")
Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 5
Activation of wild-type p53 by MDM2 inhibitors: a new strategy for lymphoma treatment
Authors Pujals A, Favre L, Gaulard P, Wiels J
Received 19 February 2015
Accepted for publication 14 April 2015
Published 29 July 2015 Volume 2015:5 Pages 93—100
DOI https://doi.org/10.2147/BLCTT.S60486
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor David Dingli
Anaïs Pujals,1–3,* Loëtitia Favre,4,* Philippe Gaulard,1–3 Joëlle Wiels4
1Department of Pathology, Assistance Publique-Hôpitaux de Paris, CHU Henri Mondor, 2Faculté de Médecine, Université Paris-Est Créteil, 3Inserm U955, Institut Mondor de Recherche Biomédicale, 4UMR 8126 CNRS, Institut Gustave Roussy, Université Paris-Sud, Villejuif, France
*These authors contributed equally to this work
Abstract: The tumor suppressor TP53 is frequently mutated or inactivated in human cancers. Mutations of TP53 are less common in lymphomas than in other tumors, but the protein is often inhibited by the overexpression of its main regulator – MDM2. In the past 10 years, major efforts have been made to develop drugs that can reactivate p53 and restore its functions. This review focuses on recent advances in the development of small inhibitors of MDM2, which are potentially relevant for the treatment of B- and T-cell lymphomas. We will describe the current state of development of these drugs and discuss their mechanism of action in these hematological malignancies.
Keywords: lymphoma, p53, nutlin, apoptosis, MDM2
Introduction
When p53 was discovered, it was described as an oncogenic cellular protein interacting with the large T-antigen in SV40-infected cells.1–3 Several years later, many clues indicated that p53 was not an oncogene, but rather a tumor suppressor. It was then suggested that mutations in the TP53 gene are involved in cancer development, by inactivating wild-type p53 and preventing it from exerting its tumor-suppressor function.4 A multitude of studies investigating its mechanisms of action have since been published in quick succession.
The structure of p53 can be divided into three parts: the amino-terminus, which contains a transactivation domain and binds to the main negative regulator of p53 – MDM2; the central core, which comprises the deoxyribonucleic acid (DNA)-binding domain that interacts with specific DNA sequences and induces the transcription of p53 target genes;5,6 and finally, the carboxy-terminal domain, which binds DNA in a non-sequence-specific manner. The carboxy- and amino-terminal regions undergo posttranslational modifications that modulate the stabilization of p53. However, most cancer-associated p53 mutations affect the core region, and thereby prevent DNA binding and the transcription of p53 target genes.7
P53 is a transcription factor involved in various cellular functions that maintain cellular integrity and defense against cancer. Several p53 transcriptional target genes have been identified, including important factors for cell-cycle arrest and apoptosis. For example, upon DNA damage or genotoxic stress, activated p53 prevents cell-cycle progression by promoting transcription of the p21 gene (CDKN1A).8,9 The p21WAF1/CIP1 protein is a cyclin-dependent kinase inhibitor, which inhibits G1–S and G2–M transitions, thus enabling damaged cells to be repaired or to undergo apoptosis.10 P53 also stimulates the transcription of the Bax gene, which triggers the intrinsic pathway of apoptosis,11 as well as that of PUMA12 and NOXA, which are apoptotic effectors mostly expressed during drug-induced cell death.13 In addition, effectors of the extrinsic apoptotic pathways, such as Fas/CD95,14 death receptor (DR)-4,15 and DR5,16 are targets of p53. Besides apoptosis and the cell cycle, p53 also controls other processes, such as senescence,17 autophagy,18,19 angiogenesis,20 and DNA repair.21
Given its key role in protecting cells, p53 activation is tightly regulated. Indeed, the half-life of p53 is short under normal conditions, and is regulated by numerous E3 and E4 ligases. Ubiquitination of p53 by these different ligases also regulates its nuclear export, mitochondrial translocation, and transcriptional activity. Additionally, deubiquitinating enzymes can reverse these effects and impact p53 function. This complex network of ubiquitin ligases and deubiquitinating enzymes that regulate p53 is well documented in a review by Pant and Lozano.22 Some of these enzymes, like MDM2 in particular, are essential, while others appear to have minor effects on p53 activities. MDM2 promotes the ubiquitin-mediated degradation of p53, and the loss of MDM2 leads to increased p53 stability/activity. MDM2 is also a p53 target gene; therefore, the p53–MDM2 interaction creates a negative-feedback loop that prevents the accumulation of p53, which in turn reduces MDM2 gene transcription, thus closing the loop.23,24 Both proteins interact through their N-terminus, with the interaction domains comprising the first ˜110 N-terminal amino-acid residues of MDM2 and the first 30 N-terminal amino acid residues of p53. For p53, this interaction relies on three hydrophobic residues: Phe19, Trp23, and Leu26.25 Upon stress (eg, DNA damage or oncogene activation), p53 is activated through posttranslational modifications, resulting in its stabilization. For example, phosphorylation of serine residues in the amino-terminal domain prevents MDM2 from binding to p53, allowing p53 to be stabilized and to exert its functions.
