")
Back to Journals » Journal of Pain Research » Volume 11
Activation of spinal nociceptin receptors induces cardiovascular depression and antinociception in an independent manner in mice
Authors Tsai CY , Poon YY, Huang YH, Chan SH
Received 24 May 2018
Accepted for publication 21 August 2018
Published 1 November 2018 Volume 2018:11 Pages 2699—2708
DOI https://doi.org/10.2147/JPR.S175259
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Katherine Hanlon
Ching-Yi Tsai,1,* Yan-Yuen Poon,1,2,* Ya-Hui Huang,1 Samuel HH Chan1
1Institute for Translational Research in Biomedicine, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan; 2Department of Anesthesiology, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan
*These authors contributed equally to this work
Purpose: The nociceptin receptor (NOP) was discovered in 1994 and was designated opioid-like receptor; activation of NOP leads to reduced neuronal excitability. Although suggested by the anatomical localization of NOP in brain or spinal cord, the cardiovascular or nociceptive effects of its endogenous ligand, nociceptin, are equivocal. Taking advantage from intrathecal application of nociceptin to simultaneously activate NOP on sympathetic preganglionic neurons in the intermediolateral column (IML) and superficial laminae of dorsal horn, we investigated whether the nociceptin-induced cardiovascular effects engage the participation of baroreflex, and whether the concurrently elicited changes in blood pressure and pain responses are interrelated.
Methods: NOPs in the thoracic spinal cord of ICR or C57BL/6 mice were identified with immunofluorescence staining and were activated through intrathecal administration of nocicetpin. The elicited changes in cardiovascular parameters and tail-flick nociceptive responses were measured.
Results: Positive immunoreactivity against NOP colocalized with neurons in the IML and superficial dorsal horn layers of thoracic spinal cord. Intrathecal administration of nociceptin (1, 2, or 5 nmol) elicited a significant and dose-dependent decrease in blood pressure or heart rate that was paralleled by reduced baroreflex-mediated sympathetic vasomotor tone and mirrored by augmented cardiac vagal baroreflex, alongside prolonged tail-flick latency with an efficacy of hypotension <<< antinociception. Coadministration of the specific NOP antagonist, UFP101 (10 nmol), blunted all nociceptin-elicited responses. However, restoring blood pressure to baseline level failed to affect the antinociceptive actions of nociceptin.
Conclusion: Activation of thoracic spinal NOP in ICR and C57BL/6 mice induces blood pressure and heart rate by decreasing the sympathetic outflow of both arms of the baroreflex arc to the blood vessels and the heart, and the antinociceptive responses to nociceptin are independent of and disproportional to its cardiovascular actions.
Keywords: nociceptin, intermediolateral column, superficial layers of dorsal horn, tail-flick test, baroreflex-mediated sympathetic vasomotor tone, cardiac vagal baroreflex
Introduction
The nociceptin receptor (NOP) was discovered in 1994 and was designated opioid-like receptor (ORL-1) because it shares a high homology of amino acid sequence with opioid receptors although its affinities for opioid ligands are greatly reduced.1 The highly selective and endogenous ligand for NOP, which was discovered in 1995, is the 17-amino acid neuropeptide nociceptin/orphanin FQ.2 Nociceptin shows only very modest affinity for μ, δ, or κ receptors,3 and naloxone does not antagonize most of the effects of nociceptin.1 At the cellular level, activation of NOP by nociceptin inhibits the formation of cyclic adenosine monophosphate, mediates a pronounced inhibition of voltage-gated Ca2+ channels, and facilitates the opening of inwardly rectifying K+ channels.4–6 The overall effect of NOP activation leads to reduced neuronal excitability and reduction of neurotransmitter release.7,8
Anatomical studies have shown that NOP is localized in brain or spinal nuclei that are related to cardiovascular or nociceptive responses.9–11 However, nociceptin has been reported to decrease or increase blood pressure (BP) or heart rate (HR).7,12–15 Likewise, nociceptin reportedly produces hyperalgesia and allodynia,2,3,16or antinociception.16,17 These equivocal cardiovascular and antinociceptive effects of nociceptin may have arisen from differences in sites of action because of the routes through which this peptide was administered. In particular, the disparate cardiovascular responses to nociceptin may have resulted from differential effects induced by this peptide on the baroreflex-mediated sympathetic vasomotor tone and cardiac vagal baroreflex, which are responsible, respectively, for the short-term and long-term regulation of BP and HR.18,19 Another possibility arises from the complex relationship between BP and pain. Whereas hypertension has generally been associated with hypoalgesia,20,21 increased pain sensitivity in chronically low BP or hypotension concomitant with antinociception has also been reported.22–24
Intrathecal application of nociceptin simultaneously acts on NOP present in the dorsal horn where primary afferents from nociceptive receptors terminate,25 and intermediolateral (IML) column where sympathetic preganglionic neurons that innervate the blood vessels and the heart reside.26 The present study exploited this advantage to decipher whether by acting on the IML, which constitutes the common sympathetic output component in the circuit for the baroreflex-mediated sympathetic vasomotor tone and cardiac vagal baroreflex, the cardiovascular effects of activation of NOPs in the spinal cord engage the participation of both arms of baroreflex. We also examined whether, on an equimolar basis, the concurrently induced changes in BP and pain responses by nociceptin are interrelated.
Materials and methods
Ethics statement
All experimental procedures carried out in this study were approved by the Institutional Animal Care and Use Committee of the Kaohsiung Chang Gung Memorial Hospital and were in compliance with the guidelines for animal care and use set forth by that committee. All efforts were made to reduce the number of animals used and to minimize animal suffering during the experiment.
Animals
Adult male ICR mice (30–35 g, n=33) or C57BL/6 mice (28–30 g; n=62) were purchased from the Experimental Animal Center of the Ministry of Science and Technology and BioLASCO, Taiwan, Republic of China. They were housed in an AAALAC International-accredited Center for Laboratory Animals under temperature control (24°C–25°C) and 12 hour light–dark cycle. Standard laboratory mouse chow and tap water were available ad libitum.
Double immunofluorescence staining
We examined the expression of NOP in the dorsal horn and IML of the thoracic spinal cord by double immunofluorescence staining.27 Mice that received an intraperitoneal injection of pentobarbital sodium (50 mg kg−1) were transcardially perfused with warm isotonic saline solution, followed by ice-cold 4% paraformaldehyde in 0.1 M PBS (pH 7.2). The thoracic spinal cord was removed, postfixed by submersion in the latter solution overnight at 4°C, and cryoprotected by 30% sucrose in 0.1 M PBS. Frozen transverse section (20 µm) of the thoracic spinal cord was cut on a cryostat (Leitz, Wetzlar, Germany) and collected in 0.1 M PBS. The staining procedures were modified from those reported previously.27 In brief, free-floating sections of the thoracic spinal cord were incubated with a rabbit polyclonal anti-NOP antiserum (orb11143; Biorbyt, Cambridge, UK), together with a mouse monoclonal anti-NeuN antiserum (MAB377, Chemicon, Temecula, CA, USA). The sections were subsequently incubated concurrently with two appropriate secondary antisera (Molecular Probes, Eugene, OR, USA), a goat anti-rabbit IgG conjugated with Alex Fluor 488 (A11034) for NOP, and a goat anti-mouse IgG conjugated with Alexa Fluor 586 (A11031) for NeuN. The specificity of NOP antiserum was validated by its omission from the staining procedure. Viewed under a Fluoview FV1000 laser scanning confocal microscope (Olympus, Tokyo, Japan), immunoreactivity for NeuN exhibited red fluorescence, and NOP exhibited green fluorescence.
