")
Back to Journals » Research and Reports in Neonatology » Volume 8
Acute bilirubin encephalopathy and its progression to kernicterus: current perspectives
Authors Usman F, Diala UM , Shapiro SM, Le Pichon JB , Slusher TM
Received 4 October 2017
Accepted for publication 20 December 2017
Published 7 March 2018 Volume 2018:8 Pages 33—44
DOI https://doi.org/10.2147/RRN.S125758
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Robert Schelonka
Fatima Usman,1,2,* Udochukwu Michael Diala,3,4,* Steven M Shapiro,5–7 Jean Baptiste Le Pichon,5–7 Tina M Slusher8,9
1Department of Pediatrics, Bayero University Kano, Kano, Nigeria; 2Department of Pediatrics, Aminu Kano Teaching Hospital, Kano, Nigeria; 3Department of Pediatrics, University of Jos, Jos, Plateau State, Nigeria; 4Department of Pediatrics, Jos University Teaching Hospital, Jos, Plateau State, Nigeria; 5Department of Paediatrics, Children’s Mercy Hospital, Kansas City, MO, USA; 6Department of Paediatrics, University of Missouri-Kansas City, Kansas City, MO, USA; 7Department of Paediatrics, University of Kansas, Kansas City, MO, USA; 8Department of Paediatrics, Division of Global Healthy, University of Minnesota, Minneapolis, MN, USA; 9Pediatric Intensive Care Faculty, Hennepin County Medical Center, University of Minnesota, Minneapolis, MN, USA
*These authors contributed equally to this work
Abstract: Acute bilirubin encephalopathy (ABE) remains a significant cause of morbidity and mortality throughout the world, especially in low-middle-income countries where it can account for up to 15% of neonatal death. The pathophysiology of this acute life-threatening event of infancy and its potential evolution to kernicterus remain poorly understood. In this review, we start by reviewing the terminology of hyperbilirubinemia and its clinical consequences, ABE and later kernicterus spectrum disorder (KSD). We then review the pathogenesis of ABE and discuss clinical factors that can contribute to its pathogenicity. We examine in detail the clinical correlates of ABE and KSD. We present a comprehensive approach to its diagnosis and conclude with a set of simple clinical interventions ranging between primary preventive and rehabilitative measures that may help reduce the incidence of this largely preventable disease.
Keywords: acute bilirubin encephalopathy, neonatal jaundice, kernicterus, kernicterus spectrum disorder, low-middle-income countries
Introduction
Bilirubin encephalopathy (BE) is an important cause of cerebral palsy, developmental delay and hearing impairment particularly in low-middle-income countries (LMICs).1,2 The actual incidence of BE is difficult to estimate in both high-income countries and LMICs. In LMICs, this is largely due to delayed diagnosis, non-recognition in ICD coding, under-reporting or even lack of organized data collection and reporting in LMICs.2 Interventions such as universal neonatal jaundice risk assessment and screening, coupled with the wide availability of treatment modalities such as Rhesus immunoglobulin (Rhogam™) and intensive phototherapy resulted in the apparent near elimination of kernicterus in many countries, as reported in a survey done in the USA between 1980 and 19963 and one done in Denmark during a similar time period.4 This was followed by an apparent re-emergence of kernicterus spectrum disorder (KSD) following a shift to early (<48 hours) hospital discharge after delivery and lack of monitoring of hyperbilirubinemia at home.5 More recent scrutiny of data proposed that the reported disappearance of KSD was most likely merely a period of underreporting.6 Limited data from two high-income countries (Canada and Denmark) reported kernicterus at a rate of 1 in 44,000 live births and 0.6 per 100,000 live births, respectively,7,8 while the Parents of Infants and Children with Kernicterus report 650–750 children with kernicterus in their network mostly from the USA and the UK (Personal Communication Susan Haas). Nonetheless despite these cases, KSD is generally rare in high-income countries with most doctors never seeing a case throughout their years of practice. The same cannot be said of the LMICs, especially Sub-Saharan Africa and South-East Asia that bear the main burden of KSD, a reflection of weak health care systems amidst a high proportion of population expressing the glucose-6-phosphate dehydrogenase (G6PD) deficiency gene.9–11 In tertiary settings in Nigeria, acute bilirubin encephalopathy (ABE) accounts for 3.4% of neonatal admissions with 21.4% of those infants dying12 and at least 15% of neonatal deaths.
This review focuses on the mechanisms of neurologic injury in ABE, risk factors for neuronal injury, clinical correlates of ABE and KSD, disease progression as well as diagnosis and briefly discusses prevention.
Defining terms
The term kernicterus finds its roots in the German word “kern” for nucleus and the Greek word “ikteros” for jaundice. Originally it was used by Christian Georg Schmorl to connote the pathologic yellow staining of the basal ganglia and cerebellum.13 However, over time, the term was adopted to refer to both acute and chronic BE. In 2004, the AAP in an attempt to clarify the use of terminologies recommended that the term ABE be used to describe acute bilirubin neurological symptoms manifesting in the first few weeks of life, while kernicterus should be reserved to describe the more chronic sequelae of ABE.14 Despite these suggestions, there continued to be considerable term confusion, prompting Le Pichon et al15 to suggest the continued use of ABE for acute manifestations of BE while replacing all previously used terms for chronic BE (including but not limited to kernicterus, bilirubin-induced neurologic dysfunction [BIND], subtle kernicterus, etc.) with the term KSD. Like all spectrum disorders, KSD ranges from less severe symptoms to very severe manifestation. At the milder end of the spectrum, children may have movement disorders, isolated hearing loss and/or auditory dysfunction including isolated auditory neuropathy.16,17 Those children with more severe manifestations will have a permanent incapacitating condition characterized by dystonia, choreoathetosis, severe neurologic hearing impairment, paralysis of upward gaze and dental enamel dysplasia.18,19
Pathogenesis of ABE
Neuroanatomy of BE
Post-mortem and animal studies have shown that BE results from neural damage of typical brain regions, namely: 1) globus pallidus, 2) sub-thalamic nuclei, 3) hippocampus 4) oculomotor nuclei, 5) ventral cochlear nuclei and 6) the Purkinje cells and dentate nuclei of the cerebellum.20 This is usually seen as yellow staining of these regions with evidence of neuronal damage.20 There are considerations that regional damage of parts of the auditory or extrapyramidal pathways may be more severe in some areas as compared to other areas and may occur in isolation, sparing other common regions of the brainstem.21 It is also noteworthy that KSD typically spares the cortex and, interestingly, the striatum and thalamus. This is noteworthy as many of these structures share the same blood supply as areas that are sensitive to hyperbilirubinemia, highlighting that there must be an underlying genetic or biochemical mechanism to explain the selective susceptibility of these regions to hyperbilirubinemia.
Magnetic resonance imaging (MRI) findings consistent with KSD include bilateral, symmetric, high-intensity signals in the globus pallidus and sub-thalamic nuclei. During the phase of ABE, the increased signal is most obvious on the T1 sequences. However, as KSD develops, the MRI abnormalities will become more obvious on T2 sequences22 (Figure 1).
