")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Activation of the P38/CREB/MMP13 axis is associated with osteoarthritis
Authors Ji B, Ma Y, Wang H, Fang X, Shi P
Received 22 March 2019
Accepted for publication 4 June 2019
Published 3 July 2019 Volume 2019:13 Pages 2195—2204
DOI https://doi.org/10.2147/DDDT.S209626
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Bin Ji,*,1,2 Yan Ma,*,1 Haimin Wang,*,1,3 Xiangqian Fang,1 Peihua Shi1
1Department of Orthopaedic Surgery, Sir Run Run Shaw Hospital, Medical College of Zhejiang University, Key Laboratory of Musculoskeletal System Degeneration and Regeneration Translational Research of Zhejiang Province, Hangzhou, Zhejiang Province 310016, People’s Republic of China; 2Department of Orthopaedic Surgery, First Affiliated Hospital of Jiaxing University, Jiaxing, Zhejiang Province 314000, People’s Republic of China; 3Orthopedics Department, Taizhou Bo Ai Hospital, Taizhou, Zhejiang Province 318050, People’s Republic of China
*These authors contributed equally to this work
Purposes: Osteoarthritis (OA) is a common joint disease characterized by the degradation of articular cartilage and joint inflammation. Interleukin-1ß induces P38/cAMP response element binding protein (CREB) pathway activation, resulting in increased expression of matrix metallopeptidase-13 (MMP13) in chondrocytes. However, the role of the P38/CREB/MMP13 axis is unclear in the progression of OA. In this study, we aimed to answer the following questions: (1) how does the P38/CREB/MMP13 axis in cartilage from patients with OA compare with control specimens? (2) Can the P38 agonist anisomycin (ANS) induce mouse OA?
Materials and methods: Surgical specimens of human cartilage were divided into OA and control groups. Surgical specimens of mouse cartilage were divided into control and ANS-induced groups. Safranin O staining of the cartilage tissues was performed to evaluate the extracellular matrix. Reverse transcription-polymerase chain reaction was performed using these tissues to investigate messenger RNA expressions of type II collagen, aggrecan, MMP13, and ADAM metallopeptidase with thrombospondin type 1 motif 5. Phosphorylated (p)-P38, p-CREB, and MMP13 were evaluated by Western blot analysis. Anisomycin was used to activate P38, and p-P38, p-CREB, and MMP13 were evaluated by immunofluorescence and Western blot analysis.
Results: Safranin O staining showed that the extracellular matrix degraded in humans with OA and ANS-induced mouse cartilage samples. The expressions of p-P38, p-CREB, and MMP13 were all upregulated in osteoarthritic cartilage or anisomycin-induced chondrocytes, suggesting that the P38/CREB/MMP13 axis may play a role in the progression of OA.
Conclusions: The P38/CREB/MMP13 axis is active in osteoarthritic chondrocytes and may cause the degeneration of cartilage. Effective new therapy directed against this pathway could be developed.
Keywords: degenerative joint disease, CREB, anisomycin, mice
Introduction
Osteoarthritis (OA) is a degenerative joint disease characterized by degradation of articular cartilage and joint inflammation. A third of the worldwide population aged more than 50 years have OA.1 Oral or topical nonsteroidal anti-inflammatory drugs can relieve joint pain and stiffness.2 However, joint replacement surgery is still the only effective treatment for terminal OA. Thus, it is urgent to research the underlying molecular pathogenesis mechanisms and explore new strategies for treating OA.
Aging, genetic predisposition, and mechanical injury are the primary causes of OA.3,4 As the only cell type in cartilage, chondrocytes regulate the balance between synthesis and degradation of extracellular matrix remodeling.5 Matrix metalloproteinases (MMPs) are considered the most important catabolic enzymes associated with digestion of collagen fibers and proteoglycans.6 In addition, pro-inflammatory cytokines, including interleukin (IL)-1ß, tumor necrosis factor (TNF)-α, and IL-6, are able to stimulate the expression of MMPs during the development of OA.7,8 Among the MMPs, the expression of MMP13 is more restricted to connective tissue.9 It targets type II collagen in cartilage for degradation and degrades proteoglycan, types IV and IX collagen, osteonectin, and perlecan in cartilage.10 Additionally, the expression of MMP13 was upregulated in cartilage obtained from the femoral heads of patients with OA.11 Studies have reported that IL-1ß induced the P38/cAMP response element binding protein (CREB) pathway activation, resulting in increased expression of MMP13 in chondrocytes.12 CREB is a transcription factor that binds to certain DNA sequences called cAMP response elements and thereby increases or decreases the transcription, and thus the expression of certain genes. CREB recruitment following a specific CpG demethylation leads to the increased expression of MMP13 in chondrocytes.13 However, the role of the P38/CREB pathway is unclear in the progression of OA.
Therefore, we aimed to answer the following questions: (1) how does the P38/CREB/MMP13 axis in cartilage from patients with OA compare with control specimens? (2) As a p38 agonist, can anisomycin (ANS) stimulate p38 phosphorylation in various cells and induce mouse OA?
Materials and methods
Human cartilage and mouse model
Human cartilage samples were collected according to protocols approved by the Ethics Committee of Sir Run Run Shaw Hospital, and the methods were performed in accordance with the approved guidelines. Written informed consent was obtained from all subjects. Control cartilage was harvested from patients of the fractured femoral neck without a history of OA at the time of surgery for total hip replacement (n=20), and the pathological cartilage was acquired from patients with end-stage symptomatic hip OA at the time of surgery for total hip replacement (n=10). All the samples came from patients without autoimmune, gouty, and infectious arthritis. Human cartilage samples were obtained from the weight-bearing area of the head of the femur. According to the Outerbridge classification, control samples were grade 1, and OA samples were grade 3 or 4. Clinical characteristics of the patients were collected, as shown in Table 1.