In general, TP53 is mutated (and thereby inactivated) in approximately 50% of all human cancers, but the frequencies of mutations vary greatly from one type of cancer to another.26,27 In hematological malignancies, TP53 mutations are relatively uncommon (around 10%), but wild-type p53 is often inactivated through the overexpression of MDM2.28 Therefore, restoration of the functions of p53, and specifically its ability to induce apoptosis by disrupting MDM2–p53 binding, may be a promising therapeutic strategy. In this review, we focus on the various small-molecule inhibitors of MDM2 that have been tested in the past 10 years in different hematological malignancies.
Antioncogenic activity of nutlin 3 in B- and T-cell lymphomas
Many small molecules inhibiting the interaction between p53 and MDM2 have recently been developed. The first discovered nonpeptidic compounds that selectively disrupt p53–MDM2 binding were developed by Hoffmann-La Roche Ltd (Basel, Switzerland) and are called nutlins. They belong to the 4,5-dihydroimidazole family. Among these molecules, nutlin 3a, which disrupts the p53/MDM2 complex, with a median inhibitory concentration value of 90 nM, appears the most promising. Vassilev et al showed that treatment with nutlin 3a results in the stabilization and activation of p53 in tumor cells harboring wild-type but not mutant p53.29 In this study, the activation of p53 target genes following nutlin 3a treatment led to the apoptosis of tumor cells in vitro but also in an in vivo model of mice xenografted with a human osteosarcoma cell line. Since its discovery, many studies have evaluated the efficacy of nutlin 3 in various solid or hematological malignancies.
B-cell lymphomas
Several studies have investigated the ability of nutlin 3 to induce apoptosis in mantle-cell lymphoma (MCL). In this malignancy, mutation of the TP53 gene is relatively rare, occurring in 6%–20% of cases, with a higher prevalence in blastoid variants at relapse.30 Activation of p53 by nutlin 3 is thus an attractive therapeutic strategy in MCL, which has one of the poorest prognoses among non-Hodgkin’s lymphomas. In MCL cell lines, p53 activation following treatment with nutlin 3 leads to cell-cycle arrest, growth inhibition, and apoptosis. Furthermore, the combination of nutlin 3 with a proteasome inhibitor, bortezomib, has synergistic effects on MCL growth in both wild-type and TP53-mutated MCL cells, though with higher doses needed for mutated cells.31 The cytotoxic synergy of nutlin 3 and bortezomib in TP53-mutated MCL cells results from mitochondrial apoptosis triggered by the transcription-independent upregulation of Noxa.32 Therefore, the nutlin 3–bortezomib combination may be an effective antitumor strategy in both wild-type and TP53-mutated MCL cells (Table 1). Interestingly, Drakos et al also demonstrated that in MCL, the activation of p53 through nutlin 3 is associated with the inhibition of the AKT/mTOR oncogenic pathway and leads to cell-cycle arrest and apoptosis.33
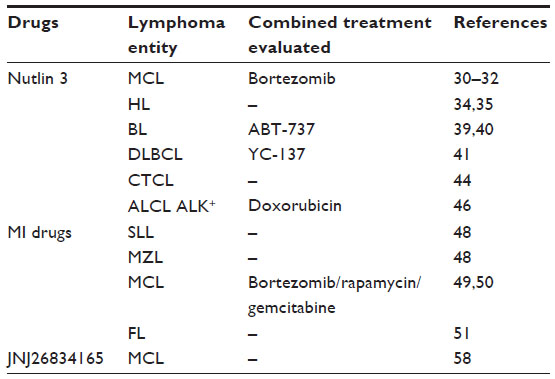 | Table 1 Small molecules used for targeting p53 in lymphomas |
