")
Back to Journals » Research Reports in Clinical Cardiology » Volume 5
Acute decompensated heart failure: current pharmacological approaches
Authors Pascual-Figal D , Pastor-Perez F, Caballero L, Garrido IP, Perez-Martinez MT
Received 6 February 2014
Accepted for publication 16 April 2014
Published 16 September 2014 Volume 2014:5 Pages 227—236
DOI https://doi.org/10.2147/RRCC.S40789
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Domingo A Pascual-Figal, Francisco Pastor-Perez, Luis Caballero, Iris P Garrido, Maria Teresa Perez-Martinez
Cardiology Department, Virgen de la Arrixaca University Hospital, School of Medicine, University of Murcia, Murcia, Spain
Abstract: Acute heart failure (AHF) represents a major burden in developed countries. However, pharmacological approaches have remained almost the same for 30 years and are still based on consensus rather than evidence, given that no medical therapy has been shown to positively affect clinical outcomes. Current pharmacological approaches are still based on decongestion by using diuretics in almost all patients, plus either vasodilators or inotropic agents to improve hemodynamics according to perfusion status. The role of loop diuretics (furosemide) and nitrates (nitroglycerin and nitroprusside) is well established, but new agents such as vasopressin and adenosine antagonists, as well as nesiritide, have failed to show any additional value. In the presence of hypoperfusion, the use of inotropics must be considered despite the lack of benefit in terms of survival, and the use of phosphodiesterase inhibitors and levosimendan has not shown any significant advantages over catecholamines (dobutamine). AHF involves a wide spectrum of patients and syndromes, and this probably accounts for the failure of trials set up to evaluate new therapeutic approaches for improving outcomes: therapies need to be tailored to specific patients. At this time, serelaxin represents a promising new agent which has a multifaceted effect, including organ protection, and has shown encouraging results when tailored for a well defined population. In addition, the role of ularitide, a synthetic form of the natriuretic peptide urodilatin, and the new cardiac myosin activators, as a new class of inotropic agents, will be established in the near future by ongoing trials. Therefore, AHF continues to be an unsolved problem and, in light of the lessons learned, new pharmacological approaches should be tailored to well defined AHF populations, incorporating concepts such as “the sooner the better”, “improve and stabilize”, and “prevent organ damage”, in order to be able to improve clinical outcomes, including both mortality and readmission rates.
Keywords: heart failure, acute, therapy, pharmacology
Acute heart failure: the unresolved problem
Acute heart failure (AHF) represents a major burden in developed countries. However, while AHF has almost doubled in the last 25 years and represents the main cause of hospitalization in those over 65 years of age, there have been few changes to the available pharmacological options.1 Patients admitted to hospital with AHF have up to 10% inhospital mortality; if they survive, there is a spike in mortality of up to 12% in the first month and up to 30% in the first year. Moreover, the risk of rehospitalization is up to 30% in the next 60 days,2,3 which accounts for the majority of related health expenditures. These numbers have not changed in recent decades,4 and no medical therapy has been shown to positively affect the clinical outcomes of patients with AHF, while some have shown the potential for harm. Numerous disappointments in large Phase II and III pharmacologic studies have occurred. Therefore, AHF currently represents an unsolved problem, and most therapeutics and guidelines are still based on consensus rather than evidence.
Current approaches
Pathophysiology
Using current guidelines patients are classified into three simple clinical types of presentation:5,6 i) patients with volume overload, manifested by pulmonary and/or systemic congestion; ii) patients with signs and symptoms of both fluid overload and impaired systemic perfusion; and iii) patients with predominant signs of depression of cardiac output and/or shock syndrome. The vast majority of patients with AHF present with dyspnea and evidence of pulmonary and/or systemic congestion (types i and ii) either as a decompensation of chronic heart failure (HF) (80%) or as a de novo presentation (20%). Patients with predominant signs of hypoperfusion (shock, type iii) are a minority and reflect a final stage of the disease or a severe acute cardiac insult; in these cases, treatment should be directed to reverse such a situation, by treating the underlying cause or replacing heart function (transplant or mechanical circulatory support).
Therefore, most patients with AHF are currently recognized by the presence of clinical congestion and, in some cases, associated signs of hypoperfusion. Accordingly, current algorithms are based on clinical evaluation of congestion and perfusion (Figure 1), and the pharmacological therapies are poorly recommended and mainly consensus-based (Table 1). Treatment for AHF consists of diuretics in almost all patients, plus either vasodilators or inotropic agents depending on perfusion status. However, the use of these agents is based on limited evidence, generally from small, single-center studies assessing the effect on acute hemodynamic parameters. Importantly, there is very limited evidence on the effect of these agents on outcomes.