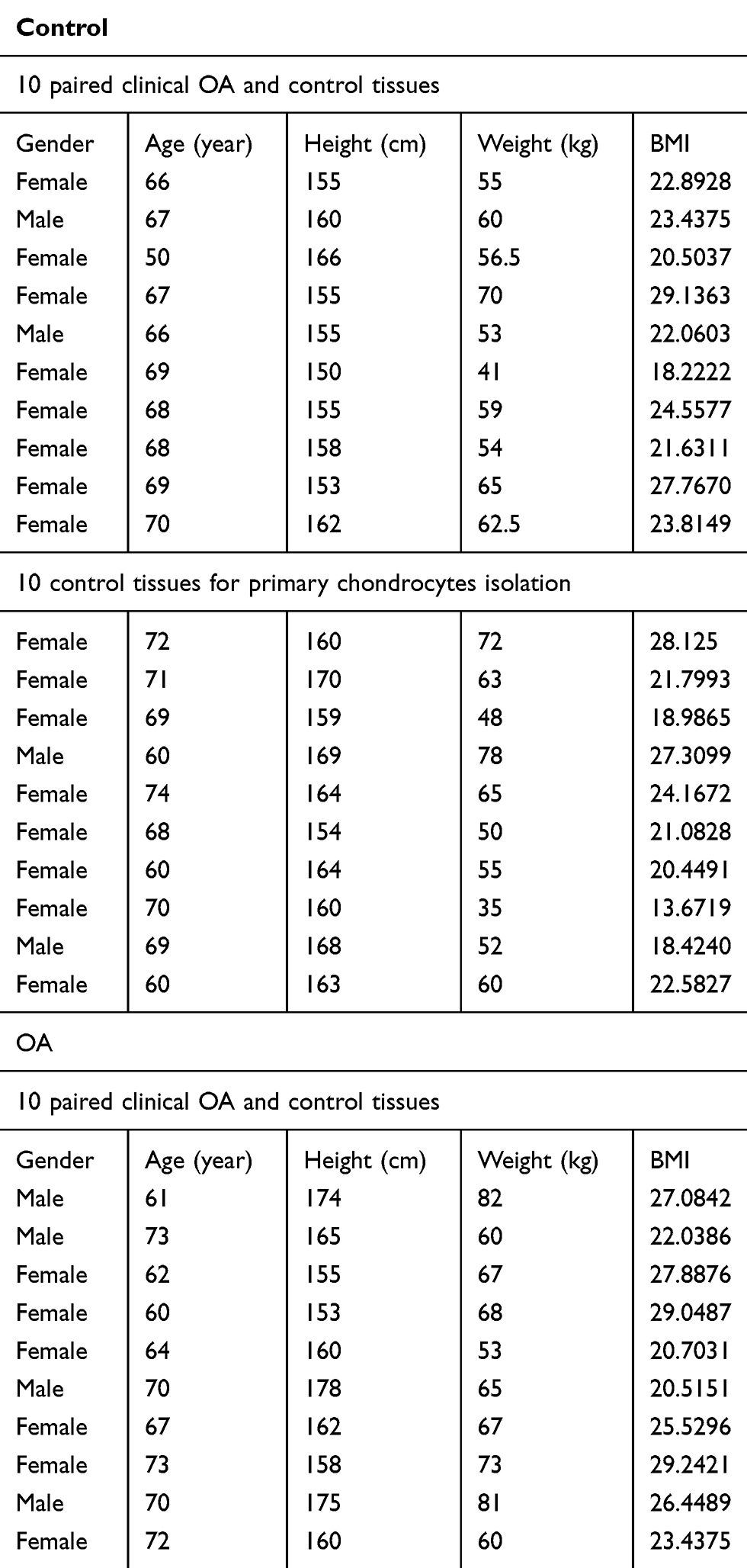 |
Table 1 Clinical characteristics of the patients |
Animal handling and experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and following approval from the Institute of Health Sciences Institutional Animal Care and Use Committee. Briefly, 30 healthy, 8-week-old male C57BL/J6 mouse were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China), and mice were divided into the control (CON) and ANS groups (n=15 per group). Mice in the ANS group received an intra-articular injection of 10 µl (100 µM) of ANS (Sigma-Aldrich, St. Louis, MO, USA) twice per week for 6 weeks. Mice in the CON group received an injection of phosphate-buffered saline (PBS) twice per week for 6 weeks. They were housed in an animal facility under controlled temperature (22–24 °C) and humidity (50–60%) conditions and a 12 hr light/dark cycle with free access to food and water. Once weekly, a professional animal keeper cleaned and equipped the home cages with new bedding. Mice were euthanized with carbon dioxide if they reached the humane end point, which was based on 20% reduction in body weight.
Histological analysis and immunohistochemistry
Human cartilage samples were obtained from the weight-bearing area of the head of the femur. Mice cartilage samples were obtained from the entire knee. Cartilage tissue specimens were fixed in 4% paraformaldehyde for paraffin embedding. Each paraffin-embedded cartilage sample was sectioned at 5 μm, and every tenth section was stained with 0.1% safranin O solution and 0.001% Fast Green solution (Sigma-Aldrich). Histological images were acquired by using the Nikon Biological Microscope Ni-U (Nikon Instruments, Inc., Melville, NY, USA) and analyzed by Image J software (National Institute of Health, Bethesda, MD, USA). Cartilage destruction of mice knee joints was scored by 2 observers who were blinded to group-identifying information using the Osteoarthritis Research Society International (OARSI) grading system.14 For immunohistochemistry, the sections were incubated at 4 °C with antibodies for HOXA1 (Abcam, Cambridge, MA, USA) overnight and for 2 hrs at room temperature with secondary antibodies (Beyotime Institute of Biotechnology, Inc., Jiangsu, China). The number of positively stained cells on the entire articular surface (including the femoral condyle and tibial plateau area) per specimen was counted, and the percentage of positive cells was calculated.15
Culturing of chondrocytes
Chondrocytes were isolated from human and mouse articular cartilage tissues as described previously.16 Chondrocytes were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (Gibco, Grand Island, NY, USA) for 24 hrs at 37 °C. The cells were filtered through a 0.075-mm cell strainer and washed before culturing or microRNA/messenger (m)-RNA isolation with sterile PBS. Primary chondrocytes at 80% confluence were used for the experiments. During the culture period, cells were incubated at 37 °C in a humidified atmosphere of 5% carbon dioxide and 95% air. Human and mice chondrocytes were grown on a 20-mm glass-bottom cell culture dish (801001; Nest Biotechnology Co., Ltd., Shanghai, China) with or without being treated with ANS (10 µM) for 24 hrs for immunofluorescence (5×105 cells/dish). Additionally, they were grown on 20-mm cell culture dish with or without being treated with ANS (10 µM) for 24 hrs for protein collection (1×106 cells/dish).