Mutations of TP53 are also uncommon (11%) in Reed–Sternberg cells,34 suggesting that nutlin 3 is a valuable therapy for patients with classical Hodgkin’s lymphoma. Janz et al demonstrated that this compound induces p53-dependent apoptosis in Hodgkin’s lymphoma cell lines, and that combined treatment with nutlin 3 and commonly used cytotoxic drugs leads to a stronger apoptotic response than any comparable single drug used alone (Table 1).35 These results were confirmed by another team, which reported that the stabilization of p53 by nutlin 3 leads to cell-cycle arrest at the G1/S checkpoint and the induction of apoptosis through the expression of Bax or impaired expression of the antiapoptotic protein Bcl-2.36
Similar studies have been performed in Burkitt’s lymphoma (BL), a B-cell lymphoma frequently associated with Epstein–Barr virus (EBV). In addition to the deregulation of c-Myc, which is essential for the pathogenesis of BL, disruption of the p53 pathway also contributes to the development of this disease. Inactivating TP53 mutations are frequently (up to 70%) found in BL cell lines and in at least 30% of BL biopsies.37 Our team and others investigated the status of TP53 and MDM2 in BL cell lines or biopsy samples, and found that in BL cells harboring a wild-type TP53 gene, the function of p53 protein is compromised by the overexpression of MDM2.38,39 We thus evaluated the efficacy of nutlin 3 treatment in a subset of TP53 wild-type BL cell lines associated or not with EBV. Irrespective of EBV status, treatment of TP53 wild-type BL cell lines with nutlin 3 resulted in p53 activation. However, while EBV– BL cell lines treated with nutlin 3 were highly susceptible to apoptosis, EBV+ cell lines expressing all the latent proteins of the virus (type III latency) were much more resistant.40 We found that Bcl-2 was overexpressed in these type III latency EBV+ cell lines and interacted with Bax, preventing its activation. Treatment with a Bcl-xL/Bcl-2 inhibitor (ABT-737) disrupted the Bax/Bcl-2 interaction, thus enabling Bax activation and the induction of apoptosis by nutlin 3 (Table 1).41 Drakos et al also evaluated the efficacy of nutlin 3 in other B-cell malignancies associated with Bcl-2 overexpression. They demonstrated that this compound causes cell-cycle arrest and apoptosis in a subset of diffuse large B-cell lymphomas (DLBCLs) harboring the t(14;18) translocation, a molecular abnormality that leads to Bcl-2 overexpression. In these cell lines, nutlin 3 promotes the accumulation of the proapoptotic proteins Bax and Puma, but also renders TP53 wild-type DLBCL cells sensitive to the activation of the intrinsic apoptotic pathway by a Bcl-2 inhibitor (YC-137) (Table 1). Furthermore, nutlin 3 also inhibits the growth of tumors in mice xenografted with t(14;18)-DLBCL containing wild-type TP53.42
T-cell lymphomas
Cutaneous T-cell lymphoma (CTCLs) are a heterogeneous group of TCLs including mycosis fungoides, Sézary syndrome, and CD30+ lymphoproliferative disorders. TP53 mutations occur rarely in CTCL, with recent studies estimating mutation rates of 2% and 5% for mycosis fungoides and CD30+ lymphoproliferative disease, respectively.43,44 The efficacy of nutlin 3 has been tested in these lymphoproliferative disorders. Manfé et al showed that nutlin 3 treatment led to the accumulation of p53 and subsequent apoptosis and senescence in several TP53 wild-type CTCL cell lines,45 whereas TP53-mutated CTCL cell lines were not sensitive to nutlin 3. Their data also indicated that the induction of apoptosis and senescence following nutlin 3 treatment does not depend on the transcriptional activity of p53.