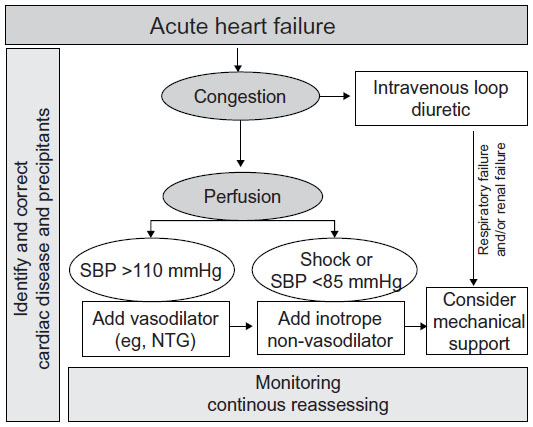 | Figure 1 General algorithm for management of acute heart failure. |
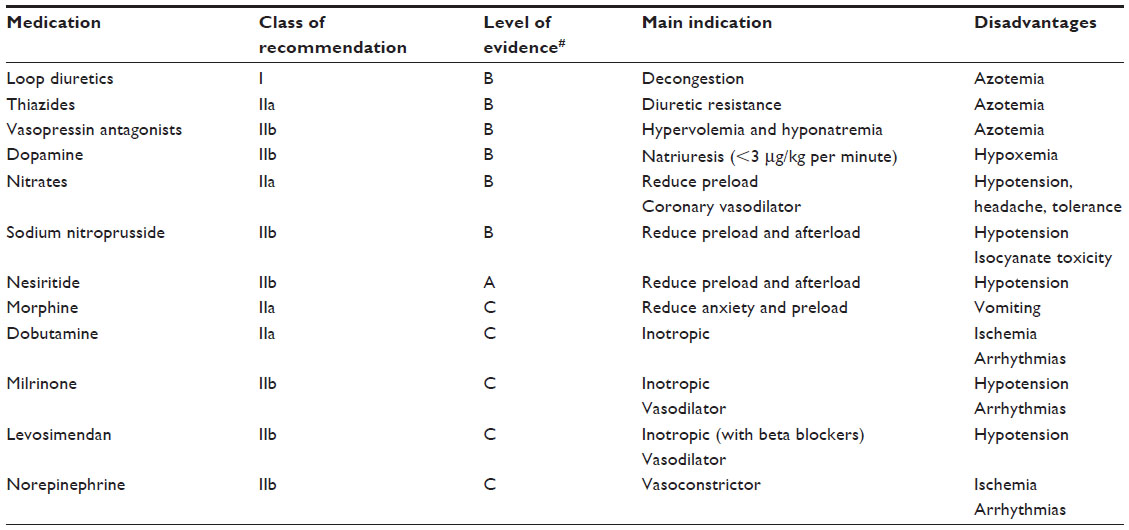 | Table 1 Current pharmacological therapies based on guideline recommendations |
Against this very simplistic view of the problem, a change toward a more complex view is necessary in order to assess the true impact of pharmacological therapy on clinical outcomes. AHF comprises a wide spectrum of patients and syndromes, which probably accounts for the failure of several therapeutic approaches for improving outcomes: “different patients and different problems need different solutions”. A more exact knowledge of the predominant pathophysiology in each patient is necessary in order to select the appropriate therapy for a given patient. In other words, therapies need to be tailored for specific patients.
Loop diuretics
Elevated filling pressures are ultimately responsible for many of the signs and symptoms of heart failure. Indeed, data from ADHERE (the Acute Decompensated Heart Failure National Registry)7 show that 90% of hospitalizations for acute decompensated heart failure are due to fluid overload in patients who have failed to respond to chronic oral diuretics. Consequently, the goals of care include relief of congestion, with diuretics being the first-line therapy for these patients. Loop diuretics (furosemide, torsemide, and bumetanide) are used preferentially because of: a superior water clearance for the same degree of natriuresis; their effectiveness despite renal impairment; and their increasing effect with increasing dose (“high ceiling” diuretics). The bioavailability of oral administration shows high interindividual and intraindividual variability, and may be considerably lower in patients with AHF. Therefore, diuretics should be initially administered intravenously as a bolus to ensure maximal bioavailability.8 Intravenous loop diuretics reduce pulmonary capillary wedge pressure and increase venous capacitance within a few minutes, before any increase in urinary output, a phenomenon that is likely to be mediated by vasodilatory prostaglandins.9 Diuretics can be given by bolus or continuous infusion and in high or low doses. These strategies were assessed recently by Felker et al in the DOSE (Diuretic Optimization Strategies Evaluation) trial10 that enrolled 308 patients with AHF and tested a high dose versus low dose of furosemide and a continuous infusion versus bolus intermittent dose, for the primary endpoint of global symptom assessment and creatinine at 72 hours. There was no significant difference between continuous infusion and bolus dosing in terms of either symptoms or renal function. However there was greater early symptom improvement with high-dose compared with low-dose diuretics, without any significant differences in renal function. A number of secondary endpoints also favored a high dose, ie, greater diuresis, more weight loss, and lower amino-terminal pro-B-type natriuretic peptide level.
Loss of diuretic potency (diuretic resistance) is a known effect of the long-term use of diuretics. Some approaches to managing resistance to these agents include increasing the dose and/or frequency of the drug, restricting sodium or water intake, administering the drug as an intravenous bolus or intravenous infusion, combining diuretics (loop and thiazide diuretics), and addition of vasodilators or inotropes.11 In another subanalysis of the DOSE study, patients receiving a high oral dose prior to the AHF episode seem to derive more benefit from a bolus strategy.12 Patients treated with diuretics should be monitored carefully for excessive urine output, development of hypotension, worsening renal function, and reductions in serum potassium and magnesium levels. Therefore, diuretic therapy must be highly individualized, based on the degree of fluid overload present and the degree of volume loss produced, to minimize these side effects.
Vasopressin and adenosine antagonists
New diuretics such as tolvaptan, named aquaretics or “vaptans”, antagonize the vasopressin type 2 receptors in the kidney and promote free water clearance without any change in electrolytes. However, the results of the EVEREST (Efficacy of Vasopressin antagonism in Heart Failure Outcome Study with Tolvaptan) trial13 were disappointing in terms of ability to reduce mortality or HF hospitalizations. In the PROTECT (Placebo-Controlled Randomized Study of the Selective A1 Adenosine Receptor Antagonist Rolofylline for Patients Hospitalized With Acute Decompensated Heart Failure and Volume Overload to Assess Treatment Effect on Congestion and Renal Function) trial,14 rolofylline, an adenosine A(1)-receptor antagonist, also did not have a favorable effect on improving clinical status and survival or preserving renal function. Therefore, at this time, these agents have no place in the pharmacological management of AHF.