RNA extraction and quantitative real-time polymerase chain reaction analysis
OA and control cartilage tissues were grinded with liquid nitrogen, and total RNA was extracted using TRIzol reagent (Invitrogen Corp., Carlsbad, CA, USA), according to the manufacturer’s instructions. RNA was stored at −80 °C. Reverse transcription was performed using 1.0 µg of the total RNA and HiFiScript complementary (c)-DNA Kit (CWBIO, Beijing, China). Amplification reactions were set using 20-µl reaction volumes containing amplification primers and UltraSYBR Mixture with ROX (CWBIO) and detected by the ABI 7500 Sequencing Detection System (Applied Biosystems, Foster City, CA, USA). A 1-µl volume of cDNA and 1-µl volume of primer (Sangon Biotech, Shanghai, China) were used in each amplification reaction. The following cycling conditions were used: 40 cycles of denaturation at 95 °C for 5 seconds and amplification at 60 °C for 24 seconds. All reactions were run in triplicate and normalized to the mRNA house-keeping gene β-actin. The primer sequences are shown in Table 2.
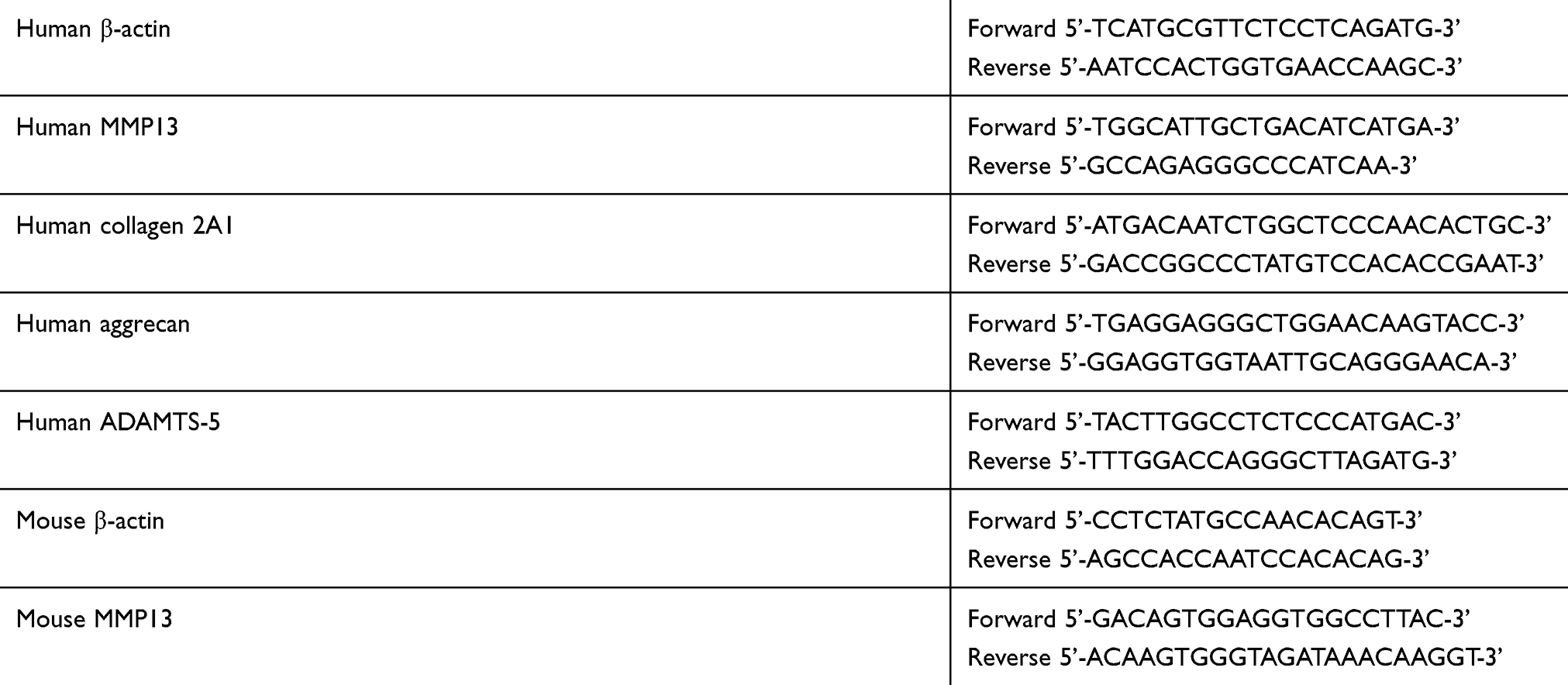 |
Table 2 Primer sequences |
Western blotting
Protein samples were isolated from cartilage tissues or chondrocytes with or without being treated with ANS (10 µM). Cartilage tissues were pulverized into powder in liquid nitrogen, and then the lysis buffer (50 mM Tris HCl [pH 7.4], 150 mM NaCl, 20 mM ethylenediaminetetraacetic acid, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], and protease inhibitors) was added and centrifuged at 160,609 g for 10 mins at 4 °C. Cell samples were washed with cold PBS and lysed in a lysis buffer. Proteins were resolved on a 10% SDS-polyacrylamide electrophoresis gel and transferred to polyvinylidene difluoride membranes (Millipore Corp., Bedford, MA, USA). After blocking with 5% nonfat milk in Tris-buffered saline plus 0.1% Tween 20, the membranes were incubated with primary antibodies against phosphorylated (p)-P38, p-CREB, MMP13, or β-actin (Abcam). After washing, primary antibodies were detected using horseradish peroxidase-conjugated secondary anti-mouse or anti-rabbit antibodies (Beyotime Institute of Biotechnology Inc.) and visualized using an enhanced chemiluminescence kit (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Immunofluorescence
Chondrocytes were grown on a 20-mm glass bottom cell culture dish (801001; Nest Biotechnology Co., Ltd., Shanghai, China) with or without being treated with ANS (10 µM) for 24 hrs. Then the cells were fixed with 4% paraformaldehyde for 15 mins, washed two times with PBS containing 0.05% Tween-20, permeabilized with 0.3% Triton X-100 for 5 mins, and then blocked with 1% bovine serum albumin for 30 mins. Next, the cells were incubated with the p-P38, p-CREB, and MMP13 antibodies (1:200 dilutions, Abcam) at 4 °C overnight. After three washes with PBS, the cells were incubated with goat anti-rabbit immunoglobulin G conjugated to fluorescent Cy5 dye (1:100 dilutions, Abcam) in PBS. The 4ʹ,6-diamidino-2-phenylindole (Life Technologies, Carlsbad, CA, USA) was used for nuclear staining. Immunofluorescent images were obtained using the Nikon Eclipse TI microscope (Nikon Instruments Inc.), Zeiss LSM780 confocal microscope (Zeiss, Jena, Germany), or Zeiss Colibri epifluorescence microscope and processed with Image J (National Institute of Health).
