats in each group. * P < 0.05, Two-way ANOVA followed by Bonferroni’s post hoc test. (B) After intraplantar injection of TNF-α (1 ng in 50 μL), PWT (in g) significantly decreased at 0.5, 1, 2 and 4 h and recovered at 24 h. The decrease of PWT induced by TNF-α was significantly attenuated when A-317491 (10 nmol in 50 μL), a specific P2X3 receptor antagonist, was pretreated to ipsilateral hind paws. n = 10 rats in each group. *P < 0.05, **p < 0.01, Bonferroni’s post hoc test, compared with vehicle + TNF-α group. |
We then blocked P2X3 receptors to explore their role in the mechanical allodynia induced by TNF-α. A higher dose of TNF-α (1 ng in 50 μL) was used in the following behavioral experiments. Intraplantar injection of 1 ng TNF-α resulted in a significant decrease in PWT within 0.5 and 4 h after injection, and recovery at 24 h (Figure 5B). If local P2X3 receptors were pharmacologically blocked by A-317491 (10 nmol in 50 μL), TNF-α-induced mechanical allodynia significantly decreased within 0.5 and 4 h after intraplantar injection of TNF-α (P < 0.05 and 0.01, Bonferroni’s post hoc test, compared with vehicle + TNF-α group, n = 10 rats; Figure 5B). Injection of A-317491 alone did not produce any effects on PWT in control rats (data not shown, n = 6 rats). The results suggested that the TNF-α-induced mechanical allodynia was mediated by P2X3 receptors, at least partially.
Discussion
We demonstrated here that a brief exposure of TNF-α rapidly potentiated P2X3 receptor-mediated ATP currents via a p38 MAPK-dependent pathway in dissociated rat DRG neurons, which might importantly underlie acute mechanical allodynia induced by TNF-α in rats.
ATP receptors include P2X1-7 subtypes. Among all subtypes, P2X3 receptor subtype is mainly located in small- and medium-sized nociceptive DRG neurons.18,20,21 In this study, the recorded ATP currents were mediated by P2X3 receptors, since they could be blocked by specific antagonist of P2X3 receptor A-317491.24 Moreover, α,β-meATP can only activate P2X3 and P2X1 receptors.31 Current studies indicated that a 5-min exposure of TNF-α can also rapidly sensitize P2X3 receptors, which was consistent with previous results showing that TNF-α can acute modulate ion channel currents, such as TRPV1 currents, TTX-R Na+ currents, and L-type Ca2+ currents.12,14,16,17 Acute application of TNF-α can dose-dependently increase α,β-meATP-activated currents in rat DRG neurons. TNF-α significantly increased the maximal current response to α,β-meATP, as shown by an upwards shift of concentration–response curve for α,β-meATP by TNF-α. But TNF-α had no effect on apparent affinity of P2X3 receptors for α,β-meATP. In addition, P2X3 receptor-mediated ATP currents were enhanced by TNF-α treatment at different membrane-holding potentials from −80 to 20 mV, indicating a voltage-independence. These electrophysiological evidences were further supported by the behavioral observations that there was a synergistic effect in TNF-α and α,β-meATP induced mechanical allodynia in rats. The rapid sensitization of P2X3 receptors by TNF-α on primary afferents might underlie TNF-α-induced mechanical allodynia. Moreover, the results that P2X3 receptor antagonist A-317491 significantly alleviated the mechanical allodynia induced by TNF-α also suggested that TNF-α-induced mechanical allodynia maybe mediated by P2X3 receptors, at least partially.
TNFR-1 and TNFR-2, two receptor subtypes of TNF-α, are located in vivo in DRG neurons.35–38 Previous studies suggest that the roles of TNF-α in pain are mostly mediated by TNFR-1.39 TNFR-1 has been shown to mediate TNF-α acute potentiation of TTX-R Na+ channel currents and hyperpolarization-activated cation currents.12,40,41 Neutralizing antibodies against TNFR-1, but not TNFR-2, attenuated thermal and mechanical hyperalgesia in a model of nerve injury.8 TNFR-1 knockout mice do not display hyperalgesia after CCI injury, while there was no difference between wild-type and TNFR-2 knockout mice.42 Recently, the expression of P2X3 receptors is identified in TNFR2-positive trigeminal ganglion neurons.43 Upregulation of P2X3 receptor in the trigeminal ganglion neurons by TNFα contributes to trigeminal neuropathic pain in rats.43 However, anyway, the TNF-α receptor subtypes in rapid sensitization of P2X3 receptors by TNF-α need to be further determined.