Subarachnoid catheterization
The design and implantation of thoracic intrathecal catheters in mice followed our previously developed procedure.28 Briefly, under pentobarbital sodium anesthesia (50 mg kg−1, intraperitoneal), the left interspinous space between T12 and T13 was exposed after the supraspinous ligament and the left interspinous ligament between T12 and T13 was removed. A 0.5 cm long PU-10 catheter (Strategic Application Inc., Libertyville, IL, USA) was inserted tangentially into the subarachnoid space along the dorsal surface of the spinal cord through a dural opening. After flushing with 3–5 µL of artificial cerebrospinal fluid (aCSF), the exterior end of the catheter was sealed by heat. The catheter was further secured on the fascia of the paravertebral muscle with suture, and the sealed end was buried under the skin. Sodium penicillin (1,000 IU) was given intramuscularly to prevent postoperative infection. The animals were returned to the animal room (temperature maintained at 24°C ± 0.5°C) for postoperative recovery. They were allowed free access to standard mouse chow and water. Only animals that showed full recovery 4–5 days after surgery and with no observable motor deficit were used in subsequent experiments.
General preparation for physiologic experiments
Anesthesia was induced in an induction chamber with 4% isoflurane (Abbot Laboratories, Abbot Park, IL, USA) in oxygen for 30 seconds at a constant flow of 1.0 L/min using a Matrx VIP 3000 vaporizer (Midmark, Orchard Park, NY, USA).29 After induction, mice were placed on an operating table and anesthesia was maintained with 2% isoflurane in 100% oxygen through a nose piece to carry out preparatory surgery. This included cannulation of the right femoral artery with a PE-10 catheter (Clay Adams, Franklin Lakes, NJ, USA) to measure arterial pressure. In some experiments, the left femoral vein was also cannulated with a PE-10 catheter (Clay Adams). After surgery, animals were placed on a mouse adaptor (Kopf, Tujunga, CA, USA) and were connected to the anesthesia machine (Univentor, Zejtun, Malta) with a mouse mask (Kopf).
Determination of BP, HR, and spontaneous baroreflex
Anesthesia was maintained at 1% isoflurane during the recording session, and mice were placed on a heating pad and their rectal temperature was maintained at 37°C ± 0.5°C. The recorded BP signals from the femoral artery were processed by an arterial BP analyzer (Notocord, Croissy-Sur-Seine, France) and the systolic blood pressure (SBP) signals were simultaneously subject to online and real-time spectral analysis (Notocord) to detect the power density of the low-frequency component of SBP spectrum (BLF; 0.15–0.6 Hz).30,31 The power density of this spectral band has been demonstrated to be a valid experimental index for spontaneous baroreflex-mediated sympathetic vasomotor tone.30–33 To evaluate the spontaneous cardiac vagal baroreflex, we employed a baroreflex sequence analyzer (Notocord) to determine baroreflex sensitivity (BRS) based on online detection of spontaneous sequences of consecutive increases or decreases in SBP associated with parallel changes in HR.31,34 With reference to Laude et al,33 the settings were analysis window: 60 seconds; synchronization delay: three cycles; and minimum sequence length: three cycles. HR was derived instantaneously from the BP signals. Concurrent temporal changes in SBP, mean arterial pressure (MAP), and HR were continuously recorded, alongside power density of the BLF band or BRS. To avoid potential bias created by nonstationary disturbances, only stationary segments of data from spectral analysis were used for statistical analysis.
Tail-flick nociceptive test
We employed the tail-flick nociceptive test because this paradigm evaluates specifically spinally-mediated pain responses.35,36 Under concurrent measurement of the cardiovascular responses as described earlier, the tail of the mice was immersed in a hot water bath maintained at 55°C ± 0.5°C. The time taken for the animal to flick its tail away from hot water was recorded, with a cutoff time of 15 seconds imposed to prevent tissue damage. The averaged value of three determinations separated by 1 minute was taken as the measured tail-flick response.
Intrathecal administration of nociceptin and its antagonist
The sealed end of the implanted subarachnoid catheter was retrieved and was flushed with 3 µL of aCSF to ensure patency. Nociceptin,37 a selective agonist of NOP (1, 2, 5 nmol; Tocris Bioscience, Bristol, UK), was given at a volume of 3 µL over 1 minute using a 25 µL Hamilton microsyringe, followed by an additional 3 µL of aCSF to flush the catheter. In experiments that engaged the specific antagonist,36 UFP101 (10 nmol; Tocris Bioscience) was similarly delivered intrathecally, together with nociceptin (5 nmol) or aCSF. The doses used were adopted from the relevant literature.36,37 The effects of drug treatments on MAP, HR, BLF power, and BRS were routinely followed for 60 minutes after administration, and the effects on tail-flick responses were evaluated 30 minutes after intrathecal injection of nociceptin or UFP101 plus nociceptin. Injection of 3 µL of aCSF served as the vehicle control for nociceptin and UFP101.
Evaluation of relationship between tail-flick responses and BP
In some experiments, after the effects of intrathecal administration of nociceptin on tail-flick responses as described in the tail-flick nociceptive test earlier were completed, the BP was titrated back to baseline level by intravenous infusion of dopamine. Tail-flick latency measurements after nociceptin administration were repeated after stabilization of the cardiovascular parameters.
Statistical analysis
Data were expressed as mean ± SEM. They were analyzed using one-way ANOVA or two-way ANOVA with repeated measures for assessment of group means, followed by the Scheffé or Dunnett post hoc multiple-range analysis for individual means. Statistical significance was accepted at P<0.05.
Results
NOP is present in the dorsal horn neurons and IML column of thoracic spinal cord
Based on the anatomical location of immunoreactivity in the spinal cord (Figure 1A), double-immunofluorescence staining revealed that NOPs were distributed in the dorsal horn and IML column of the thoracic spinal cord (Figure 1B). Viewed under a laser scanning confocal microscope, NOP immunoreactivity was found to intermingle with cells stained positively with the neuronal marker, NeuN, in the superficial layers of the dorsal horn (Figure 1C–E), or colocalized with NeuN-positive cells in the IML (Figure 1F–H) of the thoracic spinal cord.