Statistical analysis
Data are expressed as a mean ± standard deviation. The significance of differences between the 2 groups was determined using an independent-samples t-test. Statistical analyses were performed using SPSS 22.0 (IBM Corp., Armonk, NY, USA). P<0.05 or P<0.01 was considered statistically significant.
Results
P38/CREB/MMP13 is active in human osteoarthritic cartilage
Figure 1A shows an x-ray of the hip preoperatively. The result of safranin O and fast green staining showed that cartilage suffered more severe destruction and loss of integrity in osteoarthritic samples (Figure 1B). Quantitative polymerase chain reaction (qPCR) of the RNA of tissues showed that ADAM metallopeptidase with thrombospondin type 1 motif 5 (ADAMTS5) and MMP13 were upregulated in osteoarthritic cartilage, whereas Col2 and aggrecan were downregulated in osteoarthritic cartilage tissues (Figure 1C–F. Protein from osteoarthritic and control cartilage was used to evaluate the expression levels of p-P38, p-CREB, and MMP13 by Western blot analysis. The result showed that p-P38, p-CREB, and MMP13 had higher expression in osteoarthritic cartilage (Figure 1G), and the band density is calculated in Figure 1H–J.
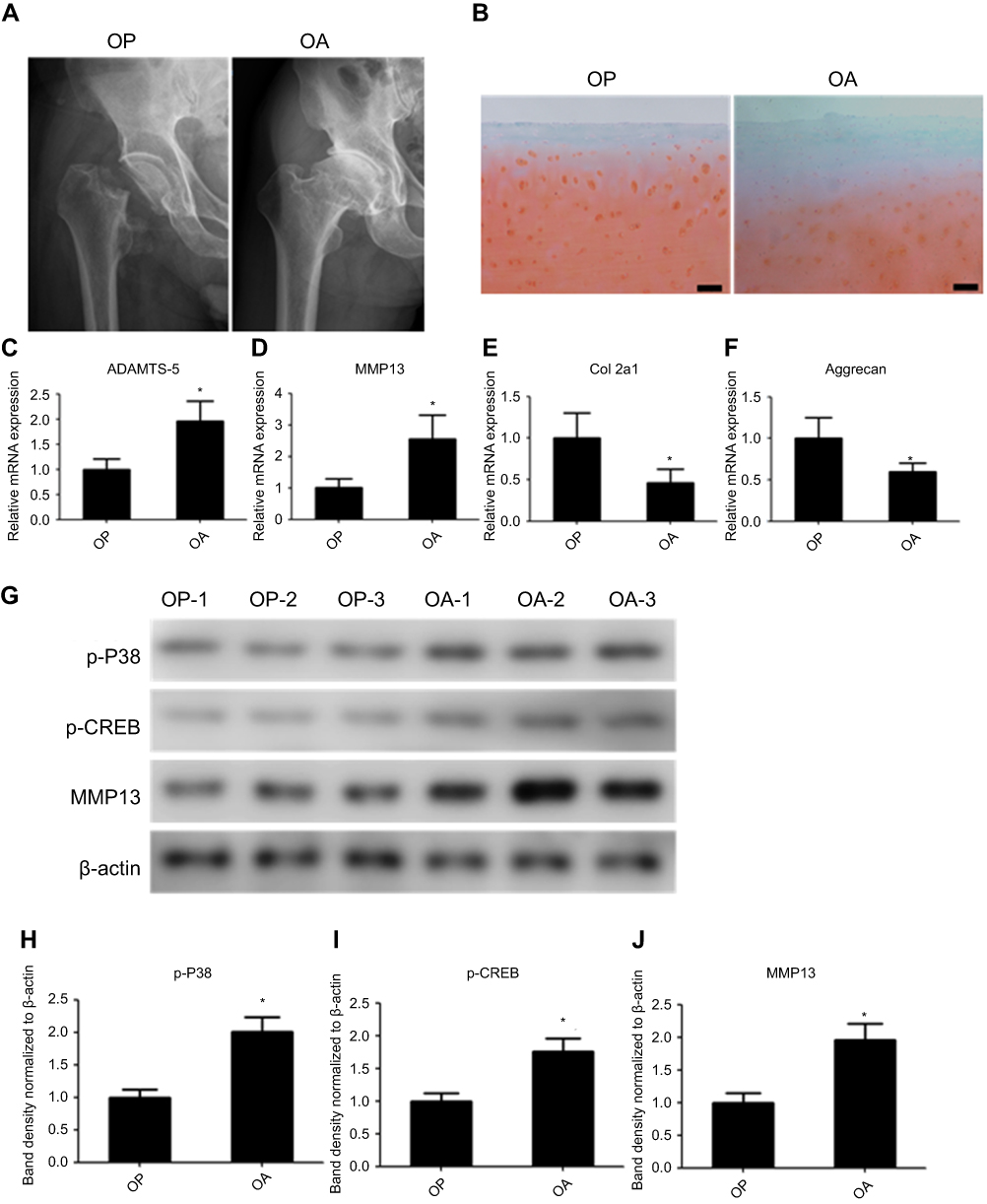 |
Figure 1 P38/CREB/MMP13 is active in human osteoarthritic chondrocytes. (A) X-rays of the hip from patients with OA and the control. (B) Safranin O and fast green staining of the cartilage tissues from patients with OA and the control. Relative messenger RNA expression of (C) ADAM metallopeptidase with thrombospondin type 1 motif 5, (D) MMP13, (E) Col2, and (F) aggrecan in human osteoarthritic and control cartilage samples. (G) Western blot analysis of p-P38, p-CREB, and MMP13 in human chondrocytes from osteoarthritic and the control cartilage. The band density analysis of (H) p-P38, (I) p-CREB, and (J) MMP13 were analyzed by Image J software and normalized to ß-actin. n=10, data represent the mean±standard error of the mean. *P<0.05. Abbreviations: CREB, cAMP response element binding protein; MMP13, matrix metallopeptidase-13; OA, osteoarthritis; p, phosphorylated. |
P38/CREB/MMP13 is active in ANS-induced human and mouse chondrocytes