TNFR-1 can activate multiple signaling pathways, including ceramide and MAPK signaling.32,33 p38 MAPK has been shown to be downstream of TNF-α and participates in the rapid modulation of TTX-R Na+ channels and hyperpolarization-activated cation channels by TNF-α.12,41 Our results indicated that p38 MAPK was necessary for the rapid sensitization of P2X3 receptors by TNF-α, because this sensitization was blocked by p38MAPK inhibitor SB202190. The rapid sensitization of P2X3 receptors by a brief (5 min) exposure of TNF-α may be the result of phosphorylation (post-translational modification) of P2X3 receptors. Previous studies have shown that Nav1.8 channels can be phosphorylated by p38 MAPK, resulting in an increase of TTX-R sodium currents.44 It remains to be studied whether activation of p38 MAPK can phosphorylate P2X3 receptors.
Studies have shown that TNF-α treatment can cause the synthesis and release of endogenous prostaglandin E2 in a COX-2-dependent manner.45,46 Prostaglandin E2 is found to increase Nav1.8 currents through PKA and PKC signaling.47 Sustained potassium currents are also inhibited by a longer treatment (15 min) of TNF-α in primary sensory neurons, which is dependent on the production of prostaglandins.48 PGE2 has been shown to enhance P2X3 receptor-mediated currents in DRG neurons via a cAMP/PKA signaling cascade.49 However, it takes about 4 h to become effective that TNF-α increases the sensitivity to capsaicin in a COX-2-dependent manner, indicating a relatively slow onset of the COX-dependent mechanisms.50 Moreover, it has been shown that acute TNF-α-mediated hypersensitivity is also COX independent.13 The present data indicated that the acute sensitization of P2X3 receptors by TNF-α was also not dependent on the COX mechanisms, because indomethacin, a potent COX inhibitor, had no effect on the enhancement of α,β-meATP-activated ATP currents by TNF-α.
TNF-α and ATP, two well-known mediators, are released during pain. Patients with pain were observed to have elevated levels of TNF-α.51,52 Levels of endogenous TNF-α also increase in some models of pain.5,6,36 ATP is associated with mechanical hypersensitivity in some pain states, such as tissue damage and inflammation.18,19 Once both TNF-α and ATP are released locally in the same inflammatory or painful area, they can sensitize and/or initiate nociceptive process by activating their cognate receptors expressed at the peripheral terminals of the nociceptive neurons. Our results showed that P2X3 receptor was a downstream regulatory target of TNF-α. The local released TNF-α could rapid sensitize P2X3 receptors via a p38 MAPK-dependent pathway in the same DRG neurons. In the present study, the characteristics of peripheral terminals were studied by using DRG neuron soma as model. The rapid sensitization of P2X3 receptors by TNF-α may also occur in peripheral nerve endings. Indeed, behavioral findings exposed that P2X3 receptors were involved in the mechanical allodynia induced by TNF-α, at least partially. TNF-α can produce more severe mechanical allodynia by sensitizing peripheral P2X3 receptors. Therefore, combination of TNF-α and α,β-meATP resulted in a synergistic effect on mechanical allodynia in rats.
Conclusion
It has been reported that intramuscular administration of α,β-meATP results in an increase in the local TNF-α and interleukin 1β levels, which contributes to mechanical hyperalgesia induced by activation of peripheral P2X3 receptors.53 Our results suggested that P2X3 receptor was a downstream regulatory target of TNF-α. A brief exposure of DRG neurons to TNF-α rapidly increased P2X3 receptor-mediated ATP currents via a p38 MAPK-dependent pathway, which constituted one mechanism by which TNF-α mediated rapid hyperalgesia. Thus, there was a positive feedback bidirectional relationship between P2X3 receptor and TNF-α, which amplified the peripheral pain signaling.
Abbreviations
α,β-methylene-ATP, α,β-meATP; ANOVA, one-way analysis of variance; APs, action potentials; COX, cyclooxygenase; DMEM, Dulbecco’s modified Eagle’s medium; DRG, dorsal root ganglion; EC50, half-maximal response; IATP, α,β-methylene-ATP activated current; I–V, current–voltage; MAPK, mitogen-activated protein kinase; PWTs, paw withdrawal thresholds; TNF-α, tumor necrosis factor-α; TNFR, tumor necrosis factor receptor; TRPV1, transient receptor potential vanilloid type 1; TTX-R, tetrodotoxin-resistant.