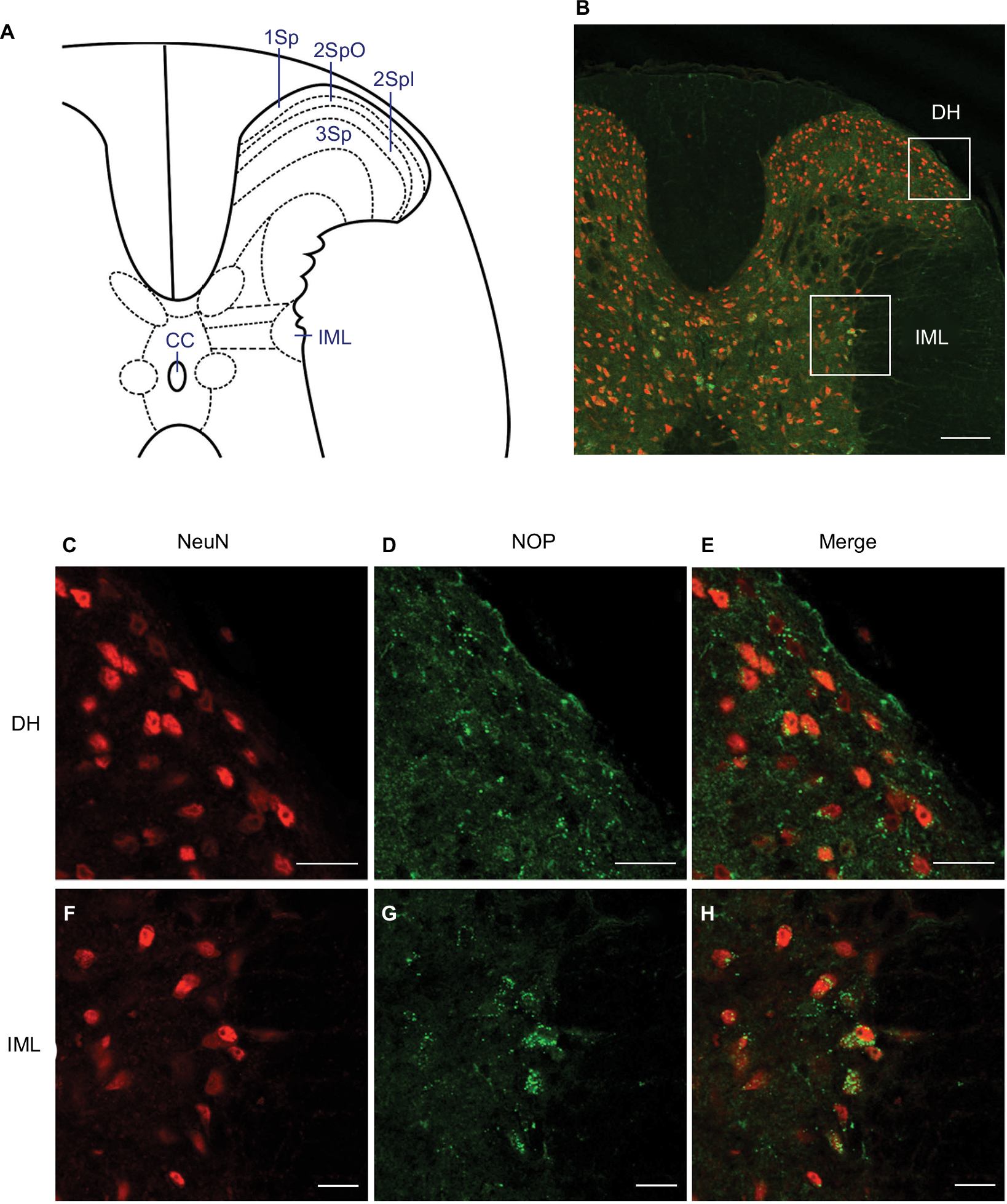 | Figure 1 Illustrative examples of expression of NOP in the thoracic spinal cord of ICR mice (n=3 animals). Notes: (A) Diagrammatic representation of mouse thoracic spinal cord. (B) Corresponding low-power laser scanning confocal microscopic image indicating our areas of interests (boxes) in the thoracic spinal cord. (C–H) High-power confocal microscopic images showing cells in the DH (C–E) or the IML (F–H) that were immunoreactive to a neuronal marker, NeuN (red fluorescence) (C, F), NOP (green fluorescence) (D, G), or both (merge) (E, H). 1Sp = lamina 1; 2SpI = lamina 2, inner; 2SpO = lamina 2, outer; 3Sp = lamina 3; CC = central canal. Scale bar =200 µm in (B) and 20 µm in (C–H). Abbreviations: IML, intermediolateral column; DH, dorsal horn; NOP, nociceptin receptor. |
Activation of spinal NOP decreases BP, HR, or baroreflex-medicated sympathetic vasomotor tone and increases cardiac vagal baroreflex in ICR and C57BL/6 mice
Intrathecal administration of nociceptin (1, 2, or 5 nmol) in ICR mice elicited a significant and dose-dependent decrease in MAP and HR over our 60 minute observation period (Figure 2). The power density of the low-frequency (BLF) component of SBP spectrum, an experimental index for spontaneous baroreflex-mediated sympathetic vasomotor tone,30–33 also underwent a dose-dependent reduction over a time course that paralleled the changes in MAP. At the same time, the BRS, an experimental index for spontaneous cardiac vagal baroreflex,31,34 exhibited a dose-dependent increase that mirrored the decrease in HR. On the other hand, intrathecal administration of aCSF (vehicle control) was ineffective.