Primary chondrocytes in the CON group were isolated from control cartilage and normal mice. ANS was used to activate the P38 signal in the chondrocytes. Then p-P38, p-CREB, and MMP13 were evaluated by immunofluorescence and Western blot analysis. The result of immunofluorescence showed that p-P38 was active as expected, and p-CREB and MMP13 were upregulated (Figure 2A). The result of Western blot analysis was in line with that of immunofluorescence (Figure 2B). The band density is calculated in Figure 2C–D.
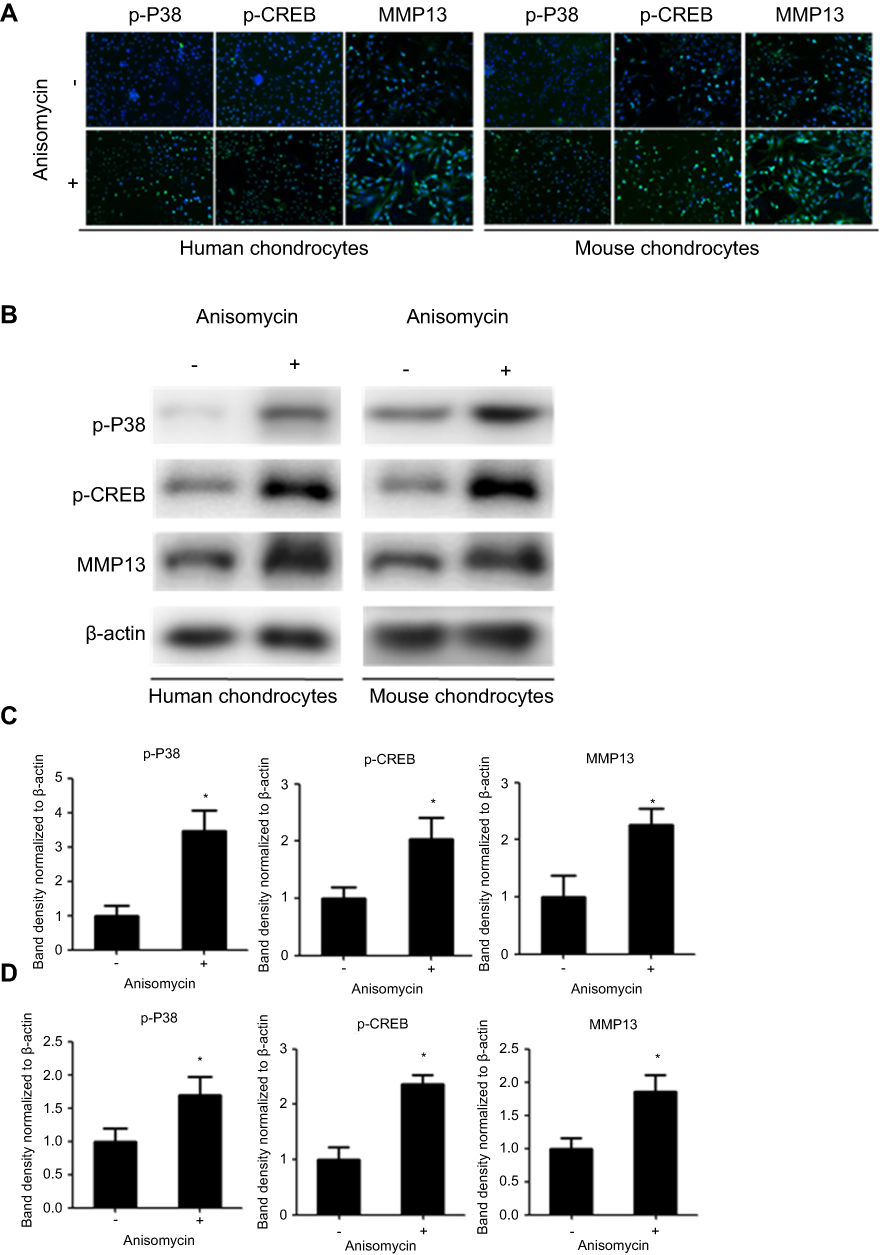 |
Figure 2 P38/CREB/MMP13 is active in ANS-induced human and mouse chondrocytes. (A) Immunofluorescence analysis of p-P38, p-CREB, and MMP13 expression in chondrocytes treated with or without ANS (10 µM) for 24 hrs. (B) Western blot analysis of p-P38, p-CREB, and MMP13 in chondrocytes treated with or without ANS (10 µM) for 24 hrs. The band density analysis of p-P38, p-CREB, and MMP13 from (C) human chondrocytes and (D) mouse chondrocytes were analyzed by Image J software and normalized to ß-actin. n=10, data are expressed as the mean±standard error of the mean. *P<0.05. Abbreviations: CREB, cAMP response element binding protein; MMP13, matrix metallopeptidase-13; p, phosphorylated; ANS, anisomycin. |
ANS-induced mouse OA is involved with activation of the P38/CREB/MMP13 axis
After mice received an intra-articular injection of ANS (10 µl [100 µm]) or PBS twice per week for 6 weeks, radiography was used to evaluated degeneration of the knee joint (Figure 3A). The result of safranin O and fast green staining suggested severe destruction and loss of integrity in mouse cartilage with ANS (Figure 3B), and the mice with ANS had a higher OARSI score (Figure 3D). Additionally, the effect size (r) was 0.825 between the two groups (Table 3). The result of immunohistochemistry showed a higher MMP13 positive cell rate in mouse cartilage with ANS (Figure 3C and E). The qPCR of the RNA of tissues showed that MMP13 was upregulated in mouse cartilage tissues with ANS (Figure 3F). Protein from mouse cartilage tissues in the ANS or CON group was used to evaluate the expression levels of p-P38, p-CREB, and MMP13 by Western blot analysis. The result showed that p-P38, p-CREB, and MMP13 had higher expressions in mice with ANS (Figure 3G), and the band density is calculated in Figure 3H–J.