Ethics Approval and Consent to Participate
The experimental protocol was approved by the animal research ethics committee of Hubei University of Science and Technology (No. 2020-08) and complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81671101).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors have no conflict of interest to declare.
References
1. Dubovy P, Jancalek R, Klusakova I, Svizenska I, Pejchalova K. Intra- and extraneuronal changes of immunofluorescence staining for TNF-alpha and TNFR1 in the dorsal root ganglia of rat peripheral neuropathic pain models. Cell Mol Neurobiol. 2006;26(7–8):1205–1217. doi:10.1007/s10571-006-9006-3
2. Wagner R, Myers RR. Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. Neuroreport. 1996;7(18):2897–2901. doi:10.1097/00001756-199611250-00018
3. Cunha FQ, Poole S, Lorenzetti BB, Ferreira SH. The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br J Pharmacol. 1992;107(3):660–664. doi:10.1111/j.1476-5381.1992.tb14503.x
4. Wacnik PW, Eikmeier LJ, Simone DA, Wilcox GL, Beitz AJ. Nociceptive characteristics of tumor necrosis factor-alpha in naive and tumor-bearing mice. Neuroscience. 2005;132(2):479–491. doi:10.1016/j.neuroscience.2004.12.035
5. Woolf CJ, Allchorne A, Safieh-Garabedian B, Poole S. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor alpha. Br J Pharmacol. 1997;121(3):417–424. doi:10.1038/sj.bjp.0701148
6. Schafers M, Geis C, Svensson CI, Luo ZD, Sommer C. Selective increase of tumour necrosis factor-alpha in injured and spared myelinated primary afferents after chronic constrictive injury of rat sciatic nerve. Eur J Neurosci. 2003;17(4):791–804. doi:10.1046/j.1460-9568.2003.02504.x
7. Lindenlaub T, Teuteberg P, Hartung T, Sommer C. Effects of neutralizing antibodies to TNF-alpha on pain-related behavior and nerve regeneration in mice with chronic constriction injury. Brain Res. 2000;866(1–2):15–22. doi:10.1016/s0006-8993(00)02190-9
8. Sommer C, Lindenlaub T, Teuteberg P, Schafers M, Hartung T, Toyka KV. Anti-TNF-neutralizing antibodies reduce pain-related behavior in two different mouse models of painful mononeuropathy. Brain Res. 2001;913(1):86–89. doi:10.1016/s0006-8993(01)02743-3
9. Junger H, Sorkin LS. Nociceptive and inflammatory effects of subcutaneous TNFalpha. Pain. 2000;85(1–2):145–151. doi:10.1016/s0304-3959(99)00262-6
10. Sorkin LS, Xiao WH, Wagner R, Myers RR. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. 1997;81(1):255–262. doi:10.1016/s0306-4522(97)00147-4
11. Sorkin LS, Doom CM. Epineurial application of TNF elicits an acute mechanical hyperalgesia in the awake rat. J Peripheral Nervous System. 2000;5(2):96–100. doi:10.1046/j.1529-8027.2000.00012.x
12. Jin X, Gereau R. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neuroscience. 2006;26(1):246–255. doi:10.1523/JNEUROSCI.3858-05.2006
13. Gudes S, Barkai O, Caspi Y, Katz B, Lev S, Binshtok AM. The role of slow and persistent TTX-resistant sodium currents in acute tumor necrosis factor-alpha-mediated increase in nociceptors excitability. J Neurophysiol. 2015;113(2):601–619. doi:10.1152/jn.00652.2014
14. Khan AA, Diogenes A, Jeske NA, Henry MA, Akopian A, Hargreaves KM. Tumor necrosis factor alpha enhances the sensitivity of rat trigeminal neurons to capsaicin. Neuroscience. 2008;155(2):503–509. doi:10.1016/j.neuroscience.2008.05.036
15. Diem R, Meyer R, Weishaupt JH, Bahr M. Reduction of potassium currents and phosphatidylinositol 3-kinase-dependent AKT phosphorylation by tumor necrosis factor-(alpha) rescues axotomized retinal ganglion cells from retrograde cell death in vivo. J Neuroscience. 2001;21(6):2058–2066. doi:10.1523/JNEUROSCI.21-06-02058.2001
16. Soliven B, Albert J. Tumor necrosis factor modulates Ca2+ currents in cultured sympathetic neurons. J Neuroscience. 1992;12(7):2665–2671. doi:10.1523/JNEUROSCI.12-07-02665.1992