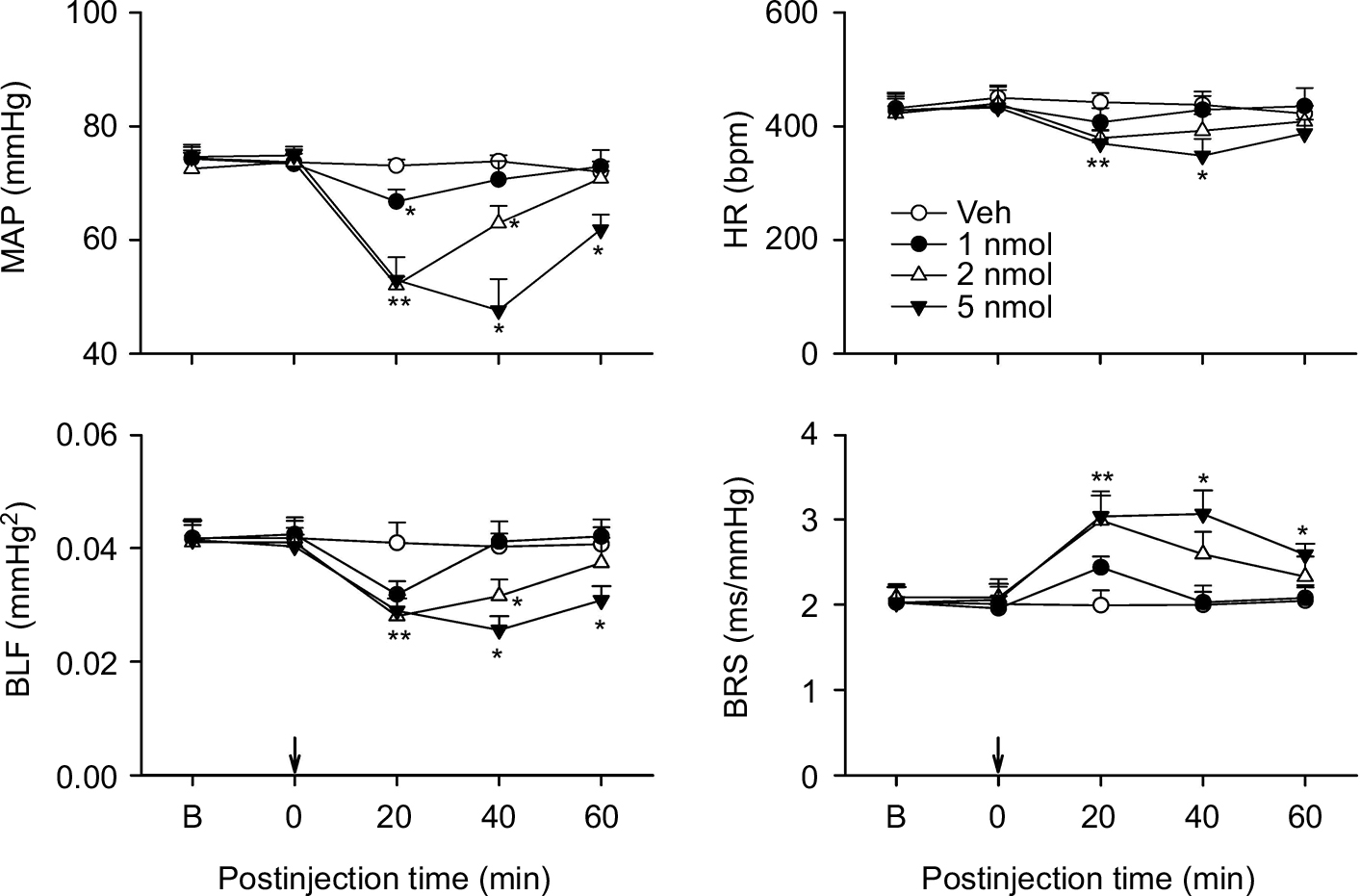 | Figure 2 Effects of activation of nociceptin receptor in the thoracic spinal cord on cardiovascular responses in ICR mice. Notes: Temporal changes in MAP, HR, BLF, an index for baroreflex-mediated sympathetic vasomotor, or BRS, an index for cardiac vagal baroreflex, in mice that received intrathecal administration of nociceptin (1, 2, or 5 nmol) or aCSF (Veh) into the thoracic spinal cord (at arrow). Values are expressed as mean ± SEM (n=7 or 8 animals per experimental group). *P<0.05 vs Veh group at corresponding time points in the post hoc Scheffé multiple-range test. Abbreviations: aCSF, artificial cerebrospinal fluid; B, baseline; BLF, power density of the low-frequency component of systolic blood pressure spectrum; BRS, baroreflex sensitivity; HR, heart rate; MAP, mean arterial pressure; Veh, vehicle control. |
Repeating this series of experiments in C57BL/6 mice to exclude strain-related responses revealed comparable results.38,39 Intrathecal administration of nociceptin (1, 2, or 5 nmol) similarly resulted in a dose-dependent reduction in MAP, HR, or BLF power and augmentation in BRS over our 60 minute observation period (Figure 3). The only difference is that the nociceptin-elicited cardiovascular responses were shorter in duration in the C57BL/6 mice. Intrathecal administration of a specific NOP antagonist, UFP101 (10 nmol), when delivered together with nociceptin (5 nmol), significantly antagonized the decrease in MAP or BLF power and increase in BRS induced by this peptide (Figure 4).