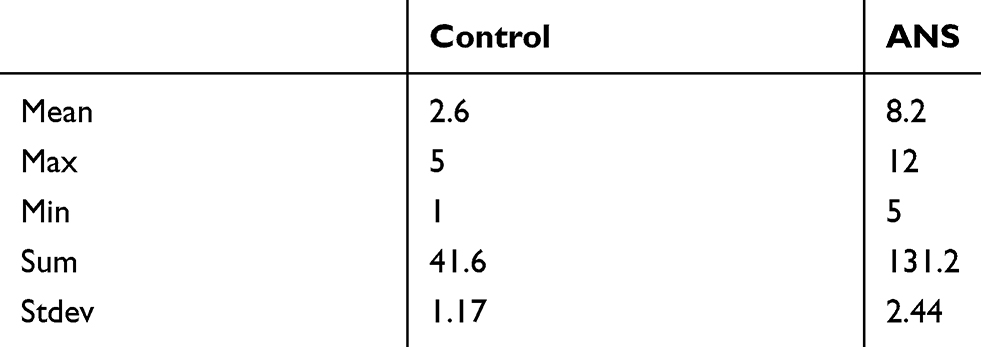 |
Table 3 Dtails about type of OARSI scoring (mean/max/sum) |
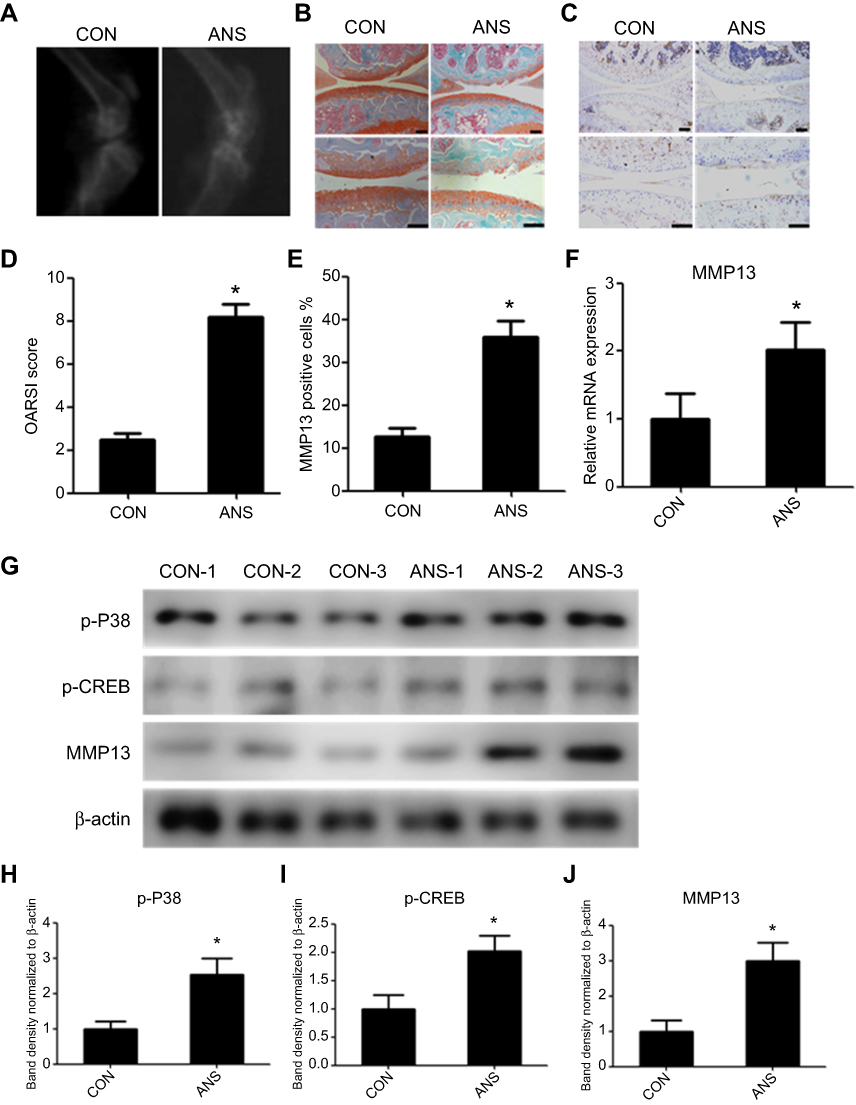 |
Figure 3 ANS-induced mouse osteoarthritis. (A) X-ray of the knee from the control ANS-induced mice. (B) Safranin O and fast green staining of cartilage tissues from mice in the CON and ANS groups. (C) Immunohistochemistry analysis of MMP13 expression in the CON group and ANS-induced mouse cartilage. (D) Osteoarthritis Research Society International scores were determined according to the safranin O and fast green staining results. (E) The percentage of MMP13-positive cells was calculated according to immunohistochemistry. (F) Relative messenger RNA expression of MMP13 in the CON group and ANS-induced mouse cartilage samples. (G) Western blot analysis of p-P38, p-CREB, and MMP13 in the CON group and ANS-induced mouse cartilage. The band density analysis of (H) p-P38, (I) p-CREB, and (J) MMP13 was performed using Image J software and normalized to β-actin. n=15, Data are expressed as the mean±standard error of the mean. *P<0.05. Abbreviations: CREB, cAMP response element binding protein; MMP13, matrix metallopeptidase-13; p, phosphorylated; ANS, anisomycin. |
Discussion
OA is a complex degenerative joint disease affected by many factors. Imbalance between synthesis and decomposition of the cartilage matrix is the primary cause of OA, which leads to cartilage destruction. MMPs are catabolic proteases capable of degrading all types of extracellular matrix proteins. In particular, MMP13 is the main protease involved in the degradation of Col2. MMP13 is crucial for OA progression, and the pharmacologic inhibition of MMP13 was an effective strategy to decelerate articular cartilage loss in a murine model of injury-induced knee OA.16,17 In the present study, we found that the P38/CREB/MMP13 axis is active in osteoarthritic chondrocytes, and P38 agonist ANS may induce mouse OA. Therefore, effective new therapy directed against this pathway could be developed.