Nitrates
Although diuretic therapy is highly effective in the initial management of mild volume overload states, patients with moderate to severe volume overload may not respond adequately to diuretic management. Vasodilators are probably most useful in patients with hypertension and should be avoided in patients with a systolic blood pressure <110 mmHg.5 A small randomized trial of primarily nitrate therapy compared with a diuretic approach suggested superiority of the vasodilator strategy.15 Moreover, a subanalysis of the ALARM-HF (Acute Heart Failure Global Survey of Standard Treatment) registry showed that patients administered a combination of diuretics and a vasodilator had lower inhospital mortality than those treated solely with diuretics.16
The nitrate agents, nitroglycerin and nitroprusside, act on arterial and venous smooth muscle to increase levels of guanosine 3′5′-cyclic monophosphate, resulting in vasodilation. Whatever the dosage, nitrates are associated with venodilatation, but arterial dilatation needs an increased nitrate dosage. The vasodilator properties are associated with decreased preload and afterload, and coronary vasodilatation. These changes decrease left ventricular filling pressure and the workload of the ventricles, thereby increasing the efficiency of cardiac function.17 Nitroglycerin is very effective in treating AHF and is the preferred nitrate given its low toxicity profile. Although nitrates can be used in different forms (sublingual, oral, transdermal, and intravenous), the most common route in AHF is intravenous, with a starting dose of 10–20 μg per minute. The use of nitrates is limited by tachyphylaxis and headache.5
Sodium nitroprusside is metabolized in erythrocytes and degraded as nitric oxide, responsible for the drug’s effects, and as cyanide, a toxic molecule rapidly converted in the liver to thiocyanate. Sodium nitroprusside induces relaxation of smooth muscle cells in arteries and veins. The starting dose should be low (ie, 0.25–0.50 μg/kg per minute). The hemodynamic effects include very efficient afterload reduction and a decrease of intracardiac filling pressures. This agent is particularly helpful for patients who present with severe pulmonary congestion associated with low cardiac output to improve ventriculoarterial coupling and to increase cardiac output.5,18 Sodium nitroprusside requires not only careful hemodynamic monitoring, often requiring indwelling catheters, but also monitoring for cyanide toxicity, especially in the presence of renal or liver dysfunction.
Natriuretic peptides: nesiritide
Natriuretic peptides provide a promising mechanism of action in the pathophysiologic background for treating congestive heart failure based on their vasodilatory and diuretic properties, and effective inhibition of the renin–angiotensin–aldosterone system. Nesiritide is a recombinant natriuretic peptide with the same 32 amino acid sequence as human B-type natriuretic peptide. Like B-type natriuretic peptide, nesiritide binds to a specific cell surface, leading to relaxation of mesangial and vascular smooth muscle cells, natriuresis, and vasodilation. This molecule was approved in 2001 for use in patients with AHF on the basis of the studies by Collucci et al19 and the VMAC (Vasodilation in the Management of Acute CHF [congestive heart failure]) trial,20 showing a reduction in pulmonary capillary wedge pressure and an improvement in dyspnea at 3 hours. However, a subsequent meta-analysis brought the safety and efficacy of the drug into question, suggesting that nesiritide, as compared with placebo, was associated with higher rates of worsening renal function (increased by a factor of 1.5) and early death (increased by a factor of 1.8), although the confidence intervals associated with these estimates were wide.21,22
The ASCEND-HF (Acute Study of Clinical Effectiveness of Nesiritide and Decompensated Heart Failure) trial was designed to prospectively answer the questions raised by the meta-analyses and to respond to the concerns of an expert panel commissioned by the drug sponsor.23 A total of 7,141 patients were randomized 1:1 to nesiritide or placebo, with concomitant standard medical therapy and without differences in the use of loop diuretics, positive inotropes, or vasodilators between the two groups after randomization.23 Self-reported dyspnea improved marginally in the nesiritide group at 6 and 24 hours, but the prespecified criterion for statistical significance was not met. Similarly, there was no significant difference in the outcome of death or heart failure hospitalization within 30 days between the treatment groups (9.4% in the nesiritide group versus 10.1% in the placebo group; P=0.31). There were also no differences in the safety endpoint between the groups with regard to the proportion of patients with a decrease in estimated glomerular filtration rate from baseline. The recently published ROSE-AHF (Renal Optimization Strategies Evaluation in Acute Heart Failure) trial22 randomized 360 hospitalized patients with AHF and renal dysfunction (estimated glomerular filtration rate 15–60 mL/min/1.73 m2) within 24 hours of admission to low doses of dopamine (2 μg/kg per minute) or low-dose of nesiritide (0.005 μg/kg per minute without bolus) and compared with placebo. The data from this study showed that neither low-dose dopamine nor low-dose nesiritide improved decongestion or renal function when added to diuretic therapy, as measured by the cumulative urine volume and change in cystatin C level. Therefore, considering the latest results of clinical trials, nesiritide is not associated with any substantial benefit compared with standard therapy, so cannot be recommended as an effective therapy in AHF.
Morphine
Although its beneficial mechanism of action in AHF is unclear, morphine is thought to produce mild venodilatation and reduction of preload. It may also impart a beneficial effect by relief of anxiety and a diminished catecholamine response.24 Given that opiates may depress respiratory drive, caution is needed.