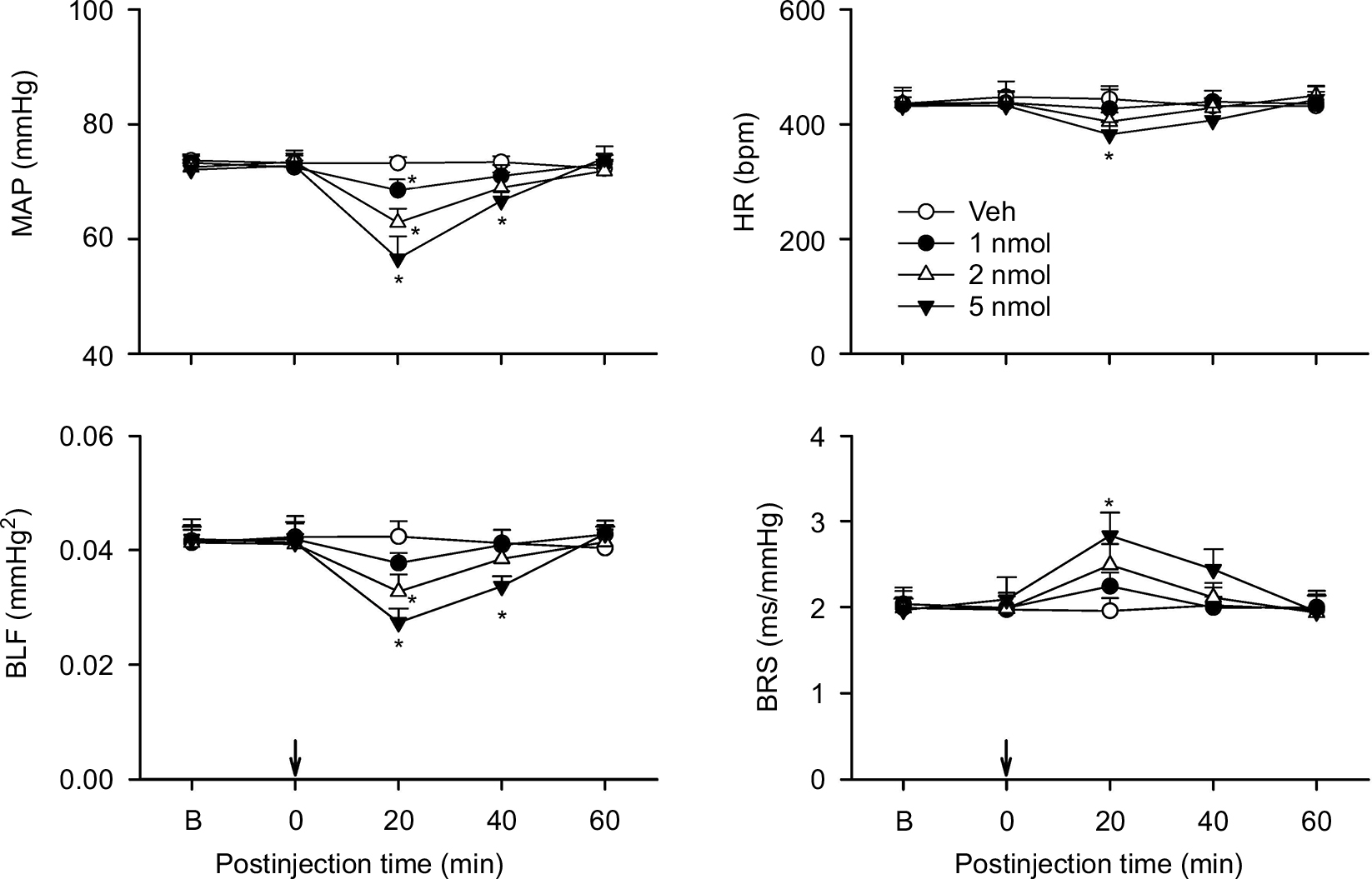 | Figure 3 Effects of activation of nociceptin receptor in the thoracic spinal cord on cardiovascular responses in C57BL/6 mice. Notes: Temporal changes in MAP, HR, BLF power, or BRS in mice that received intrathecal administration of nociceptin (1, 2, or 5 nmol) or aCSF (Veh) into the thoracic spinal cord (at arrow). Values are expressed as mean ± SEM (n=7 or 8 animals per experimental group). *P<0.05 vs Veh group at corresponding time points in the post hoc Scheffé multiple-range test. Abbreviations: aCSF, artificial cerebrospinal fluid; B, baseline; BLF power, power density of the low-frequency component of systolic blood pressure spectrum; BRS, baroreflex sensitivity; HR, heart rate; MAP, mean arterial pressure; Veh, vehicle control. |
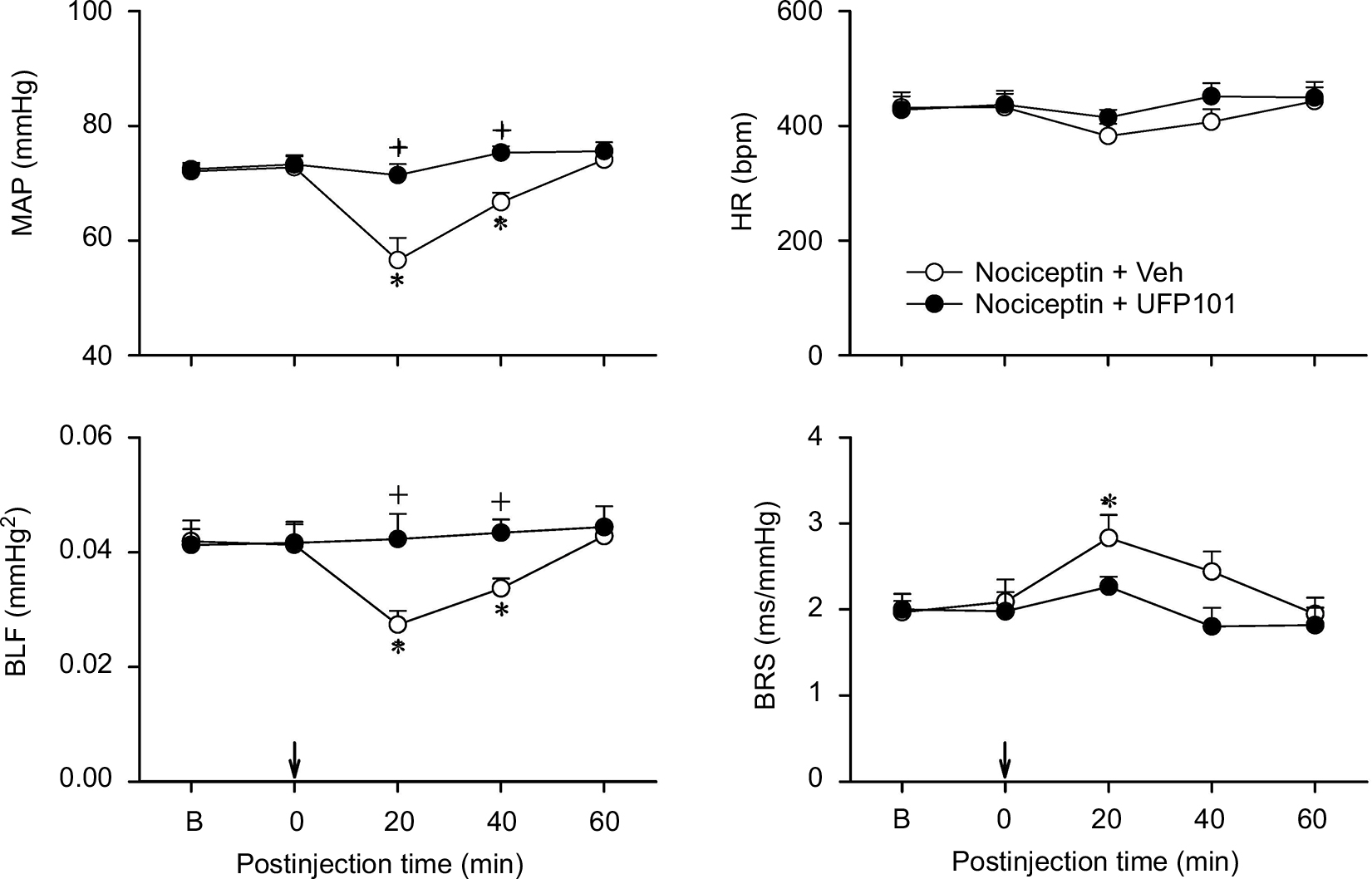 | Figure 4 Effects of nociceptin receptor antagonist on the cardiovascular responses to nociceptin. Notes: Temporal changes in MAP, HR, BLF power, or BRS in C57BL/6 mice that received intrathecal administration of nociceptin (5 nmol), together with a nociceptin receptor antagonist, UFP101 (10 nmol), or aCSF (Veh) into the thoracic spinal cord (at arrow). Values are expressed as mean ± SEM (n=6 or 7 animals per experimental group). *P<0.05 vs preinjection baseline in the post hoc Dunnett multiple-range test; and +P<0.05 vs nociceptin + Veh group at corresponding time points in the post hoc Scheffé multiple-range test. Abbreviations: aCSF, artificial cerebrospinal fluid; B, baseline; BLF power, power density of the low-frequency component of systolic blood pressure spectrum; BRS, baroreflex sensitivity; HR, heart rate; MAP, mean arterial pressure; Veh, vehicle. |
Activation of spinal NOP induces cardiovascular depression and antinociception in an independent and disproportional manner
In addition to the elicited cardiovascular responses, intrathecal administration of the three doses (1, 2, or 5 nmol) of nociceptin also simultaneously elicited a dose-dependent increase in tail-flick latency (Figure 5A), an experimental paradigm that evaluates specifically spinally mediated pain responses.35,36 This antinociceptive effect was antagonized by coadministration of UFP101 (Figure 5B). On the other hand, the dose-dependent depression of tail-flick nociceptive responses was not affected (Figure 5A) on deliberately restoring the nociceptin-suppressed MAP, HR, or BLF power and enhanced BRS to baseline level by intravenous infusion of dopamine (0.8 µg µL−1) at an infusion rate of 0.5 µL min−1 (Table 1). Of note is that, measured on an equimolar basis, the maximal increase from baseline in tail-flick latency (1 nmol, 126%; 2 nmol, 207%; 5 nmol, 536%) induced by nociceptin was discernibly larger than the maximal decrease from baseline in MAP (1 nmol, 6%; 2 nmol, 13%; 5 nmol, 21%).