Additionally, previous studies reported that activation of the p38-mitogen-activated protein kinase (MAPK) signaling pathway may lead to the expression of proinflammatory cytokines, which in turn promote the activation of P38, and this plays a key role in the development of OA.18,19 Proinflammatory agents such as IL-1ß, TNF-α, and reactive oxygen species induce the synthesis of MMPs, which lead to the degradation of cartilage.20–22 Blockage of p38 MAPK could inhibit the downstream inflammatory cytokine production and MMP expression in chondrocytes.23,24 Local treatment with a p38 MAPK inhibitor improved inflammation and joint degradation in mouse models, indicating treatment potential for human OA.25 Recent studies showed that the activation of CREB resulted in an increased expression of MMP13 in chondrocytes.12,13 Moreover, IL-1ß-induced CREB activation by phosphorylation of Ser133 is mainly dependent on p38 kinases.26,27 Herein, p-P38 and p-CREB expressions were increased in human osteoarthritic cartilage. Thus, we first drew the conclusion that the P38/CREB/MMP13 axis is active in human OA.
As a p38 agonist, ANS could stimulate p38 phosphorylation in various cells.28–32 Our results suggested that the P38/CREB/MMP13 axis was activated by ANS in human and mouse chondrocytes. In addition, p-P38, p-CREB, and MMP13 expressions were increased in ANS-induced mouse cartilage. Therefore, we concluded that the intra-articular injection of ANS could induce mouse OA, which might involve activation of the P38/CREB/MMP13 axis.
There are several limitations in our study. First, the c-Jun N-terminal kinase (JNK) pathway was also activated by ANS.30,33 Thus, ANS-stimulated chondrocytes are not limited to activation of the P38 pathway. The JNK pathway also might be involved in ANS-induced OA. All these assumptions need further research with the specific p38 or JNK agonist. Second, activation of the P38/CREB/MMP13 axis was evaluated by Western blot and immunofluorescence in our study. Thus, other methods need to be used to verify the P38/CREB/MMP13 axis, such as hybridization in situ in an in vivo study. In addition, the p38 inhibitor should be used to verify the effect of P38/CREB/MMP13 axis in reverse.
This study is the first to examine the effect of the P38/CREB/MMP13 axis in OA. We concluded that the P38/CREB/MMP13 axis is active in osteoarthritic chondrocytes, and it may cause the degeneration of cartilage. These results provide a basis for targeting the P38/CREB/MMP13 axis in the prevention and treatment of OA.
Acknowledgments
This research was supported by the Zhejiang Provincial Natural Science Foundation of China (grant number LQ18H250001) and Medical and Health Science and Technology Planning Project of Zhejiang Province (2018ky493). We thank GuoXiang Fu and Tao Zhu MD (Pathology Department, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, People's Republic of China) for their technical support with the histology and immunohistochemistry.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kinds MB, Welsing PM, Vignon EP, et al. A systematic review of the association between radiographic and clinical osteoarthritis of hip and knee. Osteoarthritis Cartilage. 2011;19(7):768–778. doi:10.1016/j.joca.2011.01.015
2. Hauk L. Treatment of knee osteoarthritis: a clinical practice guideline from the AAOS. Am Fam Physician. 2014;89(11):918–920.
3. Rogers EL, Reynard LN, Loughlin J. The role of inflammation-related genes in osteoarthritis. Osteoarthritis Cartilage. 2015;23(11):1933–1938. doi:10.1016/j.joca.2015.01.003
4. Haq SA, Davatchi F, Dahaghin S, et al. Development of a questionnaire for identification of the risk factors for osteoarthritis of the knees in developing countries. A pilot study in Iran and Bangladesh. An ILAR-COPCORD phase III study. Int J Rheum Dis. 2010;13(3):203–214. doi:10.1111/j.1756-185X.2010.01529.x
5. Smith GJ. The role of collagenolytic matrix metalloproteinases in the loss of articular cartilage in osteoarthritis. Front Biosci. 2006;11:3081–3095.
6. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. 2006;11:529–543.