17. Czeschik JC, Hagenacker T, Schafers M, Busselberg D. TNF-alpha differentially modulates ion channels of nociceptive neurons. Neurosci Lett. 2008;434(3):293–298. doi:10.1016/j.neulet.2008.01.070
18. Burnstock G. Purinergic Mechanisms and Pain. Adv Pharmacol. 2016;75:91–137. doi:10.1016/bs.apha.2015.09.001
19. Paukert M, Osteroth R, Geisler HS, et al. Inflammatory mediators potentiate ATP-gated channels through the P2X(3) subunit. J Biol Chem. 2001;276(24):21077–21082. doi:10.1074/jbc.M101465200
20. Bradbury EJ, Burnstock G, McMahon SB. The expression of P2X3 purinoreceptors in sensory neurons: effects of axotomy and glial-derived neurotrophic factor. Mol Cell Neurosci. 1998;12(4–5):256–268. doi:10.1006/mcne.1998.0719
21. Dunn PM, Zhong Y, Burnstock G. P2X receptors in peripheral neurons. Prog Neurobiol. 2001;65(2):107–134. doi:10.1016/s0301-0082(01)00005-3
22. Cockayne DA, Hamilton SG, Zhu QM, et al. Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature. 2000;407(6807):1011–1015. doi:10.1038/35039519
23. Honore P, Kage K, Mikusa J, et al. Analgesic profile of intrathecal P2X(3) antisense oligonucleotide treatment in chronic inflammatory and neuropathic pain states in rats. Pain. 2002;99(1–2):11–19. doi:10.1016/s0304-3959(02)00032-5
24. Jarvis MF, Burgard EC, McGaraughty S, et al. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci U S A. 2002;99(26):17179–17184. doi:10.1073/pnas.252537299
25. Hamilton SG, McMahon SB, Lewin GR. Selective activation of nociceptors by P2X receptor agonists in normal and inflamed rat skin. J Physiol. 2001;534(Pt. 2):437–445. doi:10.1111/j.1469-7793.2001.00437.x
26. Chen Y, Li GW, Wang C, Gu Y, Huang LM. Mechanisms underlying enhanced P2X receptor-mediated responses in the neuropathic pain state. Pain. 2005;119(1–3):38–48. doi:10.1016/j.pain.2005.09.007
27. Sharp CJ, Reeve AJ, Collins SD, et al. Investigation into the role of P2X(3)/P2X(2/3) receptors in neuropathic pain following chronic constriction injury in the rat: an electrophysiological study. Br J Pharmacol. 2006;148(6):845–852. doi:10.1038/sj.bjp.0706790
28. McGaraughty S, Wismer CT, Zhu CZ, et al. Effects of A-317491, a novel and selective P2X3/P2X2/3 receptor antagonist, on neuropathic, inflammatory and chemogenic nociception following intrathecal and intraplantar administration. Br J Pharmacol. 2003;140(8):1381–1388. doi:10.1038/sj.bjp.0705574
29. Jin Y, Qiu CY, Wei S, Han L, Liu TT, Hu WP. Potentiation of P2X3 receptor mediated currents by endothelin-1 in rat dorsal root ganglion neurons. Neuropharmacology. 2020;181:108356. doi:10.1016/j.neuropharm.2020.108356
30. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. doi:10.1016/0165-0270(94)90144-9
31. North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82(4):1013–1067. doi:10.1152/physrev.00015.2002
32. Pollock J, McFarlane SM, Connell MC, et al. TNF-alpha receptors simultaneously activate Ca2+ mobilisation and stress kinases in cultured sensory neurones. Neuropharmacology. 2002;42(1):93–106. doi:10.1016/s0028-3908(01)00163-0
33. Wu H. Assembly of post-receptor signaling complexes for the tumor necrosis factor receptor superfamily. Adv Protein Chem. 2004;68:225–279. doi:10.1016/S0065-3233(04)68007-7
34. Fehrenbacher JC, Burkey TH, Nicol GD, Vasko MR. Tumor necrosis factor alpha and interleukin-1beta stimulate the expression of cyclooxygenase II but do not alter prostaglandin E2 receptor mRNA levels in cultured dorsal root ganglia cells. Pain. 2005;113(1–2):113–122. doi:10.1016/j.pain.2004.09.031
35. Schafers M, Sorkin LS, Geis C, Shubayev VI. Spinal nerve ligation induces transient upregulation of tumor necrosis factor receptors 1 and 2 in injured and adjacent uninjured dorsal root ganglia in the rat. Neurosci Lett. 2003;347(3):179–182. doi:10.1016/s0304-3940(03)00695-5