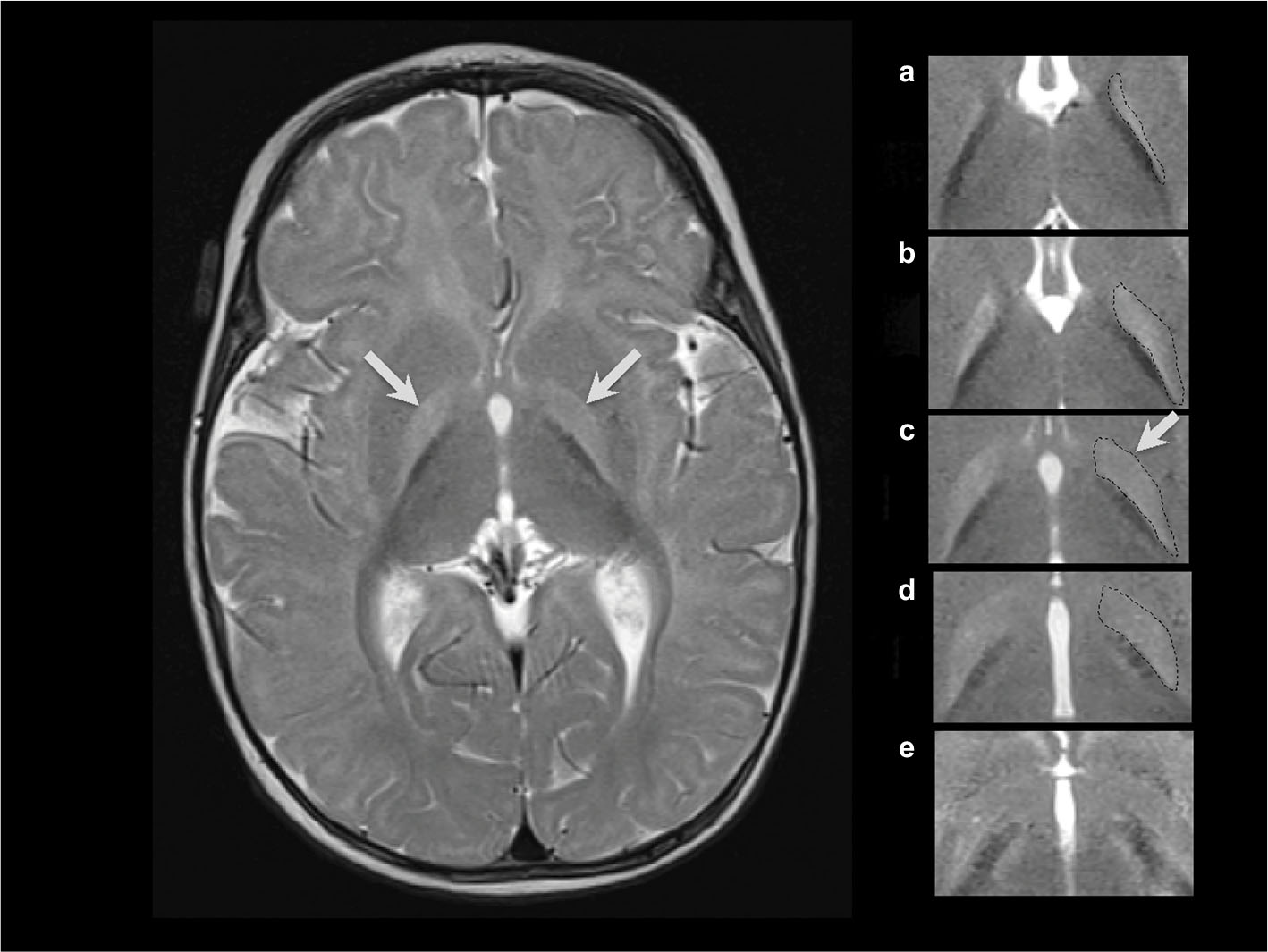 | Figure 1 T2-weighted axial MRI at 6 months of age in a male born at 40 weeks of gestation with hyperbilirubinemia due to G6PD deficiency and a total bilirubin of 39 mg/dl (665 µM) at 125 hours of age, treated with two double-volume exchange transfusions, diagnosed at 27 months of age with moderate to severe, motor-predominant kernicterus.15,23 Notes: The hyperintensity of the globus pallidus bilaterally (left panel, arrows). In the right panel, the abnormal hyperintensity is shown in 5 continuous 3 mm axial T2 slices from rostral to caudal (a–e), with the globus pallidus on the right of the figure outlined with a dotted line. Panel c (dotted line and arrow) is extracted from the image shown in the left panel, and the bottom panel (e) is below the globus pallidus and not outlined because there is no abnormal hyperintensity. Abbreviations: G6PD, glucose-6-phosphate dehydrogenase; MRI, magnetic resonance imaging. |
Mechanism of bilirubin-induced neurotoxicity
The mechanism of bilirubin-induced neuronal damage is not completely understood and appears to have several pathways including bilirubin-induced lipid peroxidation, neuroinflammation, excitotoxicity as well as sustained energy failure24 (Figure 2).
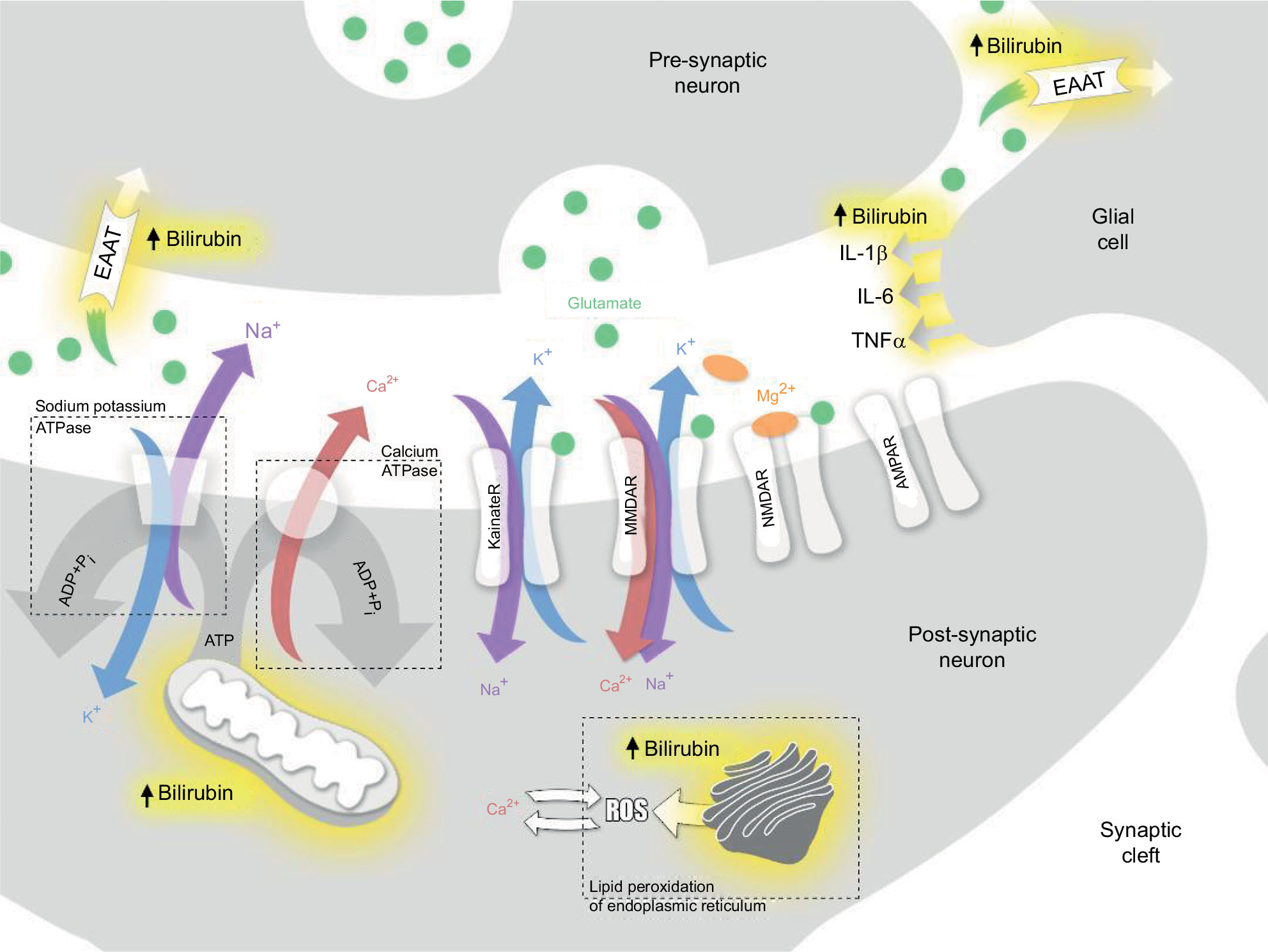 | Figure 2 Schematic overview of the proposed pathophysiological mechanisms underlying bilirubin-induced neuronal injury. Notes: Unconjugated hyperbilirubinemia exerts direct effects on the plasma membranes, mitochondria and/or endoplasmic reticulum leading to excitotoxicity, mitochondrial energy failure and increased intracellular calcium levels. Increased intracellular calcium alters enzyme activities and affects mitochondrial function. Together these mechanisms activate directly or indirectly apoptotic and or necrotic cell death. If unconjugated bilirubin exposure is of a sufficient degree and/or duration, then irreversible neuronal damage ensues. Sites of action of bilirubin are denoted by the yellow haze surrounding the area. Abbreviations: AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; EAAT, excitatory amino acid transporter; IL, interleukin; KainateR, kainate receptor; NMDAR, N-methyl-D-aspartate receptor; TNF, tumor necrosis factor. |
High levels of unbound (free) bilirubin in the brain have been suggested to be immune-stimulatory.25 Activation of microglial cells has been demonstrated in vivo leading to upregulation of pro-inflammatory cytokines such as tumor necrosis factor-α and interleukins (IL) 1β and 6 with subsequent inflammation and necrosis of the affected cells24–26 (Figure 2). This may also explain why conditions associated with systemic inflammation, such as sepsis, potentiate bilirubin-induced neuronal injury.