Reactivation of wild-type p53 by nutlin 3 treatment was also assessed in ALK+ anaplastic large-cell lymphoma (ALCL), a T-cell lymphoma characterized by translocations or inversions involving the ALK gene. Most ALK+ ALCL tumors carry a wild-type TP53 gene.46 Nutlin 3 inhibits the growth of these cells by upregulating p21 and induces apoptosis, in part through the upregulation of the proapoptotic members of the Bcl-2 protein family, Bax and Puma, but also by the activation of the extrinsic apoptotic pathway. The activation of the extrinsic pathway is caused by the upregulation of death-domain receptors like DR5 or TRAIL, which are p53 target genes, and the downregulation of the antiapoptotic molecule c-FlipS/L. Interestingly, nutlin 3 and the chemotherapeutic agent doxorubicin have a synergistic antitumor effect on p53 wild-type ALK+ ALCL cell lines (Table 1). This combined treatment was also tested in TP53-mutated cell lines, and resulted in caspase 3 activation associated with high levels of p73α expression.47
Antioncogenic activity of spiro-oxindoles in B-cell lymphoma
Shangary et al used a de novo structure-based approach to design spiro-oxindoles, which are a new class of inhibitors of the MDM2–p53 interaction.48 This class of molecules includes three compounds – MI-63, MI-219, and MI-319 – whose antioncogenic activity has been studied in several types of tumor. These molecules bind MDM2 with high affinity, which leads to the activation of the p53 pathway and subsequent inhibition of cell growth in cancer cell lines expressing wild-type TP53. MI-63 is considered unsuitable for in vivo studies because of its poor pharmacokinetic profile. However, MI-219 is an orally available compound that exhibits good pharmacokinetic and pharmacodynamics properties (oral bioavailability of 65% in rats compared to 10% for MI-63).49 In vitro and in vivo results obtained by Shangary et al show that MI-219 activates p53 and induces cell-cycle arrest in both normal and tumoral cells, but apoptosis only occurs in the latter.
The efficacy of MI-219 has been evaluated with in vitro and ex vivo experiments and compared to that of nutlin 3 in a subset of B-cell lymphomas (small lymphocytic lymphomas and marginal zone lymphoma primary cells).50 These experiments demonstrate that MI-219 significantly impairs the viability of TP53 wild-type lymphoma cells more effectively than nutlin 3 (Table 1). Experiments carried out in lymphoma cell lines indicate that this difference may be explained by the strong ability of MI-219 to induce autoubiquitination and degradation of MDM2, which correlates with the robust upregulation of p53 and subsequent transcription of some p53-dependent apoptotic genes.
In vitro and in vivo experiments have also been performed to test MI-63 in MCL cell lines. Jones et al showed that MI-63 inhibits cell proliferation by inducing both cell-cycle arrest and apoptosis in a p53-dependent manner.51 Treatment of cells with MI-63 promoted the expression of p53 target genes like P21 and PUMA, stimulated caspase 3 and 8 activities, and led to p53 phosphorylation on serine 15, 37, and 39, which coincided with DNA damage. This study also assessed the ability of MI-63 to induce apoptosis in primary cells derived from patients with blastoid MCL. MI-63 treatment resulted in the accumulation of p53 in these cells, stimulated p21 expression, and inhibited tumor-cell growth. The combination of MI-63 and others drugs (bortezomib or rapamycin) showed synergistic antiproliferative effects (Table 1). In another study, the same team evaluated the efficacy of MI-63 combined with gemcitabine, an anticancer agent that impairs DNA replication through the inhibition of ribonucleotide reductase.52 These drugs showed a synergistic effect both in MCL cell lines and primary MCL cells. They then used immunodeficient mice xenografted with MCL cell lines to evaluate the effectiveness of this combination in vivo. Mice were treated with MI-219, the in vivo formulation of MI-63 and gemcitabine. Each drug alone slowed tumor growth in this mouse model, and low doses of each compound in combination showed a cooperative effect. No side effects were observed in this study when these two drugs were used alone or in combination.
The efficacy of MI-219, MI-319, and nutlin 3 has also been tested in follicular small-cleaved B-cell lymphoma (FSCCL). This analysis was performed in vitro using cell lines or primary lymphoma cells, and in vivo, using an FSCCL systemic xenograft model.53 In this study, these three drugs induced p53 target-gene expression and limited cell growth in a p53-dependent manner, with similar efficiency in all cell lines tested, although the affinity of MI-319 for MDM2 protein was slightly higher than that of MI-219 and nutlin 3. MI-319 was the only drug tested in vivo. Mice inoculated with FSCCL and treated with the drug survived longer than control mice.