Dopamine
The pharmacodynamic effects of dopamine are dose-dependent. At ≥5 μg/kg per minute, it stimulates β-adrenergic receptors and induces release of norepinephrine from vascular neurons, thereby increasing heart rate, cardiac output, and peripheral resistance. Low-dose dopamine or a “renal dose” is generally considered to be ≤3 μg/kg per minute and may have selective renal arterial vasodilator activity promoting natriuresis. However, there is no evidence from recent randomized clinical trials that these effects translate into clinical benefit.25,26
Inotropics
Positive inotropic agents have beneficial hemodynamic effects in patients with AHF and systolic dysfunction. The current guidelines recommend inotropic agents when there is clinical evidence of hypoperfusion due to a decrease in cardiac contractility and cardiac output. However, currently we must differentiate between inotropics with and without vasodilatory properties.
Dobutamine is a synthetic catecholamine with a strong affinity for both β1 and β2 receptors, to which dobutamine binds at a 3:1 ratio. With its cardiac β1 stimulatory effects, dobutamine is a potent inotrope with weaker chronotropic activity. Vascular smooth muscle binding results in combined α1-adrenergic agonism, as well as β2 stimulation, such that the net vascular effect is often mild vasodilation, particularly at lower doses (<5 μg/kg per minute); meanwhile, doses up to 15 μg/kg per minute increase cardiac contractility without greatly affecting peripheral resistance.27 Symptomatic improvement has been demonstrated in patients with advanced heart failure after treatment with a continuous infusion of dobutamine (5–7.5 μg/kg per minute) for 3–5 days.28–30 However, there are no placebo-controlled trial data documenting improved survival as a result of either intermittent or continuous dobutamine infusion.31,32 In this setting, dobutamine appears to be as effective as milrinone.33 A nonrandomized retrospective study from ADHERE suggested that after attempted adjustment for differences in risk, milrinone and dobutamine were associated with increased mortality when compared with patients treated with nitroglycerin or nesiritide.34 Given that sicker patients were treated with the inotropes, it is difficult to know if the adjustments were sufficient. The 2013 American College of Cardiology Foundation/American Heart Association guideline concluded that long-term infusion of an inotropic agent may be useful until a definitive therapy becomes available (eg, coronary revascularization, mechanical circulatory support, or heart transplantation) or resolution of the acute precipitating problem.6 It was felt to be reasonable as a bridge therapy in patients with AHF refractory to standard therapy, who are eligible for and awaiting mechanical circulatory support or cardiac transplantation. It was also felt reasonable to consider its use as palliative therapy for symptom control in selected patients with end-stage HF despite optimal medical and device therapy.
Phosphodiesterase inhibitors, fundamentally milrinone, decrease the rate of degradation of cyclic adenosine monophosphate. The increase in cyclic adenosine monophosphate concentration results in enhanced calcium influx, a rise in cell calcium concentration, and increased contractility. However, it must be noted that these drugs cause systemic arterial and venous dilation via inhibition of peripheral phosphodiesterase.35 Administration of intravenous milrinone can provide an acute hemodynamic and symptomatic benefit in patients with advanced heart failure (New York Heart Association class III).5,6 However, the possible benefit of such therapy was not confirmed in OPTIME CHF (Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure), a controlled trial in 949 patients admitted to hospital with AHF and severe systolic dysfunction (mean left ventricular ejection fraction 23%).36 Patients were randomly assigned to milrinone or placebo for 72 hours and then followed for 2 months. Milrinone therapy showed an increment in significant hypotension and atrial fibrillation, with a nonsignificant increase in inhospital mortality (3.8% versus 2.3%) and mortality at 60 days (10.3% versus 8.9%). In a subanalysis, those patients with ischemic cardiomyopathy deteriorated on milrinone whereas a neutral/modestly beneficial effect was seen in patients with nonischemic cardiomyopathy.37
Levosimendan belongs to a group of calcium-sensitizing agents that enhance myocardial contraction via a unique mechanism of action that increases calcium sensitivity with lower intracellular calcium concentration requirements.38 The positive inotropic effect of levosimendan is based on stabilizing and prolonging the conformational change that occurs when the drug binds to calcium-saturated cardiac troponin C.39,40 Levosimendan increased myocardial contractility, improved hemodynamics, and dilated both the peripheral and coronary vessels.41–43 Several studies have evaluated the efficacy and safety of levosimendan in patients with AHF.44–48 Despite improvement in hemodynamic indices, there is no clear evidence of a short-term or long-term clinical benefit.
LIDO (the Levosimendan Infusion versus Dobutamine study), the first large trial,45 found that levosimendan was associated with a significant mortality benefit when compared with dobutamine at both 1 month (8% versus 17%, respectively) and 6 months (26% versus 38%). This result was similar to that in the RUSSLAN (Randomized stUdy on Safety and effectivenesS of Levosimendan in patients with left ventricular failure due to an Acute myocardial iNfarct) trial of 504 patients within 2 days of an acute myocardial infarction.46 Compared with placebo, a 6-hour infusion of levosimendan did not affect the primary endpoint of hypotension or clinically significant myocardial ischemia, but significantly improved the secondary endpoint of death or worsening heart failure at 1 day and 2 weeks (12% versus 20%), with a strong trend toward improvement at 6 months (23% versus 31%).