This line of reasoning is supported by evidence from Gunn rat experiments. Gunn rats lack the functional UDP glucuronosyltransferase enzyme, UGT1A1.27 In humans, deficiency of this enzyme causes Crigler-Najjar and Gilbert syndrome. Crigler-Najjar syndrome is a rare autosomal recessive disorder with severe (type I) and less severe (type II) congenital non-hemolytic jaundice and deficiency of UGT1A1. Gilbert syndrome is the most common inherited disorder of bilirubin glucuronidation due to mutations in the promoter region of the UGT1A1 leading to mild, usually asymptomatic unconjugated hyperbilirubinemia from reduced production of UGT1A1.28 Aside from being jaundiced, the Gunn rats are otherwise normal at birth, and most have lifelong mild to moderate hyperbilirubinemia with mild to moderate neurological signs. In a commonly used experimental model, Gunn rats are injected with a sulfonamide that competitively displaces bilirubin from albumin, and the resultant free bilirubin diffuses into the brain and causes ABE. In this animal model, there is strong evidence that supports chronic microglial activation leading to chronic neuroinflammation.29,30 This evidence further sheds light on the possibility of continued neuronal damage after the acute phase of BE and the possible extension of the window of opportunity for therapeutic interventions beyond the 1st week of life. Opportunities for intervention include targeting chronic neuroinflammation for an extended period of time following the acute injury; although to date there have been no clinical trials in humans or animals targeting this potential mechanism of action. Other opportunities might conceivably include specific therapies designed to promote more beneficial neurodevelopment, medications targeting neuronal hyperexcitability to suppress dysfunctional neuronal activity and transplantation of neural progenitor cells (stem cells) to replace specific subpopulations of neurons and hopefully to re-connect and re-establish beneficial function. It is important to note that all these possible therapeutic interventions beyond the 1st week are at present purely speculative.
Glutamate and N-methyl-D-aspartate (NMDA) receptor subtype is also thought to contribute to bilirubin-induced neuronal injury (Figure 2). It is known that there is a time course-dependent overexpression of NMDA receptors in the neonatal striatum (including the globus pallidus), the hippocampus, and the Purkinje cells.31 This temporal elevation of the glutamate receptors in the globus pallidus may set the stage for a glutamate driven excitotoxicity model in the immature brain.32 The excitotoxicity model proposes that excessive glutamate receptor activation ultimately results in cell death. Glutamate binds to both NMDA and non-NMDA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and kainate) receptors. Both types of receptors, when activated, allow influx of cations into the cells. NMDA receptors have a high permeability to calcium, but under normal circumstances are modulated by Mg2+, preventing the channel from opening. Exposure to bilirubin has been shown to affect several mechanisms affecting ionic equilibrium. Bilirubin reduces oxidative phosphorylation in the mitochondria. Under normal circumstances, ATPase-dependent ion pumps maintain homeostasis by pumping cations out of the cell. However, prolonged activation of the glutamate receptors results in mitochondrial failure (ATP depletion). With prolonged mitochondrial failure, the ATP-dependent ion pumps fail leading to prolonged depolarization. The depolarization in turn causes Mg2+ to dissociate from the NMDA receptors (a voltage-dependent process). This in turn leads to increased Ca2+ influx. Bilirubin is also believed to affect excitatory amino acid transporters, including glutamate transporters, resulting in prolonged exposure of glutamate in the synaptic cleft. This in turn activates the glutamate receptors, both NMDA and non-NMDA receptors. Furthermore, bilirubin in the brain contributes to oxidative stress by lipid peroxidation of membranes of the endoplasmic reticulum. In Gunn rats, bilirubin-induced neurotoxicity (including auditory brainstem response [ABR] abnormalities) was prevented by competitively inhibiting lipid peroxidation with minocycline.33,34 This mechanism is not well understood as other antioxidants (12-pyrazolinominocycline and tauroursodeoxycholic acid)35 with similar potency as bilirubin were found to also inhibit lipid peroxidation but do not prevent neuronal injury. Ultimately, these mechanisms contribute to a massive Ca2+ influx. This in turn causes edema and activation of the final common pathways for apoptosis and necrosis.28,29
Risk factors for BE
Total serum bilirubin (TSB) levels have been used in the management guidelines of neonates with hyperbilirubinemia to define critical values for interventions such as phototherapy and exchange blood transfusion. TSB measures both conjugated and unconjugated bilirubin in the blood. The unconjugated bilirubin is largely bound to albumin but a small proportion remains as free or unbound bilirubin. High TSB levels have been associated with a risk of BE in a dose-dependent pattern since at least the 1950s.36 This stems largely from the understanding that high TSB levels correlate with high free unbound unconjugated bilirubin levels at the membrane surfaces as a result of saturating the albumin binding sites. Free bilirubin is available to permeate membranes, including brain cells, and cause neuronal injury. However, reports of ABE at TSB levels considered to be non-hazardous, that is, below critical values for exchange blood transfusion (low bilirubin KSD) have highlighted the need to establish a critical value of TSB below which BE is unlikely to occur.37 Low bilirubin ABE is possible in the context of hypoalbuminemia or impaired albumin binding.38 Bilirubin is transported bound to albumin in a largely predictable way. Bilirubin bound to albumin is water soluble and does not cross the blood–brain barrier (BBB). Albumin has two types of binding sites for bilirubin – a high affinity site and other lower affinity sites. Bilirubin binding to albumin saturates when the molar ratio of bilirubin to albumin exceeds 1:1 at a pH of 7.2 and a temperature 37.5°C. Unbound (free) bilirubin is expected to be a more appropriate measure of the risk for BE than TSB. However, it is currently not practical to assay UB in clinical settings. Bilirubin:albumin (B:A) molar ratio, on the other hand, is easily assayed and has previously been proposed as a surrogate for UB and, consequently, CNS exposure to bilirubin. A threshold for commencement of treatment using B:A molar ratio was recommended;39 however, in the clinical setting, it has not been proved to be superior to TSB estimation alone in predicting neurotoxicity.39,40
Immaturity of the brain cells (especially in preterm infants) also increases susceptibility to neuronal damage even at lower bilirubin levels.41 This observation has been confirmed in animal models as well.42 Mechanisms for this increased susceptibility in the preterm include age-dependent ability of brain cells to metabolize bilirubin and lower expression levels of P-glycoprotein (6-GP).43,44 6-GP, also referred to as multidrug resistance-associated protein 1, is an ATPase membrane efflux pump present in the BBB that serves to maintain a brain–blood bilirubin gradient (about 1–2% of serum bilirubin concentration), thereby protecting the brain from excess levels of bilirubin.43,44 6-GP has a high affinity for bilirubin when compared to other substrates like leukotrienes (about 10 times higher).45 Some authors believe that its expression is lower in the brainstem auditory structures accounting at least in part for this region’s high susceptibility to bilirubin-induced neurotoxicity.
Acidosis also increases bilirubin neurotoxicity.26 Vázquez et al demonstrated that acidosis increases hydrophobic inclusion of bilirubin into erythrocyte membranes rather than aggregation of bilirubin on the surface of membranes from albumin dissociation and proposed the same mechanism in the pathogenesis of neurotoxicity with acidosis.46
Other potentiating neurotoxic factors include sepsis, drugs or other binding competitors, hypercarbia, hyperosmolality, hypoxia, asphyxia and hemolysis.26,47 Sepsis increase susceptibility to ABE by increasing blood–brain permeability and decreasing the albumin binding capacity.47 Park et al, while comparing the response of preterm neonates with or without sepsis to lipid infusion, observed poor utilization of infused fats in the sepsis group.48 Plasma free fatty acid concentration, which is known to displace bilirubin from albumin, is increased in sepsis. Hansen et al, however, concluded that the evidence in support of ABE susceptibility with sepsis was weak. They came to this conclusion on the basis of a study that demonstrated that sepsis did not increase brain mitochondrial oxidation of bilirubin, the hypothesized mechanism of bilirubin-induced neuronal injury in this context.49
In the 1950s, the wide use of sulfonamides for infection prophylaxis in neonates was associated with KSD.50 This was due to displacement of bilirubin from its binding site with albumin by sulfonamides. Several other drugs have been associated with similar displacement of bilirubin.50
Hypercarbia increases brain blood flow with resultant increase in entry of bilirubin into the brain.51 Hyperosmolality (as seen in hyperglycemia, hypernatremia and azotemia), hypoxia and asphyxia affect the integrity of the BBB leading to prolonged exposure of the brain to bilirubin (Figure 3).