Other small molecules targeting MDM2
One factor limiting the efficacy of nutlin and the MI class of drugs is the high expression of MDMX, another negative regulator of p53. MDM2 and MDMX cooperate to regulate p53, mostly because MDMX activates the MDM2-dependent degradation of p53. Although the MDM2 and MDMX genes are highly homologous, there are structural differences between the p53-binding pocket of the two proteins, which explains the poor affinity of nutlin 3 and MI drugs for MDMX. MDMX is overexpressed in many cancers, which can limit their sensitivity to MDM2 inhibitors. One study evaluating the effect of MDMX on the activation of p53 in various tumors cells showed that MDMX modulates the biological response of several BL cell lines to nutlin 3.54 In nutlin 3-sensitive BL cells, an inhibition of MDMX expression was observed after treatment, whereas in BL cells that were resistant to nutlin 3, MDMX levels were unaffected. These results suggest that in cell lines where no MDMX degradation occurred, nutlin 3 failed to induce apoptosis. This study also demonstrated that the inhibition of MDMX expression by short hairpin ribonucleic acid sensitizes MCF7 breast cancer cells to nutlin 3-induced apoptosis. Altogether, these data indicate that MDMX is an important determinant of the outcome of p53 activation. In another study performed in the Eμ-Myc mouse model of lymphoma, Garcia et al showed that restoration of p53 in lymphoma-bearing mice leads to longer overall survival in mice lacking MDMX than in control mice expressing MDMX.55 Recent effort has been made to develop dual MDM2/MDMX antagonists in light of these observations. Graves et al identified small molecules that block p53 binding to both MDM2 and MDMX. These compounds restore p53 activity and offer a therapeutic option to overcome the resistance of MDMX-overexpressing cancer cells to MDM2 antagonists.56
Another class of molecules developed to promote p53 activation in cancer cells are inhibitors of the E3 ubiquitin ligase activity of MDM2. One such molecule, JNJ-26854165 (serdemetan), a tryptamine derivative developed by Johnson & Johnson, has demonstrated preclinical efficacy in the treatment of acute myeloid and lymphoid leukemias57,58 and in various solid tumors.59 Jones et al also evaluated this molecule in models of MCL and multiple myelomas.60 They found that serdemetan inhibits the proliferation of TP53 wild-type and -mutant cell lines in a dose-dependent manner, as well as that of TP53–/– and MDM2–/– murine embryonic fibroblasts (albeit with lower efficiency), suggesting that serdemetan does not act specifically as a p53/MDM2 inhibitor (Table 1). They next investigated the mechanism of action, and showed that serdemetan inhibits cholesterol transport in MCL and multiple myelomas, and that this effect is related to the induction of cell death.
Clinical trials with MDM2 inhibitors in hematological malignancies
A large number of MDM2 inhibitors have already progressed to advanced preclinical development or early phase clinical trials (Table 2). Structure-based optimization of nutlin 3 has been done to generate RG7112 (RO5045337; Hoffmann-La Roche Ltd), which is currently under testing in clinical trials for various types of cancer (ClinicalTrials.gov identifiers NCT01164033, NCT01143740, NCT00623870, and NCT00559533). RG7112 is resistant to oxidation, which had been a problem with earlier drugs, has a reduced molecular weight, decreased metabolic liability, and binds to MDM2 with an affinity of 11 nM compared to 90 nM for nutlin 3.49 Therefore, RG7112 is three times more potent than nutlin 3 in inhibition of cell growth. The first published results of these trials come from a proof-of-mechanism study performed in patients with amplified MDM2 liposarcoma. The analysis of biopsy specimens revealed that treatment with RG7112 promoted the accumulation of p53 and p21 proteins (assessed by immunohistochemistry), as well as that of MDM2 messenger ribonucleic acid, and inhibited cellular proliferation (assessed by Ki67 labeling).61 Furthermore, there was a significant correlation between levels of MIC-1, RG7112 exposure, and tumor-cell apoptosis, suggesting that MIC-1 is a potentially valuable pharmacodynamic biomarker. Among the 20 patients enrolled, one experienced a partial response, 14 had stable disease, and five showed disease progression. However, there was a high rate of adverse reactions, including thrombocytopenia and neutropenia. A multicenter Phase I study was also performed for patients with relapsed/refractory leukemia or small-cell lymphocytic lymphomas who were treated with increasing doses of RG7112. Activation of the p53 pathway in patients treated with RG7112 was demonstrated by an increase in serum levels of MIC-1 and the induction of p53 target genes, including MDM2, P21, and PUMA. This study shows that RG7112 has single-agent clinical activity, since it significantly lowers blast counts and even leads to complete remission in some AML patients (16%).62,63 The efficacy of RO5503781, another MDM2 inhibitor developed by Hoffmann-La Roche Ltd, is currently being tested in a Phase I/IB trial as a single agent or in combination with cytarabine in patients with AML (NCT01773408). This molecule is also under evaluation in another multicenter study designed to assess the effect of posaconazole (an antifungal drug) and of food on the pharmacokinetics of RO5503781 and to test the bioavailability of new formulations in patients with solid tumors (NCT01901172). The p53 activator MI-773 developed by Sanofi SA (Paris, France; also known as SAR405838) is also currently being evaluated in a Phase I safety study in patients with liposarcoma or advanced solid tumors (NCT01636479).