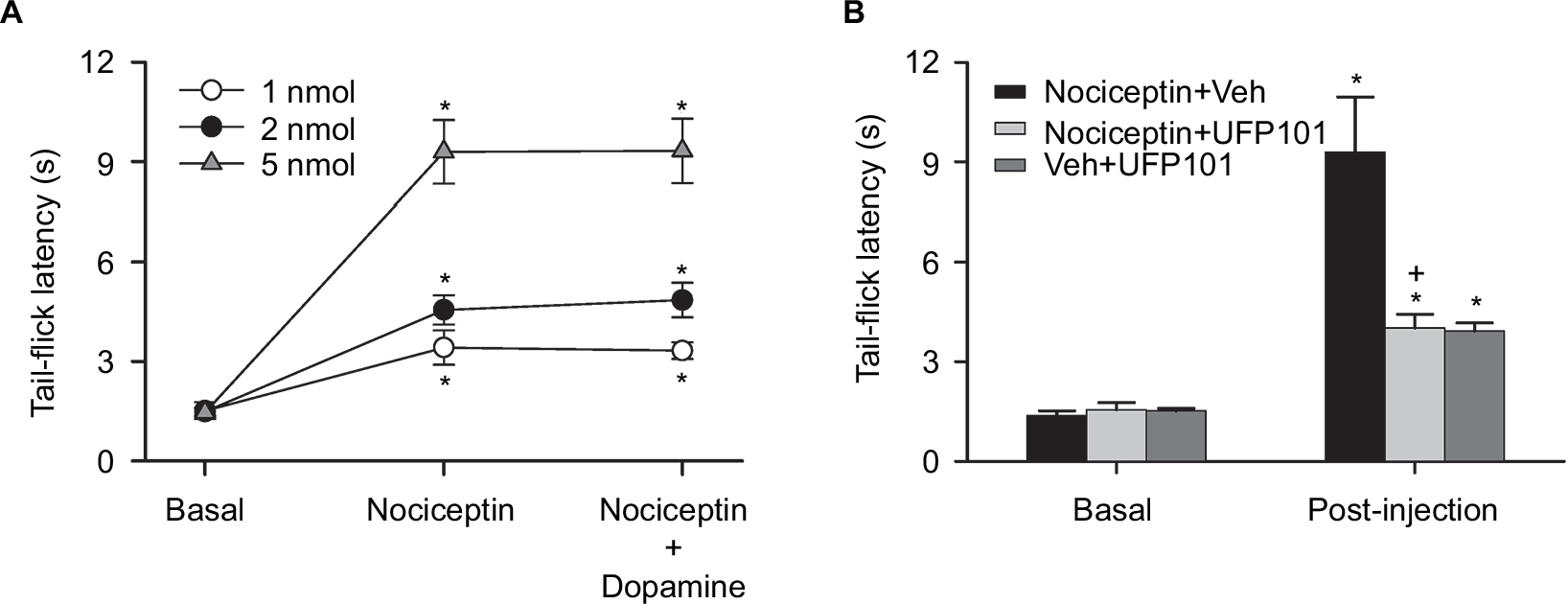 | Figure 5 Effects of activation of nociceptin receptor in the thoracic spinal cord on tail-flick latency in C57BL/6 mice. Notes: (A) Temporal changes in tail-flick latency in mice at baseline, during concurrent cardiovascular depression induced by intrathecal administration of nociceptin (1, 2, or 5 nmol), or after blood pressure was restored to baseline level by intravenous infusion of dopamine while still under the influence of nociceptin. (B) Tail-flick latency in mice that received intrathecal administration of nociceptin (5 nmol), together with UFP101 (10 nmol) or aCSF (Veh), or aCSF and UFP101 (10 nmol). In (A) and (B), values are expressed as mean ± SEM (n=6 or 7 animals per experimental group). *P<0.05 vs preinjection baseline (Basal) in the post hoc Dunnett multiple-range test; and +P<0.05 vs nociceptin + Veh group in the post hoc Scheffé multiple-range test. Abbreviations: aCSF, artificial cerebrospinal fluid; Veh, vehicle. |
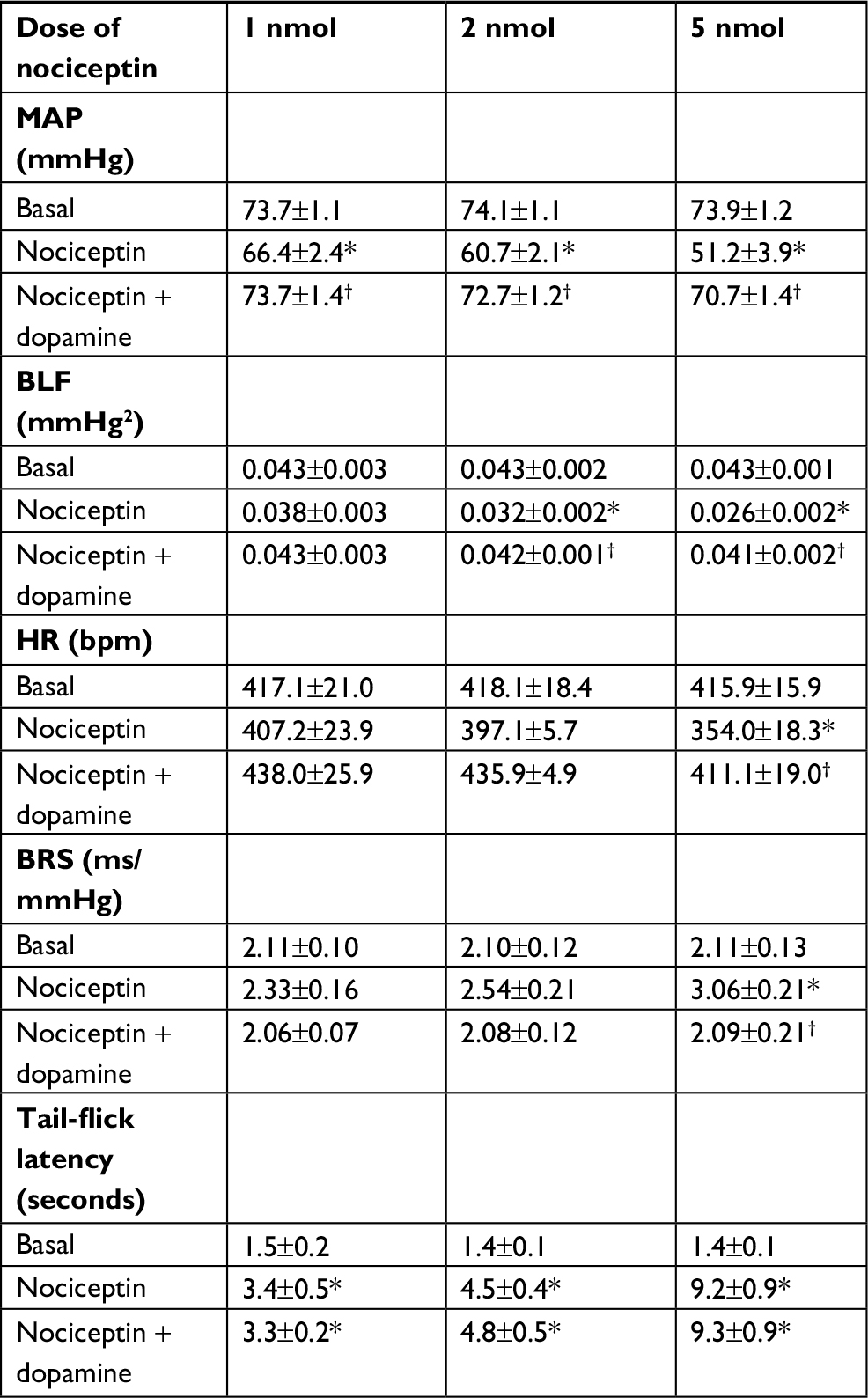 | Table 1 Dissociation of changes in tail-flick latency from cardiovascular effects on activation of nociceptin receptor in the thoracic spinal cord of C57BL/6 mice Notes: Changes in tail-flick latency in mice concurrent with cardiovascular depression induced by intrathecal administration of nociceptin (1, 2, or 5 nmol); or after restoration of cardiovascular parameters to baseline level by intravenous infusion of dopamine. Values are expressed as mean ± SEM (n=6 or 7 animals per experimental group). *P<0.05 vs preinjection baseline (basal) in the post hoc Dunnett multiple-range test and †P<0.05 vs nociceptin group in the post hoc Scheffé multiple-range test. Abbreviations: BLF, power density of the low-frequency component of systolic blood pressure spectrum BRS, baroreflex sensitivity; HR, heart rate; MAP, mean arterial pressure. |
Discussion
The present study took advantage that intrathecal application of nociceptin simultaneously acts on NOP in the IML and dorsal horn to decipher whether this endogenous opioid peptide elicits its cardiovascular depressive effects by acting on the baroreflex-mediated sympathetic vasomotor tone and cardiac vagal baroreflex, and whether the concurrently induced changes in BP and pain responses are interrelated. Our results showed that activation of spinal NOP reduces BP and HR by decreasing the sympathetic outflow of both arms of the baroreflex arc to the blood vessels and the heart, and the antinociceptive responses to nociceptin are independent of and disproportional to its cardiovascular actions.