However, in SURVIVE (Survival of Patients with Acute Heart Failure in Need of Intravenous Inotropic Support,49 the first prospective, double-blind, randomized trial utilizing mortality as the primary endpoint, levosimendan did not reduce all-cause mortality, cardiovascular mortality, or days out of hospital at 180 days when compared with dobutamine. This trial enrolled 1,327 hospitalized patients with severe AHF, left ventricular ejection fraction <30%, and a clinical need for intravenous inotropic support after intravenous diuretics and/or vasodilators. There were no differences in the proportions of patients who reported more than mild improvement in dyspnea. At 180 days, no difference in mortality was observed between patients treated with levosimendan and those treated with dobutamine (26% versus 28%, respectively; hazards ratio 0.91, confidence interval 0.74–1.13, P=0.401). No significant differences in incidence of hypotension, cardiac failure, atrial fibrillation, or ventricular tachycardia were found after treatment with levosimendan when compared with dobutamine.
Finally, in the sequential REVIVE (Randomized Multicenter Evaluation of Intravenous Levosimendan Efficacy) I and II trials,50 700 patients with acute decompensated heart failure who remained dyspneic after intravenous diuretics were randomly assigned to intravenous levosimendan or placebo for 24 hours. Although patients in the levosimendan group reported a greater improvement in symptoms and had lower B-type natriuretic peptide levels for up to 5 days, levosimendan was associated with more frequent hypotension and cardiac arrhythmia as well as a numerically higher risk of death in the two trials (40 versus 40, P=0.29) at 90 days.
Given the above findings, the long-term results in patients with AHF and systolic dysfunction treated with levosimendan clearly do not support a survival benefit. Levosimendan is currently approved for intravenous use in some countries in Europe and South America, but remains unlicensed in the USA. The 2012 European Society of Cardiology heart failure guidelines note that the efficacy and safety of levosimendan are still uncertain, although they suggest a potential pharmacological rationale for intravenous levosimendan (or a phosphodiesterase inhibitor) to reverse the effect of beta-blockade, if beta-blockade is thought to be contributing to hypoperfusion.5
New pharmacological strategies
Pathophysiological approach
Review of the literature published in recent years suggests a change in the way AHF is managed and that a desirable pharmacological approach should include the following considerations:
- symptom relief is important but should not lead to decisions by itself; instead, relief of congestion is the right way forward
- decongestion is the main objective, but should have two additional components, ie, stabilization and organ protection
- hemodynamic and neurohormonal improvements should be tailored to achieve stabilization
- organ damage (mainly myocardial, renal, and vascular) is frequent as a consequence of the AHF episode and should be minimized
- in order to avoid organ damage, no time can be lost, and an early approach could prevent it and facilitate a protective approach
- pharmacologic approaches should have a good safety profile, a low risk of hypotension, and the ability to achieve decongestion, reverse neurohormonal hyperactivation, and prevent organ damage
- finally, a reduction in the risk of mortality and new decompensation is necessary to demonstrate the efficacy of a pharmacological approach.
Serelaxin
Serelaxin is a recombinant form of human relaxin-2, a naturally occurring peptide hormone mediating the physiological cardiovascular and renal adaptations of pregnancy. The mechanism of action upregulates vascular endothelin B receptor, vascular endothelial growth factor, and production of nitric oxide, promoting systemic and renal vasodilatation and natriuresis, and also has beneficial pleiotropic effects.51 This process results in decreased systemic vascular resistance, increased cardiac output, increased renal blood flow, and an increased glomerular filtration rate. Serelaxin is also thought to inhibit angiotensin II and endothelin. In contrast with targeted therapy using vasodilators and inotropes, the multifaceted effects of serelaxin may make it a valuable option in the treatment of AHF, a complex disease.
In contrast with other promising therapies, serelaxin has been tailored for a well defined population of patients with AHF, ie, those presenting with dyspnea, radiographic evidence of pulmonary congestion, increased brain natriuretic peptide, mild-to-moderate chronic renal failure, and systolic blood pressure >125 mmHg. In these patients, serelaxin was started during the first 16 hours after admission and administered for 24 hours. The results obtained have been encouraging, and the US Food and Drug Administration has labeled it as a “breakthrough” intravenous therapy for AHF.
In Pre-RELAX-AHF (Relaxin for the Treatment of Acute Heart Failure), a multicenter-randomized Phase IIb trial,52 administration of serelaxin versus placebo showed improved dyspnea, a decrease in days of hospitalization, and reduced cardiovascular mortality and hospital readmissions for heart or kidney failure at 60 days, all associated with a good safety profile. The recently published, randomized, placebo-controlled Phase III trial, RELAX-AHF,53 has further confirmed the favorable clinical effects of serelaxin 30 μg/kg/day for 48 hours in 1,161 patients with the same clinical profile. Serelaxin improved dyspnea and reduced worsening heart failure events, signs and symptoms of congestion, length of stay, and time spent in intensive care. The most favorable effect was a significant reduction in all-cause and cardiovascular mortality at 180 days. However, the study was not powered to address this secondary outcome and the reduction in rate of rehospitalizations for heart failure did not reach statistical significance. Subanalyses of this trial showed that the effect of serelaxin appeared to be similar across subgroups of patients.54 Moreover, administration of serelaxin improved markers of cardiac, renal, and hepatic damage, consistent with the notion that prevention of organ damage may be related to the observed benefits.55
An important issue with serelaxin is the good safety profile, in that even worsening renal function was significantly lower in the serelaxin group than in the placebo group. Patients with vasoconstriction, congestion (such as those receiving background loop diuretics), and mild-to-moderate renal impairment early in the course of an exacerbation of heart failure are most likely to benefit from this medication. However, more data are necessary in order to confirm if single use of this drug is able to reduce short-term and long-term mortality, and a significant reduction in the rate of rehospitalizations would be expected to explain such clinical benefit. These findings will be confirmed or not in the ongoing RELAX-AHF 2 trial (Efficacy, Safety and Tolerability of Serelaxin When Added to Standard Therapy in AHF, ClinicalTrials.gov identifier NCT01870778). This study is powered to detect a survival benefit, it is planned to recruit more than 6,000 patients with AHF and it is expected to be completed in June 2016.