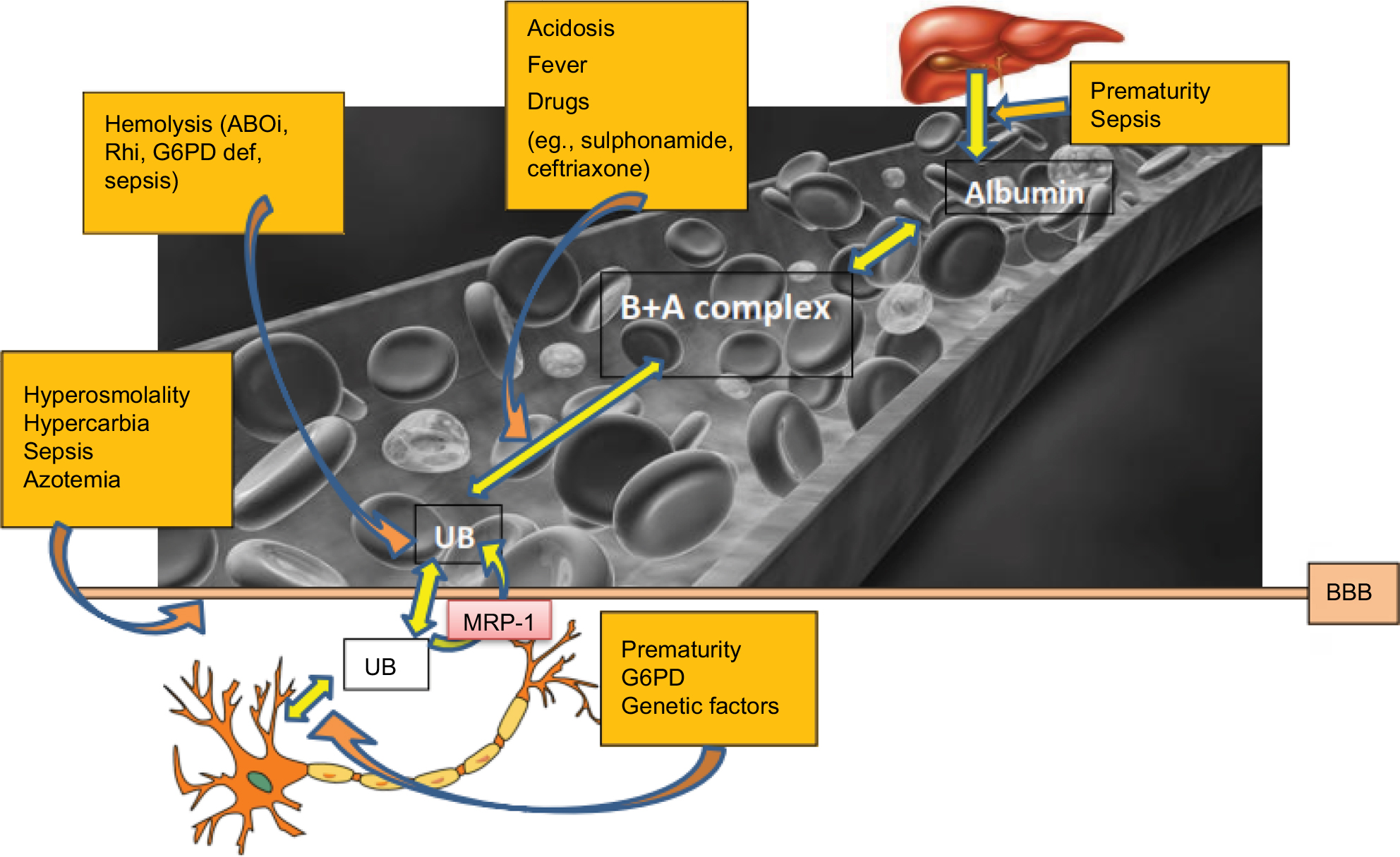 | Figure 3 Schematic of factors contributing to bilirubin-albumin hemodynamic. Abbreviations: ABOi, ABO incompatibility; B+A complex, bilirubin:albumin complex; BBB, blood–brain barrier; BE, bilirubin encephalopathy; G6PD def, glucose-6-phosphate dehydrogenase deficiency; MRP-1, multidrug resistant protein-1; Rhi, Rh (Rhesus) blood group incompatibility. |
Hemolysis increases the risk of BE mainly due to the rapid increase in bilirubin production, most notably in the context of delayed treatment.52 During rapid hemolysis, TSB levels may rise more quickly than it can be conjugated or isomerized following transport into the skin53 leading to neuronal injury. This rapid elevation of TSB results in a proportional elevation of free bilirubin (as bilirubin binding to albumin follows Michaelis–Menten kinetics). Consequently, the brain is exposed to higher levels of free bilirubin resulting in increased risk for BE. G6PD deficiency is one of the most common causes of non-immune hemolytic jaundice. Eleven million neonates delivered each year are G6PD deficient globally.2 Studies from Nigeria, Oman, Singapore and the USA have demonstrated at least a fourfold increase in the incidence of KSD in G6PD-deficient neonates compared to non-G6PD-deficient neonates.3,9,10,54,55 As stated by Kaplan and Hammerman, this increased sensitivity observed in infants with G6PD is likely a result of a combination of factors, including excessive bilirubin production, diminished conjugation and other genetic and environmental pressures.56
More recently, Hegyi et al showed that increasing doses of intra-lipids in infants born preterm had a direct effect on the free bilirubin concentration.57 Presumably, this occurs as with increasing concentration of intra-lipids, free fatty acids, known to competitively compete with the bilirubin albumin binding site, displace bilirubin. This is especially noteworthy that at higher doses of free fatty acids, the free bilirubin concentration no longer follows a linear relationship to the total bilirubin. It follows that the total bilirubin concentration probably underestimates the free bilirubin and consequently the risk for BE.
Many unanswered questions in the pathogenesis of ABE remain. For example, it is not clear why some term and near-term neonates with extreme hyperbilirubinemia (500–600 µmoles/l or 30–35 mg/dl) escape unharmed, while other neonates develop KSD even at total bilirubin levels below exchange transfusion threshold.58 This opens the possibility that there may be other genetic factors at play that predispose certain neonates and protect others from unbound bilirubin.
Clinical correlates
ABE
The signs and symptoms of ABE may be subtle requiring a high index of suspicion, or apparent with overt neurologic abnormalities.59 The spectrum of manifestation has been categorized into 3 phases with variable times of onset for each phase.60 Phase 1 (early ABE) manifests early usually at 3–5 days of life with decreased alertness, poor feeding, hypotonia and weak Moro. Phase 2 (intermediate ABE) has variable onset and duration, usually presenting in latter in the 1st week but can be latter with stupor, irritability, hypertonia of extensor muscles, which may alternate with hypotonia, opisthotonos, retrocollis and high-pitched cry. Phase 3 (advanced ABE) often presents after the 1st week and is typically characterized by hypotonia. Other features include coma, pronator spasm of upper extremities, sun setting eyes, fever, inability to feed and apnea.53,56 Mortality may be at least as high as 21%,12 usually due to respiratory failure or refractory seizures.
The clinical features of encephalopathy in preterm neonates are the same as term neonates albeit more subtle, mainly due to neuronal immaturity and masking clinical conditions.61
KSD
KSD is a more chronic and permanent clinical sequel of bilirubin toxicity in neonates who survive ABE. It evolves slowly over several years in the affected children.62 In the early phase, which occurs in the 1st year of life, it usually presents with hypotonia, hyper-reflexia, persistence of tonic neck reflex and delayed milestones.62 After the 1st year, the manifestation is more variable, with a tetrad of symptoms including auditory, visual and dental abnormalities, and extrapyramidal disturbances. Auditory abnormalities usually present as a neural hearing loss, often mischaracterized as sensorineural hearing loss (SNHL), but best characterized by auditory neuropathy spectrum disorder (ANSD), since the auditory dysfunction is localized to auditory brainstem nuclei ± possibly the auditory nerve, with no evidence of a primary sensory (i.e., hair cell) involvement. This is one of the earliest features of ABE and KSDs and it may be seen even in the absence of other clinical manifestations of BE.18 Visual abnormalities include paralyses of upward gaze, horizontal gaze dysfunction and a blank stare or “scared appearance” caused by the combination of upward gaze paresis and facial dystonia.63 Dental abnormalities include dental enamel hypoplasia of the deciduous (baby) teeth and green stained teeth. Extrapyramidal disturbances consist of dystonia, the commonest manifestation, and athetosis, although chorea may also occur. Bulbar functions may also be impacted.62,64 The cognitive function of individuals with KSD is relatively speared.11 Some studies suggest otherwise;58 however, it is likely that these studies underestimate the cognitive abilities of the affected subjects due to the significant motor limitations they suffer from.
Diagnosis
In the past, it was erroneously believed that the diagnosis of BE could only be made by autopsy. Using history, a focused physical examination, the BIND score,16,54 and electrophysiological and neuroimaging studies, the diagnosis can be ascertained with reasonable certainty. Assessment of the encephalopathy must be individualized, taking into account predisposing risk factors.