36. Shubayev VI, Myers RR. Axonal transport of TNF-alpha in painful neuropathy: distribution of ligand tracer and TNF receptors. J Neuroimmunol. 2001;114(1–2):48–56. doi:10.1016/s0165-5728(00)00453-7
37. Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 1995;5(10):392–399. doi:10.1016/s0962-8924(00)89088-1
38. Leo M, Argalski S, Schafers M, Hagenacker T. Modulation of Voltage-Gated Sodium Channels by Activation of Tumor Necrosis Factor Receptor-1 and Receptor-2 in Small DRG Neurons of Rats. Mediators Inflamm. 2015;2015:124942. doi:10.1155/2015/124942
39. Parada CA, Yeh JJ, Joseph EK, Levine JD. Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci. 2003;17(9):1847–1852. doi:10.1046/j.1460-9568.2003.02626.x
40. Zhang H, Dougherty PM. Acute inhibition of signalling phenotype of spinal GABAergic neurons by tumour necrosis factor-alpha. J Physiol. 2011;589(Pt 18):4511–4526. doi:10.1113/jphysiol.2011.215301
41. Zhang H, Nei H, Dougherty PM. A p38 mitogen-activated protein kinase-dependent mechanism of disinhibition in spinal synaptic transmission induced by tumor necrosis factor-alpha. J Neurosci. 2010;30(38):12844–12855. doi:10.1523/JNEUROSCI.2437-10.2010
42. Vogel C, Stallforth S, Sommer C. Altered pain behavior and regeneration after nerve injury in TNF receptor deficient mice. J Peripheral Nervous System. 2006;11(4):294–303. doi:10.1111/j.1529-8027.2006.00101.x
43. Koizumi M, Asano S, Furukawa A, et al. P2X3 receptor upregulation in trigeminal ganglion neurons through TNFalpha production in macrophages contributes to trigeminal neuropathic pain in rats. J Headache Pain. 2021;22(1):31. doi:10.1186/s10194-021-01244-4
44. Hudmon A, Choi JS, Tyrrell L, et al. Phosphorylation of sodium channel Na(v)1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neuroscience. 2008;28(12):3190–3201. doi:10.1523/JNEUROSCI.4403-07.2008
45. Maier JA, Hla T, Maciag T. Cyclooxygenase is an immediate-early gene induced by interleukin-1 in human endothelial cells. J Biol Chem. 1990;265(19):10805–10808. doi:10.1016/S0021-9258(19)38515-1
46. Mark KS, Trickler WJ, Miller DW. Tumor necrosis factor-alpha induces cyclooxygenase-2 expression and prostaglandin release in brain microvessel endothelial cells. J Pharmacol Exp Ther. 2001;297(3):1051–1058.
47. Gold MS, Levine JD, Correa AM. Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J Neuroscience. 1998;18(24):10345–10355. doi:10.1523/JNEUROSCI.18-24-10345.1998
48. Liu BG, Dobretsov M, Stimers JR, Zhang JM. Tumor Necrosis Factor-alpha Suppresses Activation of Sustained Potassium Currents in Rat Small Diameter Sensory Neurons. Open Pain J. 2008;1:1. doi:10.2174/1876386300801010001
49. Wang C, Li GW, Huang LY. Prostaglandin E2 potentiation of P2X3 receptor mediated currents in dorsal root ganglion neurons. Mol Pain. 2007;3:22. doi:10.1186/1744-8069-3-22
50. Nicol GD, Lopshire JC, Pafford CM. Tumor necrosis factor enhances the capsaicin sensitivity of rat sensory neurons. J Neuroscience. 1997;17(3):975–982. doi:10.1523/JNEUROSCI.17-03-00975.1997
51. Fernandes JC, Martel-Pelletier J, Pelletier JP. The role of cytokines in osteoarthritis pathophysiology. Biorheology. 2002;39(1–2):237–246.
52. Emshoff R, Puffer P, Rudisch A, Gassner R. Temporomandibular joint pain: relationship to internal derangement type, osteoarthrosis, and synovial fluid mediator level of tumor necrosis factor-alpha. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(4):442–449. doi:10.1067/moe.2000.108801
53. Schiavuzzo JG, Teixeira JM, Melo B, et al. Muscle hyperalgesia induced by peripheral P2X3 receptors is modulated by inflammatory mediators. Neuroscience. 2015;285:24–33. doi:10.1016/j.neuroscience.2014.11.020
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.