Ularitide
In addition to nesiritide, ularitide is another natriuretic peptide studied in AHF. Ularitide is a synthetic form of urodilatin, a human natriuretic peptide produced in the kidneys. It induces natriuresis and diuresis, relaxing smooth muscle cells and leading to vasodilation and increased renal blood flow. Urodilatin significantly decreases systolic blood pressure and central venous pressure and increases diuresis and urinary sodium excretion. The safety and efficacy of ularitide was tested in 24 patients with acute decompensated heart failure in the SIRIUS (Safety and Efficacy of an Intravenous Placebo-Controlled Randomized Infusion of Ularitide in a Prospective Double-blind Study in Patients With Symptomatic, Decompensated Chronic Heart Failure)-I trial,56 where ularitide was infused over 24 hours and compared with placebo. After 6 hours, urodilatin significantly decreased pulmonary capillary wedge pressure in a dose-dependent manner and tended to improve dyspnea. The subsequent SIRIUS-II study assessed the clinical effects of ularitide more precisely in a greater number of patients (n=221) with AHF.57 Ularitide achieved a significant reduction in pulmonary capillary wedge pressure in all three dosage groups (7.5, 15, and 30 ng/kg per minute), a reduction in systemic vascular resistance, and increased cardiac index at higher doses, along with an improvement in dyspnea. TRUE-AHF (Efficacy and Safety of Ularitide for the Treatment of Acute Decompensated Heart Failure, ClinicalTrials.gov identifier NCT01661634) is an ongoing randomized Phase III clinical trial designed to evaluate the role of ularitide as an intravenous infusion in addition to conventional therapy in patients with AHF.58 This trial is planned to recruit approximately 2,116 patients with AHF, and is expected to be completed in September 2014.
Myosin activators
Cardiac myosin activators represent a new class of cardioselective compounds that work directly on sarcomeric proteins within cardiac myocytes, influencing the cross-bridge cycle. These molecules accelerate the rate of actin-dependent phosphate release from the weakly bound actin-myosin cross-bridge (the rate-limiting step of the cross-bridge cycle) promoting transition to the force-producing on-state of the cross-bridge.59 As a consequence, more cross-bridges enter the force-producing state, more cross-bridges are activated per unit of time, and contractile force increases. Several of these compounds have been investigated in recent years,60 and found to stimulate myosin-adenosine triphosphatase and increase the fractional shortening of myocytes without increasing intracellular calcium transients. The most studied molecule so far is omecamtiv mecarbil (formerly CK-1827452), a cardioselective allosteric activator of the myosin S1 domain.61,62 In different animal models, it has been shown to improve cardiac contractility by prolonging systolic ejection time, without altering myocardial oxygen consumption, myocyte calcium levels, or the rate of development of left ventricular pressure.61,63 In Phase I–II trials,64,65 it has demonstrated dose-dependent and concentration-dependent increases in several parameters of contractility and systolic function. Because of the concern that omecamtiv might adversely impact diastolic filling time and increase the risk of coronary ischemia, another study evaluated patients with documented ischemic cardiomyopathy, exercise-induced angina, reduced ejection fraction, and symptomatic heart failure.66 Omecamtiv mecarbil was generally well tolerated as a 20-hour infusion, and did not limit exercise capacity. ATOMIC-HF (Acute Treatment with Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure) is an ongoing, randomized, controlled, multicenter Phase IIb trial (ClinicalTrials.gov identifier NCT01300013) that has recruited 600 patients with systolic dysfunction and hospitalized for AHF to evaluate the safety and efficacy of intravenous infusion of omecamtiv mecarbil.67
Conclusion
AHF remains an unsolved problem and represents a major burden in developed countries. Current therapies remain almost the same as they were 30 years ago, and therapeutic approaches are still based on consensus rather than evidence, given that no medical therapy has been shown to positively affect the clinical outcomes for patients with AHF. In order to improve symptoms and eliminate volume overload based on diuretics, and to improve hemodynamics based on vasodilators or inotropic agents, we need to incorporate new approaches and pharmacologic agents able to improve clinical outcomes. AHF comprises a wide spectrum of patients and syndromes, which probably accounts for the failure of several therapeutic approaches to improve outcomes, so therapies need to be tailored for specific patients. In this way, new approaches in selected populations such as “the sooner the better”, “stabilize”, and “prevent organ damage” could make it possible to improve clinical outcomes, including mortality and readmission rates.
Disclosure
The authors report no conflicts of interest in this work.