Relevant to the history is the severity and duration of hyperbilirubinemia. Other risk factors such as prematurity, sepsis, acidosis and Rh disease should be assessed. It is also important to elicit a history of abnormal neonatal neurologic signs or symptoms known to associate with ABE (abnormal tone, abnormal cry, posturing, abnormal eye movements or positions). Finally, later in life, one should enquire about a history of delayed speech, gross or fine motor development, dental enamel dysplasia of deciduous teeth or hearing abnormalities, all consistent with KSD.59
On examination, dystonia, choreoathetosis, ataxia, variable hypo/hypertonia, spasticity, incoordination, gaze abnormalities, staining or flaking of deciduous teeth, dental enamel hypoplasia, dysarthria, hearing impairment or difficulty localizing sound may be detected.59
The BIND score60 is a tool to objectify and facilitate a clinical diagnosis of ABE as well as to monitor the neonatal neurological exam in infants with progressive hyperbilirubinemia as a predecessor to encephalopathy. The BIND score uses the mental state, muscle tone and cry pattern to categorize neonates into three levels of increasing abnormality (subtle, moderate, advanced). A score ranging between zero and three is given for each clinical parameter assessed based on severity. A score of zero indicates a normal neonate. A value ranging between 1 and 3 signifies subtle encephalopathy. Moderate encephalopathy and advanced encephalopathy correspond to scores of 4–6 and 7–9, respectively. Following the publication of the classic BE facies, now called the “kernicteric facies”63 (Figure 4), the BIND score was modified for use primarily in low-resource settings often without ancillary testing such as ABRs and MRIs. The modified bilirubin-induced neurologic dysfunction scale (M-BIND), a 12-point score, incorporates eye abnormalities such as a divergent gaze, paralysis of upward gaze, anxious appearance and nystagmus, and serves to better discriminate degrees of BIND severity and to aid in distinguishing ABE from other common causes of neonatal morbidity and mortality such as tetanus still seen in low-resources settings.65 The reliability of using the M-BIND algorithm in resource limited setting was assessed and was found to have a positive predictive value of 88.9%, a negative predictive value of 98.2% and a weighted kappa coefficient of 0.7969 (substantial agreement) between the scores obtained by the consultants and residents.65
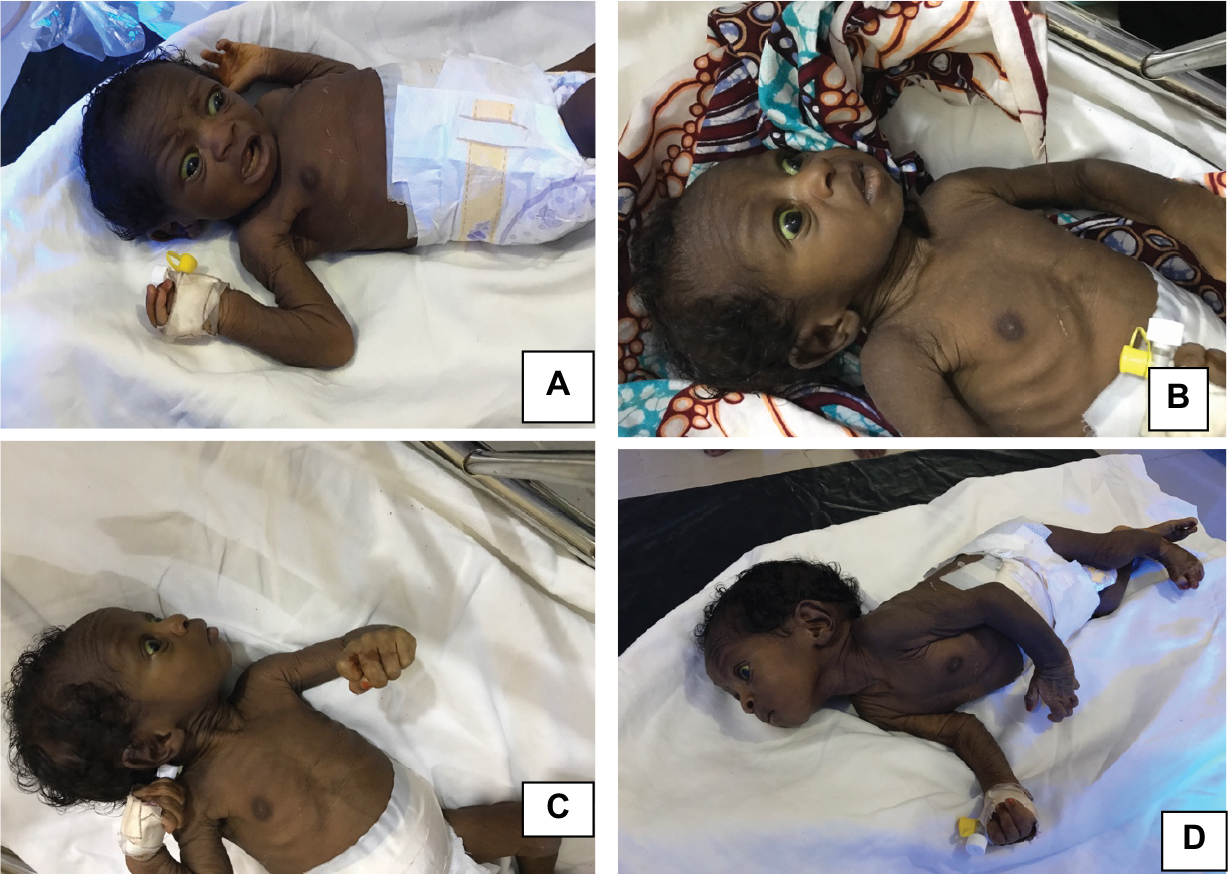 | Figure 4 A baby with advanced bilirubin encephalopathy and secondary failure to thrive. Notes: (A and B) Anxious looking, paralysis of upward gaze with lid retraction (collier sign) and facial dystonia. (C) Bicycling/windmill movement of upper limbs. (D) Neck retraction and scissoring of lower limbs. Note: Photographs courtesy of Dr Fatima Usman and used with maternal permission. |
The tool named the brainstem auditory evoked response (BAER), also referred to as ABR, is an effective, non-invasive screening tool with a sensitivity of 100% and specificity of 99.7%66 in detecting hearing impairment. This test can be used in neonates to predict impending encephalopathy, identify subtle BE that could be reversed and detect early hearing abnormalities.67 Emerging evidence suggests that the auditory neural pathways are the most sensitive system to overt bilirubin toxicity, with the most consistent feature being ANSD ± hearing loss.18,68 Thus, auditory evaluation may improve detection of bilirubin-induced neurotoxicity in neonates.69 The BAER recording is a series of vertex positive waves of which wave I through wave V are evaluated. In bilirubin neurotoxicity, the BAER is absent or abnormal (prolonged inter-wave intervals and/or diminished amplitudes) indicating damage to the auditory nerve (wave l) and/or more likely auditory brainstem nuclei (waves III and V).70–73 These changes correlate significantly with bilirubin level.70 In neonates with ANSD secondary to ABE, abnormal BAERs with normal cochlear microphonic (CMs) responses and initially normal otoacoustic emissions (OAEs) are observed.61,73,75 With ANSD, OAEs, initially normal, often disappear over time, whereas CMs never disappear. Note that with SNHL, which is more common than ANSD, both OAEs and CMs are abnormal or absent to the same extent as the abnormal or absent BAER.
Similarly, cranial magnetic resonance imaging (cMRI) can be used to detect bilirubin neurotoxicity. In ABE, MRI may show characteristic T1-hyperintense involvement of the globus pallidus and subthalamic nuclei, while KSD usually demonstrates increased signal intensity on T2-weighted images of the same regions, especially in children with classical and motor-predominant kernicterus.68,75 Although cMRI has an important role in the diagnosis of ABE and KSD, it does not always correlate with clinical and laboratory findings.75 Various studies have shown inconsistent findings in infants with bilirubin toxicity, reporting normal cMRI results despite abnormal neurologic findings.76–78 One should note that it is easy to confuse a benign appearance of high T1 signal intensity in the basal ganglia of newborn and premature infants as a result of incomplete myelination.75 This point serves to emphasize the importance of relying on the expertise of a radiologist well versed in MRI of the newborn infant and of not relying on a single test to make a diagnosis of ABE.
Based on the presence of abnormal neurologic findings, BAER and cMRI, Shapiro proposed the use of the terms “certain”, “probable” and “possible” to define neonates with KSD.16 In the recent paper by Le Pichon et al,15 he further clarified these terms and further encourages using two Kernicterus Diagnostic Toolkits to help make a diagnosis of KSD in infants born at or near term. These toolkits are designed to be used by health care professionals, caregivers and families to assess the probability, severity and type of a KSD. Each toolkit is scored and provides a quantifiable measure that can be comparable across assessors. The authors propose validating the toolkits in hopes that a standard diagnostic algorithm for KSDs will be useful to families, providers and researchers.