 | Table 2 Inhibitors of MDM2 currently in preclinical and clinical trials |
Three more MDM2 inhibitors have recently entered clinical trials: DS-3032b (Daiichi Sankyo Company Ltd, Tokyo, Japan), which is being tested in a Phase I ascending study in patients with advanced solid tumors or lymphomas (NCT01877382); CGM097 (Novartis International AG, Basel, Switzerland), which has been under evaluation since 2013 in a Phase I escalation study in patients with advanced solid tumors (NCT01760525); and finally SCH 900242 (MK-8242; Merck & Co Inc., Whitehouse Station, NJ, USA), which is being tested in two Phase I clinical trials as a single agent in patients with advanced solid tumors (NCT01463696) or in combination with cytarabine in patients with AML (NCT01451437). The results of all these Phase I clinical trial are not yet published.
Conclusion
Since its discovery in 1979, p53 has been the subject of numerous studies, and the mechanisms involved in its tumor-suppressor functions are now well understood. However, the translation of fundamental research carried out on p53 into clinical practice remains challenging. In the past 10 years, major efforts have been made to develop small molecules able to correct a dysfunctional p53-regulatory pathway. Many MDM2 inhibitors, which block p53–MDM2 interactions, have been synthesized and tested in a large panel of tumors, including lymphomas. Results obtained in vitro with nutlins or MI drugs on various types of B- and T-cell lymphomas are promising, and clinical trials are ongoing to test these inhibitors in different hematological malignancies. However, some important issues remain to be addressed to improve the benefit of these drugs for patients. For example, biomarkers that can accurately predict the response of patients to MDM2 inhibitors await discovery. An understanding of the mechanisms of intrinsic or acquired resistance to these drugs should also lead to better patient management. Various mutations, occurring in the DNA-binding domain of p53, have already been shown to confer resistance to MDM2 inhibitors. Furthermore, a mutation that abrogates p53 transcriptional activity has been observed in cell lines after prolonged exposure to nutlin 3 treatment.64,65 These data indicate that seeking the TP53 status in cohorts of patients before inclusion in clinical trials and after relapse could help strategize treatment plans for patients and could improve our understanding about these resistance-conferring mutations.
Many studies have demonstrated that MDM2 inhibitors may be used either as a single agent or in combination with other anticancer drugs. These combined approaches seem to be successful, and constitute promising treatments for patients with lymphomas that retain wild-type TP53.
Disclosure
The authors report no conflicts of interest in this work.
References
Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–263. | |
Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol. 1979;31:472–483. | |
Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52. | |
Baker SJ, Fearon ER, Nigro JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–221. | |
Bargonetti J, Friedman PN, Kern SE, Vogelstein B, Prives C. Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell. 1991;65:1083–1091. | |
Kern SE, Kinzler KW, Bruskin A, et al. Identification of p53 as a sequence-specific DNA-binding protein. Science. 1991;252:1708–1711. | |
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. | |
Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249:912–915. | |
el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. | |
Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci U S A. 1992;89:4495–4499. | |
Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. | |
Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. | |
Villunger A, Michalak EM, Coultas L, et al. p53- and Drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science. 2003;302:1036–1038. | |
Müller M, Wilder S, Bannasch D, et al. p53 Activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 1998;188:2033–2045. | |