References
Fang J, Mensah GA, Croft JB, Keenan NL. Heart failure-related hospitalization in the US, 1979 to 2004. J Am Coll Cardiol. 2008;52:428–434. | |
Fonarow GC, Heywood JT, Heidenreich PA, Lopatin M, Yancy CW. Temporal trends in clinical characteristics, treatments, and outcomes for heart failure hospitalizations, 2002 to 2004: findings from Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J. 2007;153:1021–1028. | |
Solomon SD, Dobson J, Pocock S, et al. Influence of nonfatal hospitalization for heart failure on subsequent mortality in patients with chronic heart failure. Circulation. 2007;116:1482–1487. | |
Lee DS, Mamdani MM, Austin PC, et al. Trends in heart failure outcomes and pharmacotherapy: 1992 to 2000. Am J Med. 2004;116:581–589. | |
McMurray JJ, Adamopoulos S, Anker SD, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33:1787–1847. | |
Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128:e240–e327. | |
Adams KF Jr, Fonarow GC, Emerman CL, et al. Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J. 2005;149:209–216. | |
Brater DC. Diuretic therapy. N Engl J Med. 1998;339:387–395. | |
Kramer BK, Schweda F, Riegger GA. Diuretic treatment and diuretic resistance in heart failure. Am J Med. 1999;106:90–96. | |
Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364:797–805. | |
Felker GM. Loop diuretics in heart failure. Heart Fail Rev. 2012;17:305–311. | |
Shah RV, McNulty S, O’Connor CM, Felker GM, Braunwald E, Givertz MM. Effect of admission oral diuretic dose on response to continuous versus bolus intravenous diuretics in acute heart failure: an analysis from diuretic optimization strategies in acute heart failure. Am Heart J. 2012;164:862–868. | |
Konstam MA, Gheorghiade M, Burnett JC Jr, et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297:1319–1331. | |
Massie BM, O’Connor CM, Metra M, et al. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N Engl J Med. 2010;363:1419–1428. | |
Cotter G, Metzkor E, Kaluski E, et al. Randomised trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary oedema. Lancet. 1998;351:389–393. | |
Follath F, Yilmaz MB, Delgado JF, et al. Clinical presentation, management and outcomes in the Acute Heart Failure Global Survey of Standard Treatment (ALARM-HF). Intensive Care Med. 2011;37:619–626. | |
Imhof PR, Ott B, Frankhauser P, Chu LC, Hodler J. Difference in nitroglycerin dose-response in the venous and arterial beds. Eur J Clin Pharmacol. 1980;18:455–460. | |
Monrad ES, Baim DS, Smith HS, Lanoue AS. Milrinone, dobutamine, and nitroprusside: comparative effects on hemodynamics and myocardial energetics in patients with severe congestive heart failure. Circulation. 1986;73:III168–III174. | |
Colucci WS, Elkayam U, Horton DP, et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N Engl J Med. 2000;343:246–253. | |
Publication Committee for the VMAC Investigators (Vasodilatation in the Management of Acute CHF). Intravenous nesiritide versus nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA. 2002;287:1531–1540. | |
Sackner-Bernstein JD, Skopicki HA, Aaronson KD. Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation. 2005;111:1487–1491. | |
Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA. 2005;293:1900–1905. | |
O’Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365:32–43. | |
Vismara LA, Leaman DM, Zelis R. The effects of morphine on venous tone in patients with acute pulmonary edema. Circulation. 1976;54:335–337. | |
Chen HH, Anstrom KJ, Givertz MM, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA. 2013;310:2533–2543. | |
Triposkiadis FK, Butler J, Karayannis G, et al. Efficacy and safety of high dose versus low dose furosemide with or without dopamine infusion: the Dopamine in Acute Decompensated Heart Failure II (DAD-HF II) Trial. Int J Cardiol. 2014;172:115–121. | |
Ruffolo RR Jr. The pharmacology of dobutamine. Am J Med Sci. 1987;294:244–248. | |
Unverferth DV, Magorien RD, Lewis RP, Leier CV. Long-term benefit of dobutamine in patients with congestive cardiomyopathy. Am Heart J. 1980;100:622–630. | |
Leier CV, Webel J, Bush CA. The cardiovascular effects of the continuous infusion of dobutamine in patients with severe cardiac failure. Circulation. 1977;56:468–472. | |
Liang CS, Sherman LG, Doherty JU, Wellington K, Lee VW, Hood WB Jr. Sustained improvement of cardiac function in patients with congestive heart failure after short-term infusion of dobutamine. Circulation. 1984;69:113–119. | |
Sindone AP, Keogh AM, Macdonald PS, McCosker CJ, Kaan AF. Continuous home ambulatory intravenous inotropic drug therapy in severe heart failure: safety and cost efficacy. Am Heart J. 1997;134:889–900. | |
Oliva F, Latini R, Politi A, et al. Intermittent 6-month low-dose dobutamine infusion in severe heart failure: DICE multicenter trial. Am Heart J. 1999;138:247–253. | |
Yamani MH, Haji SA, Starling RC, et al. Comparison of dobutamine-based and milrinone-based therapy for advanced decompensated congestive heart failure: hemodynamic efficacy, clinical outcome, and economic impact. Am Heart J. 2001;142:998–1002. | |
Abraham WT, Adams KF, Fonarow GC, et al. In-hospital mortality in patients with acute decompensated heart failure requiring intravenous vasoactive medications: an analysis from the Acute Decompensated Heart Failure National Registry (ADHERE). J Am Coll Cardiol. 2005;46:57–64. | |
Colucci WS, Wright RF, Jaski BE, Fifer MA, Braunwald E. Milrinone and dobutamine in severe heart failure: differing hemodynamic effects and individual patient responsiveness. Circulation. 1986;73:III175–III183. | |
Cuffe MS, Califf RM, Adams KF Jr, et al. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA. 2002;287:1541–1547. | |