Acute encephalopathy progressing to KSD
A considerable amount of research has focused on how best to predict the likelihood of bilirubin neurotoxicity, but to date no study has identified a direct causal factor as an absolute predictor of ABE with subsequent progression to KSD.79 Kaplan and Hammerman reported ABE progression to KSD in 84% of affected infants.61 There have been several reports of resolution of ABE without progression to KSD in infants with moderate to severe disease following treatment.80
Observational literature supports the assertion that genetic factors are relevant modulators to bilirubin neurotoxicity in addition to previously described modifiers such as prematurity, high levels of bilirubin with long duration of exposure, presence of comorbid conditions that predispose to hemolysis and increase propensity for injury, factors that alter BBB permeability to bilirubin, and variations in albumin concentration and/or its bilirubin-binding capacity.14,47,81 There is persuasive theoretic evidence backing variable gene expression as a significant determinant for bilirubin susceptibility and subsequent progression to KSD. It is likely that regional differential genetic expression accounts, at least in part, for the selective brain vulnerability. While there have been numerous genes linked to the development of hyperbilirubinemia, relatively few studies have reported on the genes and pathways important to determine the newborn’s susceptibility to the toxic effect of bilirubin. Additionally, it is well recognized that the precise level at which bilirubin is neurotoxic is unpredictable.18,82–84 It is therefore reasonable to postulate that a complex nature-nurture interplay underlies the mechanisms predisposing individuals to ABE and KSD.
Several lines of evidence point toward environmental factors as significant risks to disease progression. Delayed feeding, use of icterogenic substances, delay in seeking timely and appropriate care including failed recognition of the onset of severe hyperbilirubinemia and poor perception of its severity have been linked with advancing disease.59,68 Conversely, prompt and aggressive treatment with exchange blood transfusion and adjunct intensive phototherapy reverses neurotoxicity largely by reducing the duration of exposure of brain cells to UB.85–87 Reversal of abnormal ABR has been reported following exchange blood transfusion,86,87 supporting the recommendation to treat aggressively infants with moderate to advanced ABE, even when the TSB is below the threshold for exchange transfusion.14 Another potentiating factor in disease progression is the clinical stage of disease at presentation. Infants presenting with mild disease are more likely to have complete reversal of neuronal injury with aggressive treatment.87 Phase 1 ABE is reversible with prompt and appropriate treatment, while the outcome in phase 2 is variable.60 Individualized and timely intervention may prevent further brain damage and minimize the severity of the sequelae in the advanced phase of the disease.60 One might summarize this entire discussion by stating the following: the faster and the more aggressive the treatment, the less the likelihood of progression and the better the outcome. As such, treatment should not be withheld in any infant due to late presentation.
Prevention of ABE and KSD
An anticipatory and individualized approach with the goal of avoiding excessive hyperbilirubinemia is the key to preventing severe neonatal jaundice, ABE87 and its subsequent progression to KSD. Using a systematic tiered approach, targeted preventive strategies are essential at each level during the assessment of newborn infants to prevent these complications. Obstacles to preventive strategies and need for solutions to overcome obstacles at each point have been elucidated in manuscripts such as the ones by Olusanya et al91 and Ogunfowora and Daniel.89 At the primary level, programs aimed at promoting and supporting successful breast feeding, documentation of the mother’s blood group during antenatal care with cheap and available and appropriately used Rhesus immunoglobulin (Rhogam™), meticulous risk assessment and providing the parents with written and oral information about jaundice are paramount.90 Included in this counseling must be advice to avoid products such as camphor, henna, naphthalene and mentholatum in neonatal care.91 Data presented at a recent scientific meeting highlighted a significant reduction in ABE with maternal education alone.92 Similarly, recognizing that visual estimation of the severity of jaundice may be misleading93 and establishing protocols for the identification and evaluation of hyperbilirubinemia are of prime importance. Improvement on older screening methods are needed and being effectively pursued. It is essential to screen for jaundice and G6PD deficiency as part of a systemic evaluation on all babies for the risk of severe hyperbilirubinemia before discharge.14,96 A shortened hospital stay of <48 hours after delivery especially for preterm babies should be discouraged.14 Regardless of the place of birth or duration of hospitalization, infants should be screened by the most reliable method available around day 3–4 of life and of course sooner if jaundice in the first 24 hours of life or jaundice of the eyes, hands or feet is diagnosed.14
At the secondary level, measuring bilirubin levels (TSB or TcB) in jaundiced babies, interpreting all bilirubin levels based on hour-specific modified country-specific Bhutani-type nomogram is advocated.95 Additionally, providing appropriately timed and effective treatment using gestational age, weight appropriate threshold for risk assessment and country-specific guidelines is urgently needed.14,91 Effective treatment means a minimum conventional phototherapy with an irradiance of at least 10-W/nm/cm2 but preferably intensive at 30-W/nm/cm2 or greater for those with serious hyperbilirubinemia including need for exchange transfusion and ABE.14,91 With appropriate studies and location-specific protocols, newer solutions providing effective phototherapy should be considered such as filtered sunlight phototherapy using selectively tested window tinting and/or solar and/or battery powered units should be considered in locations without consistent electricity.91,96 Combined with effective phototherapy, health care facilities also need to be able to quickly refer to tertiary centers that are able to do emergent exchange blood transfusions when needed.87,88 Finally, close post-discharge follow-up strategies are critical in preventing bilirubin neurotoxicity.88
Follow-up
Those who unfortunately fall through the cracks in our health care systems and develop KSD need to have appropriate rehabilitation services provided. Limitation of disabilities and rehabilitation of survivors is multi-disciplinary and determined by disease severity. The use of cued speech, hearing aids and cochlear implants for those with hearing impairment and physical and occupational therapy with or without pharmacotherapy (trihexyphenidyl, baclofen, benzodiazepines and botulinum toxin injections) for motor involvement may be helpful. Deep brain stimulation may be beneficial in some cases.15 Unfortunately, most of these options are priced out of the range of infants and children in LMICs. Our goal, however, should be to bring these therapies to those who need them the most regardless of their country or socioeconomic status maximizing the potential of these vulnerable infants to maximize their potential as the grow and develop.
Conclusion
The relationship between ABE and subsequent development of KSD has always been an area of controversy, especially when determining the risk of progression. Indeed, except for the advanced stage of ABE, other stages do not connote the development of KSD. In order to predict this risk of progression, appropriate epidemiologic studies to document the incidence of KSD using precise indicators of long-term neurodevelopmental outcome as well as comparing predisposing factors are necessary.
Acknowledgment
The authors are grateful to Marie Le Pichon and Addison Amiri for their work in conceptualization and design of Figure 2.
Author contributions
Fatima Usman co-wrote the first draft and edited the final version. Udochukwu Michael Diala co-wrote the first draft, conceptualized Figures 2 and 3, and edited the final version. Steven M Shapiro edited and provided expertise on BAER, MRI, ABE, Gunn rats and KSD. Jean Baptiste Le Pichon co-conceptualized Figure 2, edited and provided expertise on ABE and KSD. Tina M Slusher edited throughout and provided expertise on ABE, KSD, BIND-M, diagnosis, prevention and treatment in LMICs. All authors contributed toward data analysis, drafting and critically reviewing and editing this paper and agree with the final document. Drs Usman and Diala contributed equally as co-first authors.
Disclosure
The authors report no conflicts of interest in this work.