Liu X, Yue P, Khuri FR, Sun SY. p53 Upregulates death receptor 4 expression through an intronic p53 binding site. Cancer Res. 2004;64:5078–5083. | |
Wu GS, Burns TF, McDonald ER 3rd, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17:141–143. | |
Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–884. | |
Rosenfeldt MT, O’Prey J, Morton JP, et al. p53 Status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. | |
Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–185. | |
Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265:1582–1584. | |
Sengupta S, Harris CC. p53: Traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol. 2005;6:44–55. | |
Pant V, Lozano G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev. 2014;28:1739–1751. | |
Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–468. | |
Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–1132. | |
Kussie PH, Gorina S, Marechal V, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. | |
Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. | |
Leroy B, Fournier JL, Ishioka C, et al. The TP53 website: an integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 2013;41:D962–D969. | |
Watanabe T, Ichikawa A, Saito H, Hotta T. Overexpression of the MDM2 oncogene in leukemia and lymphoma. Leuk Lymphoma. 1996;21:391–397. | |
Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. | |
Greiner TC, Moynihan MJ, Chan WC, et al. p53 Mutations in mantle cell lymphoma are associated with variant cytology and predict a poor prognosis. Blood. 1996;87:4302–4310. | |
Tabe Y, Sebasigari D, Jin L, et al. MDM2 antagonist nutlin-3 displays antiproliferative and proapoptotic activity in mantle cell lymphoma. Clin Cancer Res. 2009;15:933–942. | |
Jin L, Tabe Y, Kojima K, et al. MDM2 antagonist nutlin-3 enhances bortezomib-mediated mitochondrial apoptosis in TP53-mutated mantle cell lymphoma. Cancer Lett. 2010;299:161–170. | |
Drakos E, Atsaves V, Li J, et al. Stabilization and activation of p53 downregulates mTOR signaling through AMPK in mantle cell lymphoma. Leukemia. 2009;23:784–790. | |
Montesinos-Rongen M, Roers A, Küppers R, Rajewsky K, Hansmann ML. Mutation of the p53 gene is not a typical feature of Hodgkin and Reed-Sternberg cells in Hodgkin’s disease. Blood. 1999;94:1755–1760. | |
Janz M, Stühmer T, Vassilev LT, Bargou RC. Pharmacologic activation of p53-dependent and p53-independent apoptotic pathways in Hodgkin/Reed-Sternberg cells. Leukemia. 2007;21:772–779. | |
Drakos E, Thomaides A, Medeiros LJ, et al. Inhibition of p53-murine double minute 2 interaction by nutlin-3A stabilizes p53 and induces cell cycle arrest and apoptosis in Hodgkin lymphoma. Clin Cancer Res. 2007;13:3380–3387. | |
Gaidano G, Ballerini P, Gong JZ, et al. p53 Mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 1991;88:5413–5417. | |
Capoulade C, Bressac-de Paillerets B, Lefrère I, et al. Overexpression of MDM2, due to enhanced translation, results in inactivation of wild-type p53 in Burkitt’s lymphoma cells. Oncogene. 1998;16:1603–1610. | |
Wilda M, Bruch J, Harder L, et al. Inactivation of the ARF-MDM-2-p53 pathway in sporadic Burkitt’s lymphoma in children. Leukemia. 2004;18:584–588. | |
Renouf B, Hollville E, Pujals A, Tétaud C, Garibal J, Wiels J. Activation of p53 by MDM2 antagonists has differential apoptotic effects on Epstein-Barr virus (EBV)-positive and EBV-negative Burkitt’s lymphoma cells. Leukemia. 2009;23:1557–1563. | |
Pujals A, et al. Treatment with a BH3 mimetic overcomes the resistance of latency III EBV (+) cells to p53-mediated apoptosis. Cell Death Dis. 2011;2:e184. | |
Drakos E, Singh RR, Rassidakis GZ, et al. Activation of the p53 pathway by the MDM2 inhibitor nutlin-3a overcomes BCL2 overexpression in a preclinical model of diffuse large B-cell lymphoma associated with t(14;18)(q32;q21). Leukemia. 2011;25:856–867. | |
Dereure O, Levi E, Vonderheid EC, Kadin ME. Infrequent Fas mutations but no Bax or p53 mutations in early mycosis fungoides: a possible mechanism for the accumulation of malignant T lymphocytes in the skin. J Invest Dermatol. 2002;118:949–956. | |