7. Aida Y, Maeno M, Suzuki N, Shiratsuchi H, Motohashi M, Matsumura H. The effect of IL-1beta on the expression of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in human chondrocytes. Life Sci. 2005;77(25):3210–3221. doi:10.1016/j.lfs.2005.05.052
8. Kobayashi M, Squires GR, Mousa A, et al. Role of interleukin-1 and tumor necrosis factor alpha in matrix degradation of human osteoarthritic cartilage. Arthritis Rheum. 2005;52(1):128–135. doi:10.1002/art.20776
9. Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002;4(3):157–164. doi:10.1186/ar401
10. Shiomi T, Lemaitre V, D’Armiento J, Okada Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol Int. 2010;60(7):477–496. doi:10.1111/j.1440-1827.2010.02547.x
11. Roach HI, Yamada N, Cheung KS, et al. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005;52(10):3110–3124. doi:10.1002/art.21300
12. Ha YJ, Choi YS, Kang EH, et al. SOCS1 suppresses IL-1beta-induced C/EBPbeta expression via transcriptional regulation in human chondrocytes. Exp Mol Med. 2016;48:e241. doi:10.1038/emm.2016.47
13. Bui C, Barter MJ, Scott JL, et al. cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. Faseb J. 2012;26(7):3000–3011. doi:10.1096/fj.12-206367
14. Pritzker KP, Gay S, Jimenez SA, et al. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage. 2006;14(1):13–29. doi:10.1016/j.joca.2005.07.014
15. Chen P, Xia C, Mei S, et al. Intra-articular delivery of sinomenium encapsulated by chitosan microspheres and photo-crosslinked GelMA hydrogel ameliorates osteoarthritis by effectively regulating autophagy. BIOMATERIALS. 2016;81:1–13. doi:10.1016/j.biomaterials.2015.12.006
16. Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc. 2008;3(8):1253–1260. doi:10.1038/nprot.2008.95
17. Wang M, Sampson ER, Jin H, et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res Ther. 2013;15(1):R5. doi:10.1186/ar4133
18. Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford). 2008;47(4):409–414. doi:10.1093/rheumatology/kem297
19. Geng Y, Valbracht J, Lotz M. Selective activation of the mitogen-activated protein kinase subgroups c-Jun NH2 terminal kinase and p38 by IL-1 and TNF in human articular chondrocytes. J Clin Invest. 1996;98(10):2425–2430. doi:10.1172/JCI119056
20. Zayed N, Afif H, Chabane N, et al. Inhibition of interleukin-1beta-induced matrix metalloproteinases 1 and 13 production in human osteoarthritic chondrocytes by prostaglandin D2. Arthritis Rheum. 2008;58(11):3530–3540. doi:10.1002/art.23958
21. Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7(1):33–42. doi:10.1038/nrrheum.2010.196
22. Kanwar JR, Kanwar RK, Burrow H, Baratchi S. Recent advances on the roles of NO in cancer and chronic inflammatory disorders. Curr Med Chem. 2009;16(19):2373–2394.
23. Sun HY, Hu KZ, Yin ZS. Inhibition of the p38-MAPK signaling pathway suppresses the apoptosis and expression of proinflammatory cytokines in human osteoarthritis chondrocytes. CYTOKINE. 2017;90:135–143. doi:10.1016/j.cyto.2016.11.002
24. Joos H, Albrecht W, Laufer S, Brenner RE. Differential effects of p38MAP kinase inhibitors on the expression of inflammation-associated genes in primary, interleukin-1beta-stimulated human chondrocytes. Br J Pharmacol. 2010;160(5):1252–1262. doi:10.1111/j.1476-5381.2010.00760.x
25. Maudens P, Seemayer CA, Pfefferle F, Jordan O, Allemann E. Nanocrystals of a potent p38 MAPK inhibitor embedded in microparticles: therapeutic effects in inflammatory and mechanistic murine models of osteoarthritis. J Control Release. 2018;276:102–112. doi:10.1016/j.jconrel.2018.03.007
26. Frost RA, Nystrom GJ, Lang CH. Stimulation of insulin-like growth factor binding protein-1 synthesis by interleukin-1beta: requirement of the mitogen-activated protein kinase pathway. ENDOCRINOLOGY. 2000;141(9):3156–3164. doi:10.1210/endo.141.9.7641
27. Funding AT, Johansen C, Kragballe K, Iversen L. Mitogen- and stress-activated protein kinase 2 and cyclic AMP response element binding protein are activated in lesional psoriatic epidermis. J Invest Dermatol. 2007;127(8):2012–2019. doi:10.1038/sj.jid.5700821
28. Ross S, Chen T, Yu V, et al. High-content screening analysis of the p38 pathway: profiling of structurally related p38alpha kinase inhibitors using cell-based assays. Assay Drug Dev Technol. 2006;4(4):397–409. doi:10.1089/adt.2006.4.397
29. Ma J, Ma Y, Liu X, et al. Gambogic acid inhibits osteoclast formation and ovariectomy-induced osteoporosis by suppressing the JNK, p38 and Akt signalling pathways. Biochem J. 2015;469(3):399–408. doi:10.1042/BJ20150151
30. Liu Y, Ge J, Li Q, et al. Anisomycin induces apoptosis of glucocorticoid resistant acute lymphoblastic leukemia CEM-C1 cells via activation of mitogen-activated protein kinases p38 and JNK. NEOPLASMA. 2013;60(1):101–110. doi:10.4149/neo_2013_014
31. Zhao TC, Zhang L, Liu JT, Guo TL. Disruption of Nox2 and TNFRp55/p75 eliminates cardioprotection induced by anisomycin. Am J Physiol Heart Circ Physiol. 2012;303(10):H1263–H1272. doi:10.1152/ajpheart.00306.2012
32. Shafer LM, Slice LW. Anisomycin induces COX-2 mRNA expression through p38(MAPK) and CREB independent of small GTPases in intestinal epithelial cells. Biochim Biophys Acta. 2005;1745(3):393–400. doi:10.1016/j.bbamcr.2005.07.002
33. Liu Y, Ge J, Li Q, et al. Low-dose anisomycin sensitizes glucocorticoid-resistant T-acute lymphoblastic leukemia CEM-C1 cells to dexamethasone-induced apoptosis through activation of glucocorticoid receptor and p38-MAPK/JNK. Leuk Lymphoma. 2014;55(9):2179–2188. doi:10.3109/10428194.2013.866664
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.