The baroreflex-mediated sympathetic vasomotor tone and cardiac vagal baroreflex represent the most fundamental regulatory mechanisms for short-term and long-term maintenance of BP and HR.18,19 To sustain a reduction in BP and HR, it is, therefore, imperative that the baroreflex-mediated sympathetic vasomotor tone must be depressed and the cardiac vagal baroreflex augmented. Our present results revealed that this essential modus operandi indeed underlies the cardiovascular depressive effects of activation of spinal NOP. We found that on intrathecal administration of nociceptin, the decrease in BLF power and increase in BRS, our experimental indices for the two arms of the baroreflex, were accompanied by concomitant reduction in MAP and HR. Lai et al8 showed in rat slices that nociceptin concentration-dependently suppresses the excitatory postsynaptic potentials evoked by focal stimulation and hyperpolarizes a population of antidromically identified sympathetic preganglionic neurons. It is, therefore, conceivable that activation of NOP on the IML neurons sustains a decrease in BP by lessening the efficacy of the baroreflex-mediated sympathetic vasomotor tone via a reduction in sympathetic outflow to the blood vessels. On the other hand, by inhibiting the IML neurons, nociceptin sustains a decrease in HR by potentiating the cardiac vagal baroreflex via a reduction in sympathetic excitatory outflow to the heart that normally balances the cardiac inhibitory effects of the vagal outflow from the nucleus ambiguus.
Direct injection of nociceptin into the nucleus tractus solitarii increases BP and HR,15 and microinjection of this peptide into the rostral ventrolateral medulla causes hypotension and bradycardia.14 Electrophysiologic results from rat slices also showed that nociceptin decreases GABAergic neurotransmission to cardiac vagal neurons in the nucleus ambiguus.40 Collectively, these observations are consistent with the notion that nociceptin may act on key components of the baroreflex circuits to depress baroreflex-mediated sympathetic vasomotor tone and enhance cardiac vagal baroreflex. It is, therefore, possible that our present results may have arisen from inadvertent diffusion of the intrathecally delivered nociceptin to the brain stem. This possibility is deemed unlikely because we have demonstrated that given intrathecally at a volume of 3 µL using our method, the highest levels of the spinal cord that the injected agents may reach are T1–T2.28
The relationship between BP and pain is equivocal at best. Hypoalgesia has been described in animal models of hypertension.20,41,42 An inverse relationship between BP and pain sensitivity has also been reported.23 Based on concurrent evaluation of changes in BP and pain responses, our results revealed that the cardiovascular actions of spinally-applied nociceptin are independent of and disproportional to its antinociceptive effects. We demonstrated that restoring the reduced MAP, HR, or BLF power and augmented BRS to baseline levels with the infusion of dopamine did not affect the increase in tail-flick latency induced by nociceptin. Intriguingly, for a given dose, there was a disproportional degree of inhibition in the order of antinociception >>> hypotension. Similar observations have been reported previously for clonidine and guanabenz.22,24 Nevertheless, our study design does not allow us to offer a mechanistic explanation. Speculatively, on an equimolar basis, the much larger areas and hence presumably higher presence of NOP in the dorsal horn when compared to the IML may account for the differential cardiovascular depressive and antinociceptive effects of nociceptin. Nociceptin has been shown to produce biphasic effects in the hole-board exploration test in mice.43 Perhaps its depressive effects on nociception and cardiovascular function are similarly manifested. By showing that nociceptin induced dose-dependent cardiovascular depression and antinociception (Figures 2, 3, and 5A), the possibility for nociceptin to exhibit biphasic dose-related responses is deemed minimal.
Denayer et al44 pointed to the importance of selecting the appropriate mouse strain for mechanistic investigation. Mattson38 and Mogil et al39 also drew attention to potentially strain-related differences in cardiovascular and antinociceptive responses. By showing comparable results from an outbred strain (ICR mice) and inbred strain (C57BL/6 mice), we confirmed that our observed cardiovascular responses to nociceptin were not strain-specific. We recognize that our results on tail-flick latency were obtained under 1% isoflurane and may hence be confounded by the use of anesthesia. However, as our experimental design calls for simultaneous evaluation of the cardiovascular responses, an appropriate level of sedation is a mandatory requirement by our Institutional Animal Care and Use Committee. We reason that as the tail-flick latency is a specific measurement of spinally mediated pain response,35,36 and intrathecal administration of nociceptin elicited a dose-related increase in this nociceptive response that is independent of its cardiovascular effects, the influence of anesthesia on our results is deemed minimal. The doses of nociceptin we used (1, 2, or 5 nmol=1.8, 3.6, or 9 µg) in anesthetized mice compared favorably with the intrathecal antinociceptive half-maximal effective dose value (8.2 µg) in awake mice.45
Conclusion
Based on the concurrent activation of NOP on the IML and dorsal horn via the intrathecal route, the present study revealed that nociceptin reduces BP and HR by decreasing the sympathetic outflow of both arms of the baroreflex arc from the IML to the blood vessels and the heart. We further found that the cardiovascular depressive actions of nociceptin are independent of its antinociceptive responses and were in the order of hypotension <<< antinociception. The latter finding may have promising therapeutic implications, because it is ideal to have a drug that is high in antinociceptive potency and has much lower depressive effects on hemodynamic functions.
Acknowledgments
This work was supported in part by research grants from the Chang Gung Medical Foundation CMRPG8A0851 (to Y.Y. Poon) and OMRPG8C0021 (to S.H.H. Chan). The funding body plays no role in the design of the study, data collection, analysis, interpretation of the data, or in writing the manuscript. The abstract of this paper was presented at the 18th World Congress of Basic and Clinical Pharmacology held at Kyoto/Japan, July 1–6, 2018, as a poster with interim findings. The abstract is available online at https://www.micenavi.jp/wcp2018/search/detail_program/id:1816.
Disclosure
The authors report no conflicts of interest in this work.