References
Olusanya BO, Osibanjo FB, Mabogunje CA, Slusher TM, Olowe SA. The burden and management of neonatal jaundice in Nigeria: a scoping review of the literature. Niger J Clin Pract. 2016;19(1):1–17. | ||
Bhutani VK, Zipursky A, Blencowe H, et al. Neonatal hyperbilirubinemia and Rhesus disease of the newborn: incidence and impairment estimates for 2010 at regional and global levels. Pediatr Res. 2013;74 Suppl 1:86–100. | ||
Burke BL, Robbins JM, Bird TM, Hobbs CA, Nesmith C, Tilford JM. Trends in hospitalizations for neonatal jaundice and kernicterus in the United States, 1988–2005. Pediatrics. 2009;123(2):524–532. | ||
Ebbesen F, Andersson C, Verder H, et al. Extreme hyperbilirubinaemia in term and near-term infants in Denmark. Acta Paediatr. 2005;94(1):59–64. | ||
Bhutani VK, Johnson L. Kernicterus in the 21st century: frequently asked questions. J Perinatol. 2009;29 Suppl 1:S20–S24. | ||
Brooks JC, Fisher-Owens SA, Wu YW, Strauss DJ, Newman TB. Evidence suggests there was not a “resurgence” of kernicterus in the 1990s. Pediatrics. 2011;127(4):672–679. | ||
Ebbesen F, Bjerre JV, Vandborg PK. Relation between serum bilirubin levels ≥450 μmol/L and bilirubin encephalopathy; a Danish population-based study. Acta Paediatr. 2012;101(4):384–389. | ||
Sgro M, Campbell DM, Kandasamy S, Shah V. Incidence of chronic bilirubin encephalopathy in Canada, 2007–2008. Pediatrics. 2012;130(4): e886–e890. | ||
Williams O, Gbadero D, Edowhorhu G, Brearley A, Slusher T, Lund TC. Glucose-6-phosphate dehydrogenase deficiency in Nigerian children. PLoS One. 2013;8(7):e68800. | ||
Owa JA, Ogunlesi TA. Why we are still doing so many exchange blood transfusion for neonatal jaundice in Nigeria. World J Pediatr. 2009;5(1):51–55. | ||
Arain YH, Bhutani VK. Prevention of Kernicterus in South Asia: role of neonatal G6PD deficiency and its identification. Indian J Pediatr. 2014;81(6):599–607. | ||
Adebami O. Factors associated with the incidence of acute bilirubin encephalopathy in Nigerian population. J Pediatr Neurol. 2011;9(20):347–353. | ||
Hansen TW. Pioneers in the scientific study of neonatal jaundice and kernicterus. Pediatrics. 2000;106(2):E15. | ||
American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114(1):297–316. | ||
Le Pichon JB, Riordan SM, Watchko JF, Shapiro SM. The neurological sequelae of neonatal hyperbilirubinemia: definitions, diagnosis and treatment of the Kernicterus Spectrum Disorders (KSDs). Curr Pediatr Rev. 2017;13(3):199–209. | ||
Shapiro SM. Hyperbilrubinemia and the risk for brain injury. In: Perlman J, Polin RA, editors. Neurology: Neonatology Questions and Controversies. Philadelphia, PA: Saunders/Elsevier; 2008:195–209. | ||
Hansen TW. Kernicterus in term and near-term infants--the specter walks again. Acta Paediatr. 2000;89(10):1155–1157. | ||
Shapiro SM. Definition of the clinical spectrum of kernicterus and bilirubin-induced neurologic dysfunction (BIND). J Perinatol. 2005;25(1):54–59. | ||
De Vries LS, Lary S, Whitelaw AG, Dubowitz LM. Relationship of serum bilirubin levels and hearing impairment in newborn infants. Early Hum Dev. 1987;15(5):269–277. | ||
Ahdab-Barmada M. The neuropathology of kernicterus: definitions and debate. In: Maisels MJ, Watchko JF, editors. Neonatal Jaundice. Amsterdam: Harwood Academic Publishers; 2000:75–88. | ||
Volpe JJ. Bilirubin and brain injury. In: Volpe JJ, editor. Neurology of the Newborn. 5th ed. Philadelphia, PA: Saunders Elsevier; 2008:635–637. | ||
Sarı S, Yavuz A, Batur A, Bora A, Caksen H. Brain magnetic resonance imaging and magnetic resonance spectroscopy findings of children with kernicterus. Pol J Radiol. 2015;80:72–80. | ||
Watchko JF. Kernicterus and the molecular mechanisms of bilirubin-induced CNS injury in newborns. Neuromolecular Med. 2006;8(4):513–529. | ||
Shapiro SM. Chronic bilirubin encephalopathy: diagnosis and outcome. Semin Fetal Neonatal Med. 2010;15(3);157–163. | ||
Brites D. The evolving landscape of neurotoxicity by unconjugated bilirubin: role of glial cells and inflammation. Front Pharmacol. 2012;3:88. | ||
Brites D, Brito MA. Bilirubin toxicity. In: Stevenson DK, Maisels MJ, Watchko JF, editors. Care of the Jaundiced Neonate. New York: McGraw Hill; 2012:115–143. | ||
Baron MS, Chaniary KD, Rice AC, Shapiro SM. Multi-neuronal recordings in the basal ganglia in normal and dystonic rats. Front Syst Neurosci. 2011;5:67. | ||
Bosma PJ, Chowdhury JR, Bakker C, et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N Engl J Med. 1995;333(18):1171–1175. | ||
Liaury K, Miyaoka T, Tsumori T, et al. Morphological features of microglial cells in the hippocampal dentate gyrus of Gunn rat: a possible schizophrenia animal model. J Neuroinflammation. 2012;9:56. | ||
Liaury K, Miyaoka T, Tsumori T, et al. Minocycline improves recognition memory and attenuates microglial activation in Gunn rat: a possible hyperbilirubinemia-induced animal model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2014;50:184–190. | ||
Greenamyre T, Penney JB, Young AB, Hudson C, Silverstein FS, Johnston MV. Evidence for transient perinatal glutamatergic innervation of globus pallidus. J Neurosci. 1987;7(4):1022–1030. | ||
McDonald JW, Shapiro SM, Silverstein FS, Johnston MV. Role of glutamate receptor-mediated excitotoxicity in bilirubin-induced brain injury in the Gunn rat model. Exp Neurol. 1998;150(1):21–29. | ||
Geiger AM, Petitti DB, Yao JF. Rehospitalisation for neonatal jaundice: risk factors and outcomes. Paediatr Perinat Epidemiol. 2001;15(4):352–358. | ||
Geiger AS, Rice AC, Shapiro SM. Minocycline blocks acute bilirubin-induced neurological dysfunction in jaundiced Gunn rats. Neonatology. 2007;92(4):219–226. | ||
Daood MJ, Hoyson M, Watchko JF. Lipid peroxidation is not the primary mechanism of bilirubin-induced neurologic dysfunction in jaundiced Gunn rat pups. Pediatr Res. 2012;72(5):455–459. | ||
Hsia DY, Allen FH Jr, Gellis SS, Diamond LK. Erythroblastosis fetalis. VIII. Studies of serum bilirubin in relation to Kernicterus. N Engl J Med. 1952;247(18):668–671. | ||
Odutolu Y, Emmerson AJ. Low bilirubin kernicterus with sepsis and hypoalbuminaemia. BMJ Case Rep. 2013;2013. pii: bcr2012008042. | ||
Watchko JF, Maisels MJ. The enigma of low bilirubin kernicterus in premature infants: why does it still occur, and is it preventable? Semin Perinatol. 2014;38(7):397–406. | ||
Hulzebos CV, Dijk PH, van Imhoff DE, et al; BARTrial Study Group. The bilirubin albumin ratio in the management of hyperbilirubinemia in preterm infants to improve neurodevelopmental outcome: a randomized controlled trial--BARTrial. PLoS One. 2014;9(6):e99466. | ||
Iskander I, Gamaleldin R, El Houchi S, et al. Serum bilirubin and bilirubin/albumin ratio as predictors of bilirubin encephalopathy. Pediatrics. 2014;134(5):e1330–e1339. | ||
Govaert P, Lequin M, Swarte R, et al. Changes in globus pallidus with (pre)term kernicterus. Pediatrics. 2003;112(6 Pt 1):1256–1263. | ||
Conlee JW, Shapiro SM. Development of cerebellar hypoplasia in jaundiced Gunn rats treated with sulfadimethoxine: a quantitative light microscopic analysis. Acta Neuropathol. 1997;93(5):450–460. | ||
Tsai CE, Daood MJ, Lane RH, Hansen TW, Gruetzmacher EM, Watchko JF. P-glycoprotein expression in mouse brain increases with maturation. Biol Neonate. 2002;81(1):58–64. | ||
Hansen TW, Allen JW. Oxidation of bilirubin by brain mitochondrial membranes-- dependence on cell type and postnatal age. Biochem Mol Med. 1997;60(2):155–160. | ||
Rigato I, Pascolo L, Fernetti C, Ostrow JD, Tiribelli C. The human multidrug-resistance-associated protein MRP1 mediates ATP-dependent transport of unconjugated bilirubin. Biochem J. 2004;383(Pt 2):335–341. | ||
Vázquez J, García-Calvo M, Valdivieso F, Mayor F, Mayor F Jr. Interaction of bilirubin with the synaptosomal plasma membrane. J Biol Chem. 1988;263(3):1255–1265. | ||
Hansen TW. Mechanisms of bilirubin toxicity: clinical implications. Clin Perinatol. 2002;29(4):765–778, viii. | ||
Park W, Paust H, Schröder H. Lipid infusion in premature infants suffering from sepsis. JPEN J Parenter Enteral Nutr. 1984;8(3):290–292. | ||
Hansen TW, Maynard EC, Cashore WJ, Oh W. Endotoxemia and brain bilirubin in the rat. Biol Neonate. 1993;63(3):171–176. | ||