Felker GM, Benza RL, Chandler AB, et al. Heart failure etiology and response to milrinone in decompensated heart failure: results from the OPTIME-CHF study. J Am Coll Cardiol. 2003;41:997–1003. | |
Kass DA, Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006;113:305–315. | |
Figgitt DP, Gillies PS, Goa KL. Levosimendan. Drugs. 2001;61:613–627. | |
Papp Z, Csapo K, Pollesello P, Haikala H, Edes I. Pharmacological mechanisms contributing to the clinical efficacy of levosimendan. Cardiovasc Drug Rev. 2005;23:71–98. | |
Haikala H, Kaheinen P, Levijoki J, Linden IB. The role of cAMP- and cGMP-dependent protein kinases in the cardiac actions of the new calcium sensitizer, levosimendan. Cardiovasc Res. 1997;34:536–546. | |
Pagel PS, Harkin CP, Hettrick DA, Warltier DC. Levosimendan (OR-1259), a myofilament calcium sensitizer, enhances myocardial contractility but does not alter isovolumic relaxation in conscious and anesthetized dogs. Anesthesiology. 1994;81:974–987. | |
Udvary E, Papp JG, Vegh A. Cardiovascular effects of the calcium sensitizer, levosimendan, in heart failure induced by rapid pacing in the presence of aortic constriction. Br J Pharmacol. 1995;114:656–661. | |
De Luca L, Colucci WS, Nieminen MS, Massie BM, Gheorghiade M. Evidence-based use of levosimendan in different clinical settings. Eur Heart J. 2006;27:1908–1920. | |
Follath F, Cleland JG, Just H, et al. Efficacy and safety of intravenous levosimendan compared with dobutamine in severe low-output heart failure (the LIDO study): a randomised double-blind trial. Lancet. 2002;360:196–202. | |
Moiseyev VS, Poder P, Andrejevs N, et al. Safety and efficacy of a novel calcium sensitizer, levosimendan, in patients with left ventricular failure due to an acute myocardial infarction. A randomized, placebo-controlled, double-blind study (RUSSLAN). Eur Heart J. 2002;23:1422–1432. | |
Nieminen MS, Akkila J, Hasenfuss G, et al. Hemodynamic and neurohumoral effects of continuous infusion of levosimendan in patients with congestive heart failure. J Am Coll Cardiol. 2000;36:1903–1912. | |
Slawsky MT, Colucci WS, Gottlieb SS, et al. Acute hemodynamic and clinical effects of levosimendan in patients with severe heart failure. Study Investigators. Circulation. 2000;102:2222–2227. | |
Mebazaa A, Nieminen MS, Packer M, et al. Levosimendan vs dobutamine for patients with acute decompensated heart failure: the SURVIVE randomized trial. JAMA. 2007;297:1883–1891. | |
Packer M, Colucci W, Fisher L, et al. Effect of levosimendan on the short-term-clinical course of patients with acutely decompensated heart failure. JACC Heart Fail. 2013;1:103–111. | |
Teichman SL, Unemori E, Teerlink JR, Cotter G, Metra M. Relaxin: review of biology and potential role in treating heart failure. Curr Heart Fail Rep. 2010;7:75–82. | |
Teerlink JR, Metra M, Felker GM, et al. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet. 2009;373:1429–1439. | |
Teerlink JR, Cotter G, Davison BA, et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013;381:29–39. | |
Metra M, Ponikowski P, Cotter G, et al. Effects of serelaxin in subgroups of patients with acute heart failure: results from RELAX-AHF. Eur Heart J. 2013;34:3128–3136. | |
Metra M, Cotter G, Davison BA, et al. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX-AHF) development program: correlation with outcomes. J Am Coll Cardiol. 2013;61:196–206. | |
Mitrovic V, Luss H, Nitsche K, et al. Effects of the renal natriuretic peptide urodilatin (ularitide) in patients with decompensated chronic heart failure: a double-blind, placebo-controlled, ascending-dose trial. Am Heart J. 2005;150:1239. | |
Mitrovic V, Seferovic PM, Simeunovic D, et al. Haemodynamic and clinical effects of ularitide in decompensated heart failure. Eur Heart J. 2006;27:2823–2832. | |
US National Institutes of Health. Efficacy and Safety of Ularitide for the Treatment of Acute Decompensated Heart Failure (TRUE-AHF). Available from: http://clinicaltrials.gov/ct2/show/NCT01661634. Accessed May 14, 2014. | |
Vale RD, Milligan RA. The way things move: looking under the hood of molecular motor proteins. Science. 2000;288:88–95. | |
Teerlink JR. A novel approach to improve cardiac performance: cardiac myosin activators. Heart Fail Rev. 2009;14:289–298. | |
Malik FI, Hartman JJ, Elias KA, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331:1439–1443. | |
Malik FI, Morgan BP. Cardiac myosin activation part 1: from concept to clinic. J Mol Cell Cardiol. 2011;51:454–461. | |
Shen YT, Malik FI, Zhao X, et al. Improvement of cardiac function by a cardiac myosin activator in conscious dogs with systolic heart failure. Circ Heart Fail. 2010;3:522–527. | |
Teerlink JR, Clarke CP, Saikali KG, et al. Dose-dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecamtiv mecarbil: a first-in-man study. Lancet. 2011;378:667–675. | |
Cleland JG, Teerlink JR, Senior R, et al. The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: a double-blind, placebo-controlled, crossover, dose-ranging Phase II trial. Lancet. 2011;378:676–683. | |
Greenberg BH, Chou W, Escandon R. Phase II safety study evaluating the novel cardiac myosin activator, CK-1827452, in patients with ischemic cardiomyopathy and angina. J Card Fail. 2009;15:S67. | |
US National Institutes of Health. Study to evaluate the safety and efficacy of intravenous infusion treatment with omecamtiv mecarbil in subjects with left ventricular systolic dysfunction hospitalized for acute heart failure. Available from: http://clinicaltrials.gov/ct2/show/NCT01300013. Accessed May 13, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.