Kapur S, Menke MA, Tiemann M, Schubert C, Parwaresch R. Early mycosis fungoides: molecular analysis for its diagnosis and the absence of p53 gene mutations in cases with progression. J Dermatol Sci. 2001;26:36–45. | |
Manfé V, Biskup E, Johansen P, et al. MDM2 inhibitor nutlin-3a induces apoptosis and senescence in cutaneous T-cell lymphoma: role of p53. J Invest Dermatol. 2012;132:1487–1496. | |
Rassidakis GZ, Thomaides A, Wang S, et al. p53 Gene mutations are uncommon but p53 is commonly expressed in anaplastic large-cell lymphoma. Leukemia. 2005;19:1663–1669. | |
Drakos E, Atsaves V, Schlette E, et al. The therapeutic potential of p53 reactivation by nutlin-3a in ALK+ anaplastic large cell lymphoma with wild-type or mutated p53. Leukemia. 2009;23:2290–2299. | |
Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A. 2008;105:3933–3938. | |
Zhao Y, Bernard D, Wang S. Small molecule inhibitors of MDM2-p53 and MDMX-p53 interaction as new cancer therapeutics. BioDiscovery. 2013;4:1–15. | |
Sosin AM, Burger AM, Siddiqi A, Abrams J, Mohammad RM, Al-Katib AM. HDM2 antagonist MI-219 (spiro-oxindole), but not nutlin-3 (cis-imidazoline), regulates p53 through enhanced HDM2 autoubiquitination and degradation in human malignant B-cell lymphomas. J Hematol Oncol. 2012;5:57. | |
Jones RJ, Chen Q, Voorhees PM, et al. Inhibition of the p53 E3 ligase HDM-2 induces apoptosis and DNA damage – independent p53 phosphorylation in mantle cell lymphoma. Clin Cancer Res. 2008;14:5416–5425. | |
Jones RJ, Baladandayuthapani V, Neelapu S, et al. HDM-2 inhibition suppresses expression of ribonucleotide reductase subunit M2, and synergistically enhances gemcitabine-induced cytotoxicity in mantle cell lymphoma. Blood. 2011;118:4140–4149. | |
Mohammad RM, Wu J, Azmi AS, et al. An MDM2 antagonist (MI-319) restores p53 functions and increases the life span of orally treated follicular lymphoma bearing animals. Mol Cancer. 2009;8:115. | |
Wade M, Wong ET, Tang M, Stommel JM, Wahl GM. Hdmx modulates the outcome of p53 activation in human tumor cells. J Biol Chem. 2006;281:33036–33044. | |
Garcia D, Warr MR, Martins CP, Brown Swigart L, Passegué E, Evan GI. Validation of MdmX as a therapeutic target for reactivating p53 in tumors. Genes Dev. 2011;25:1746–1757. | |
Graves B, Thompson T, Xia M, et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc Natl Acad Sci U S A. 2012;109:11788–11793. | |
Kojima K, Burks JK, Arts J, Andreeff M. The novel tryptamine derivative JNJ-26854165 induces wild-type p53- and E2F1-mediated apoptosis in acute myeloid and lymphoid leukemias. Mol Cancer Ther. 2010;9:2545–2557. | |
Smith MA, Gorlick R, Kolb EA, et al. Initial testing of JNJ-26854165 (Serdemetan) by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;59:329–332. | |
Chargari C, Leteur C, Angevin E, et al. Preclinical assessment of JNJ-26854165 (Serdemetan), a novel tryptamine compound with radiosensitizing activity in vitro and in tumor xenografts. Cancer Lett. 2011;312:209–218. | |
Jones RJ, Gu D, Bjorklund CC, et al. The novel anticancer agent JNJ-26854165 induces cell death through inhibition of cholesterol transport and degradation of ABCA1. J Pharmacol Exp Ther. 2013;346:381–392. | |
Ray-Coquard I, Blay JY, Italiano A, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13:1133–1140. | |
Andreeff M, Kojima K, Padmanabhan S, et al. A Multi-Center, Open-Label, Phase I Study of Single Agent RG7112, A First In Class p53-MDM2 Antagonist, In Patients with Relapsed/Refractory Acute Myeloid and Lymphoid Leukemias (AML/ALL) and Refractory Chronic Lymphocytic Leukemia/Small Cell Lymphocytic Lymphomas (CLL/SCLL). American Society of Hematology. 2010. Abstract. | |
Andreeff M, Kelly KR, Yee K, et al. Results of the Phase 1 Trial of RG7112, a Small-Molecule MDM2 Antagonist, in Acute Leukemia. American Society of Hematology. 2012. Abstract. | |
Aziz MH, Shen H, Maki CG. Acquisition of p53 mutations in response to the non-genotoxic p53 activator nutlin-3. Oncogene. 2011;30:4678–4686. | |
Michaelis M, Rothweiler F, Barth S, et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011;2:e243. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.