Maruyama K, Harada S, Nishigori H, Iwatsuru M. Classification of drugs on the basis of bilirubin-displacing effects on human serum albumin. Chem Phram Bull. 1984;32(6):2414–2420. | ||
Hansen TW. Bilirubin entry into and clearance from rat brain during hypercarbia and hyperosmolality. Pediatr Res. 1996;39(1):72–76. | ||
MacDonald MG. Hidden risks: early discharge and bilirubin toxicity due to glucose 6-phosphate dehydrogenase deficiency. Pediatrics. 1995;96(4 Pt 1):734–738. | ||
Kaplan M, Bromiker R, Hammerman C. Hyperbilirubinemia, hemolysis, and increased bilirubin neurotoxicity. Semin Perinatol. 2014;38(7):429–437. | ||
Ogunlesi TA, Ogunfowora OB. Predictors of acute bilirubin encephalopathy among Nigerian term babies with moderate-to-severe hyperbilirubinaemia. J Trop Pediatr. 2011;57(2):80–86. | ||
Nair PA, Al Khusaiby SM. Kernicterus and G6PD deficiency--a case series from Oman. J Trop Pediatr. 2003;49(2):74–77. | ||
Kaplan M, Hammerman C. Glucose-6-phosphate dehydrogenase deficiency and severe neonatal hyperbilirubinemia: a complexity of interactions between genes and environment. Semin Fetal Neonatal Med. 2010;15(3):148–156. | ||
Hegyi T, Kleinfeld A, Huber A, et al. Effects of soybean lipid infusion on unbound free fatty acids and unbound bilirubin in preterm infants. J Pediatr. 2017;184:45.e1–50.e1. | ||
Riordan SM, Bittel DC, Le Pichon JB, et al. A hypothesis for using pathway genetic load analysis for understanding complex outcomes in bilirubin encephalopathy. Front Neurosci. 2016;10:376. | ||
Dennery PA, Seidman DS, Stevenson DK. Neonatal hyperbilirubinemia. N Engl J Med. 2001;344(8):581–590. | ||
Johnson L, Brown AK, Bhutani VK. BIND-a clinical score for bilirubin induced neurologic dysfunction in newborns. Pediatrics. 1999;104(3):746. | ||
Bhutani VK, Johnson LH, Shapiro SM. Kernicterus in sick and preterm infants (1999–2002): a need for an effective preventive approach. Semin Perinatol. 2004;28(5):319–325. | ||
Kaplan M, Hammerman C. Understanding severe hyperbilirubinemia and preventing kernicterus: adjuncts in the interpretation of neonatal serum bilirubin. Clin Chim Acta. 2005;356(1–2):9–21. | ||
Slusher TM, Owa JA, Painter MJ, Shapiro SM. The kernicteric facies: facial features of acute bilirubin encephalopathy. Pediatr Neurol. 2011;44(2):153–154. | ||
Newman TB, Klebanoff MA. Neonatal hyperbilirbinemia and long-term outcome: another look at the Collaborative Perinatal Project. Pediatrics. 1993;92(5):651–657. | ||
Radmacher PG, Groves FD, Owa JA, et al. A modified bilirubin-induced neurologic dysfunction (BIND-M) algorithm is useful in evaluating severity of jaundice in a resource-limited setting. BMC Pediatr. 2015;15:28. | ||
Hall JW 3rd, Smith SD, Popelka GR. Newborn hearing screening with combined otoacoustic emissions and auditory brainstem responses. J Am Acad Audiol. 2004;15(6):414–425. | ||
Cabra MA, Whitfield J. The challenge of preventing neonatal bilirubin encephalopathy: a new nursing protocol in the well newborn nursery. Proc (Bayl Univ Med Cent). 2005;18(3):217–219. | ||
Shapiro SM, Bhutani VK, Johnson L. Hyperbilirubinemia and kernicterus. Clin Perinatol. 2006;33(2):387–410. | ||
Shapiro SM, Nakamura H. Bilirubin and the auditory system. J Perinatol. 2001;21 Suppl 1:S52–S55; discussion S59–S62. | ||
Nakamura H, Takada S, Shimabuku R, Matsuo M, Matsuo T, Negishi H. Auditory nerve and brainstem responses in newborn infants with hyperbilirubinemia. Pediatrics. 1985;75(4):703–708. | ||
Rhee CK, Park HM, Jang YJ. Audiologic evaluation of neonates with severe hyperbilirubinemia using transiently evoked otoacoustic emissions and auditory brainstem responses. Laryngoscope. 1999;109(12):2005–2008. | ||
Agrawal VK, Shukla R, Misra PK, Kapoor RK, Malik GK. Brainstem auditory evoked response in newborns with hyperbilirubinemia. Indian Pediatr. 1998;35(6):513–518. | ||
Mohammadi M, Ashrafi M, Shabanian R. Auditory brainstem response in hyperbilirubinemic newborns. Med J Islam Repub Iran. 2002;16(2):63–66. | ||
Baradaranfar MH, Atighechi S, Dadgarnia MH, et al. Hearing status in neonatal hyperbilirubinemia by auditory brain stem evoked response and transient evoked otoacoustic emission. Acta Med Iran. 2011;49(2):109–112. | ||
Wisnowski JL, Panigrahy A, Painter MJ, Watchko JF. Magnetic resonance imaging of bilirubin encephalopathy: current limitations and future promise. Semin Perinatol. 2014;38(7):422–428. | ||
Katar S, Akay HO, Taskesen M, Devecioglu C. Clinical and cranial magnetic resonance imaging (MRI) findings of 21 patients with serious hyperbilirubinemia. J Child Neurol. 2008;23(4):415–417. | ||
Yilmaz Y, Alper G, Kiliçoglu G, Celik L, Karadeniz L, Yilmaz-Değirmenci S. Magnetic resonance imaging findings in patients with severe neonatal indirect hyperbilirubinemia. J Child Neurol. 2001;16(6):452–455. | ||
Tatli MM, Karadağ A, Ödemiş E, Sarraoğlu S, Yorubulut M. The role of magnetic resonance imaging in the prediction of the neurodevelopmental outcome of acute bilirubin encephalopathy in newborn. Turk J Med Sci. 2009;39(4):507–511. | ||
Trikalinos TA, Chung M, Lau J, Ip S. Systematic review of screening for bilirubin encephalopathy in neonates. Pediatrics. 2009;124(4):1162–1171. | ||
Johnson LH, Bhutani VK, Brown AK. System-based approach to management of neonatal jaundice and prevention of kernicterus. J Pediatr. 2002;140(4):396–403. | ||
Wallenstein MB, Bhutani VK. Jaundice and kernicterus in the moderately preterm infant. Clin Perinatol. 2013;40(4):679–688. | ||
Soorani-Lunsing I, Woltil HA, Hadders-Algra M. Are moderate degrees of hyperbilirubinemia in healthy term neonates really safe for the brain? Pediatr Res. 2001;50(6):701–705. | ||
Harris MC, Bernbaum JC, Polin JR, Zimmerman R, Polin RA. Developmental follow-up of breastfed term and near-term infants with marked hyperbilirubinemia. Pediatrics. 2001;107(5):1075–1080. | ||
Farouk ZL, Muhammed A, Gambo S, Mukhtar-Yola M, Umar Abdullahi S, Slusher TM. Follow-up of children with Kernicterus in Kano, Nigeria. J Trop Pediatr. Epub 2017 June 12. | ||
Deorari AK, Singh M, Ahuja GK, et al. One year outcome of babies with severe neonatal hyperbilirubinemia and reversible abnormality in brainstem auditory evoked responses. Indian Pediatr. 1994;31(8):915–921. | ||
Nwaesei CG, Van Aerde J, Boyden M, Perlman M. Changes in auditory brainstem responses in hyperbilirubinemic infants before and after exchange transfusion. Pediatrics. 1984;74(5):800–803. | ||
Hansen TW. The role of phototherapy in the crash-cart approach to extreme neonatal jaundice. Semin Perinatol. 2011;35(3):171–174. | ||
Olusanya BO, Ogunlesi TA, Slusher TM. Why is kernicterus still a major cause of death and disability in low-income and middle-income countries? Arch Dis Child. 2014;99(12):1117–1121. | ||
Ogunfowora OB, Daniel OJ. Neonatal jaundice and its management: knowledge, attitude and practice of community health workers in Nigera. BMC Public Health. 2006;6:19. | ||
Zipursky A, Bhutani VK. Impact of Rhesus disease on the global problem of bilirubin-induced neurologic dysfunction. Semin Fetal Neonatal Med. 2015;20(1):2–5. | ||
Olusanya BO, Ogunlesi TA, Kumar P, et al. Management of late-preterm and term infants with hyperbilirubinaemia in resource-constrained settings. BMC Pediatr. 2015;15:39. | ||
Imam Z, Farouk Z, Abdulkadir I, et al. Empowering mothers prevents kernicterus in Nigeria. Platform Presented at: PAS; May 1; 2016; Baltimore, MD. Presentation number 750558. | ||
Riskin A, Tamir A, Kugelman A, Hemo M, Bader D. Is visual assessment of jaundice reliable as a screening tool to detect significant neonatal hyperbilirubinemia? J Pediatr. 2008;152(6):782–787, 787.e1–e2. | ||
Barrington KJ, Sankaran K; Canadian Pediatric Society, Fetus and Newborn Committee abridged version. Guidelines for detection, management and prevention of hyperbilirubinemia in term and late preterm newborn infants. Pediatr Child Health. 2007;12(5):1–14. | ||
Bhutani VK, Johnson L, Sivieri EM. Predictive ability of a predischarge hour-specific serum bilirubin for subsequent significant hyperbilirubinemia in healthy term and near-term newborns. Pediatrics. 1999;103(1):6–14. | ||
Slusher TM, Day LT, Ogundele T, Woolfield N, Owa JA. Filtered sunlight, solar powered phototherapy and other strategies for managing neonatal jaundice in low-resource settings. Early Hum Dev. 2017;114:11–15. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.