References
Mollereau C, Parmentier M, Mailleux P, et al. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994;341(1):33–38. | ||
Meunier JC, Mollereau C, Toll L, et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377(6549):532–535. | ||
Reinscheid RK, Nothacker HP, Bourson A, et al. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270(5237):792–794. | ||
Hawes BE, Graziano MP, Lambert DG. Cellular actions of nociceptin: transduction mechanisms. Peptides. 2000;21(7):961–967. | ||
New DC, Wong YH. The ORL1 receptor: molecular pharmacology and signalling mechanisms. Neurosignals. 2002;11(4):197–212. | ||
Vaughan CW, Christie MJ. Increase by the ORL1 receptor (opioid receptor-like1) ligand, nociceptin, of inwardly rectifying K conductance in dorsal raphe nucleus neurones. Br J Pharmacol. 1996;117(8):1609–1611. | ||
Brailoiu GC, Lai CC, Chen CT, Hwang LL, Lin HH, Dun NJ. Sympathoinhibitory action of nociceptin in the rat spinal cord. Clin Exp Pharmacol Physiol. 2002;29(3):233–237. | ||
Lai CC, Wu SY, Chen CT, Dun NJ. Nociceptin inhibits rat sympathetic preganglionic neurons in situ and in vitro. Am J Physiol Regul Integr Comp Physiol. 2000;278(3):R592–R597. | ||
Anton B, Fein J, To T, Li X, Silberstein L, Evans CJ. Immunohistochemical localization of ORL-1 in the central nervous system of the rat. J Comp Neurol. 1996;368(2):229–251. | ||
Dun NJ, Dun SL, Hwang LL. Nociceptin-like immunoreactivity in autonomic nuclei of the rat spinal cord. Neurosci Lett. 1997;234(2–3):95–98. | ||
Gouardères C, Tafani JA, Meunier JC, Jhamandas K, Zajac JM. Nociceptin receptors in the rat spinal cord during morphine tolerance. Brain Res. 1999;838(1–2):85–94. | ||
Champion HC, Czapla MA, Kadowitz PJ. Nociceptin, an endogenous ligand for the ORL1 receptor, decreases cardiac output and total peripheral resistance in the rat. Peptides. 1997;18(5):729–732. | ||
Chitravanshi VC, Sapru HN. Microinjections of nociceptin into the nucleus ambiguus elicit tachycardia in the rat. Brain Res. 2005;1051(1–2):199–204. | ||
Chu X, Xu N, Li P, Mao L, Wang JQ. Inhibition of cardiovascular activity following microinjection of novel opioid-like neuropeptide nociceptin (orphanin FQ) into the rat rostral ventrolateral medulla. Brain Res. 1999;829(1–2):134–142. | ||
Mao L, Wang JQ. Pharmacological activation of nociceptin receptors in the nucleus tractus solitarius inhibits baroreceptor reflex in pentobarbital-anesthetized rats. Neuroscience. 2000;101(2):435–440. | ||
Yamamoto T, Nozaki-Taguchi N, Sakashita Y, Kimura S. Nociceptin/orphanin FQ: role in nociceptive information processing. Prog Neurobiol. 1999;57(5):527–535. | ||
Hao JX, Xu IS, Wiesenfeld-Hallin Z, Xu XJ. Anti-hyperalgesic and anti-allodynic effects of intrathecal nociceptin/orphanin FQ in rats after spinal cord injury, peripheral nerve injury and inflammation. Pain. 1998;76(3):385–393. | ||
Cowley AW Jr, Liard JF, Guyton AC. Role of baroreceptor reflex in daily control of arterial blood pressure and other variables in dogs. Circ Res. 1973;32(5):564–576. | ||
Thrasher TN. Baroreceptors and the long-term control of blood pressure. Exp Physiol. 2004;89(4):331–335. | ||
Ghione S. Hypertension-associated hypalgesia. Evidence in experimental animals and humans, pathophysiological mechanisms, and potential clinical consequences. Hypertension. 1996;28(3):494–504. | ||
Saccò M, Meschi M, Regolisti G, et al. The relationship between blood pressure and pain. J Clin Hypertens (Greenwich). 2013;15(8):600–605. | ||
Chan SH. Differential effects of clonidine on pain, arterial blood pressure, and heart rate in the cat: lack of interactions with naloxone. Exp Neurol. 1984;84(2):338–346. | ||
Duschek S, Dietel A, Schandry R, del Paso GA. Increased sensitivity to heat pain in chronic low blood pressure. Eur J Pain. 2009;13(1):28–34. | ||
Lim HC, Chong OK, Chan SH. Concomitant hypotensive and antinociceptive effects of guanabenz in conscious rats: involvement of nucleus reticularis gigantocellularis. Exp Neurol. 1988;99(1):133–141. | ||
Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 2010;11(12):823–836. | ||
Appel NM, Elde RP. The intermediolateral cell column of the thoracic spinal cord is comprised of target-specific subnuclei: evidence from retrograde transport studies and immunohistochemistry. J Neurosci. 1988;8(5):1767–1775. | ||
Poon YY, Chang AY, Chan SH. Differential contribution of N-methyl-D-aspartate and non-N-methyl-D-aspartate receptors in the intermediolateral cell column of the thoracic spinal cord to sympathetic vasomotor tone during experimental endotoxemia in the rat. Shock. 2006;26(4):372–378. | ||
Poon YY, Chang AY, Ko SF, Chan SH. Catheterization of the thoracic spinal subarachnoid space in mice. J Neurosci Methods. 2011;200(1):36–40. | ||
Szczesny G, Veihelmann A, Massberg S, Nolte D, Messmer K. Long-term anaesthesia using inhalatory isoflurane in different strains of mice-the haemodynamic effects. Lab Anim. 2004;38(1):64–69. | ||
Poon YY, Tsai CY, Cheng CD, Chang AY, Chan SH. Endogenous nitric oxide derived from NOS I or II in thoracic spinal cord exerts opposing tonic modulation on sympathetic vasomotor tone via disparate mechanisms in anesthetized rats. Am J Physiol Heart Circ Physiol. 2016;311(3):H555–H562. | ||
Su CH, Tsai CY, Chang AY, Chan JY, Chan SH. MRI/DTI of the brain stem reveals reversible and irreversible disruption of the baroreflex neural circuits: clinical implications. Theranostics. 2016;6(6):837–848. | ||
Baudrie V, Laude D, Elghozi JL. Optimal frequency ranges for extracting information on cardiovascular autonomic control from the blood pressure and pulse interval spectrograms in mice. Am J Physiol Regul Integr Comp Physiol. 2007;292(2):R904–R912. | ||
Laude D, Baudrie V, Elghozi JL. Applicability of recent methods used to estimate spontaneous baroreflex sensitivity to resting mice. Am J Physiol Regul Integr Comp Physiol. 2008;294(1):R142–R150. | ||
Tsai CY, Su CH, Baudrie V, et al. Visualizing oxidative stress-induced depression of cardiac vagal baroreflex by MRI/DTI in a mouse neurogenic hypertension model. Neuroimage. 2013;82:190–199. | ||
Liu CH, Cherng CH, Lin SL, et al. N-methyl-D-aspartate receptor antagonist MK-801 suppresses glial pro-inflammatory cytokine expression in morphine-tolerant rats. Pharmacol Biochem Behav. 2011;99(3):371–380. | ||
Nazzaro C, Rizzi A, Salvadori S, et al. UFP-101 antagonizes the spinal antinociceptive effects of nociceptin/orphanin FQ: behavioral and electrophysiological studies in mice. Peptides. 2007;28(3):663–669. | ||
Inoue M, Shimohira I, Yoshida A, et al. Dose-related opposite modulation by nociceptin/orphanin FQ of substance P nociception in the nociceptors and spinal cord. J Pharmacol Exp Ther. 1999;291(1):308–313. | ||
Mattson DL. Comparison of arterial blood pressure in different strains of mice. Am J Hypertens. 2001;14(5 Pt 1):405–408. | ||
Mogil JS, Smith SB, O’Reilly MK, Plourde G. Influence of nociception and stress-induced antinociception on genetic variation in isoflurane anesthetic potency among mouse strains. Anesthesiology. 2005;103(4):751–758. | ||
Venkatesan P, Wang J, Evans C, Irnaten M, Mendelowitz D. Nociceptin inhibits gamma-aminobutyric acidergic inputs to cardiac parasympathetic neurons in the nucleus ambiguus. J Pharmacol Exp Ther. 2002;300(1):78–82. | ||
Sitsen JM, de Jong W. Hypoalgesia in genetically hypertensive rats (SHR) is absent in rats with experimental hypertension. Hypertension. 1983;5(2):185–190. | ||
Wendel OT, Bennett B. The occurrence of analgesia in an animal model of hypertension. Life Sci. 1981;29(5):515–521. | ||
Kamei J, Matsunawa Y, Miyata S, Tanaka S, Saitoh A. Effects of nociceptin on the exploratory behavior of mice in the hole-board test. Eur J Pharmacol. 2004;489(1–2):77–87. | ||
Denayer T, Stöhr T, Van Roy M. Animal models in translational medicine: validation and prediction. New Horiz Transl Med. 2014;2(1):5–11. | ||
Kolesnikov YA, Pasternak GW. Peripheral orphanin FQ/nociceptin analgesia in the mouse. Life Sci. 1999;64(22):2021–2028. | ||
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.