")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer's disease and schizophrenia
Authors Foster D, Choi D, Conn PJ, Rook J
Received 28 September 2013
Accepted for publication 18 October 2013
Published 28 January 2014 Volume 2014:10 Pages 183—191
DOI https://doi.org/10.2147/NDT.S55104
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Daniel J Foster, Derrick L Choi, P Jeffrey Conn, Jerri M Rook
Department of Pharmacology and Vanderbilt Center for Neuroscience Drug Discovery, Vanderbilt University Medical Center, Nashville, TN, USA
Abstract: Alzheimer's disease (AD) and schizophrenia (SZ) are neurological disorders with overlapping symptomatology, including both cognitive deficits and behavioral disturbances. Current clinical treatments for both disorders have limited efficacy accompanied by dose-limiting side effects, and ultimately fail to adequately address the broad range of symptoms observed. Novel therapeutic options for AD and SZ are needed to better manage the spectrum of symptoms with reduced adverse-effect liability. Substantial evidence suggests that activation of muscarinic acetylcholine receptors (mAChRs) has the potential to treat both cognitive and psychosis-related symptoms associated with numerous central nervous system (CNS) disorders. However, use of nonselective modulators of mAChRs is hampered by dose-limiting peripheral side effects that limit their clinical utility. In order to maintain the clinical efficacy without the adverse-effect liability, efforts have been focused on the discovery of compounds that selectively modulate the centrally located M1 and M4 mAChR subtypes. Previous drug discovery attempts have been thwarted by the highly conserved nature of the acetylcholine site across mAChR subtypes. However, current efforts by our laboratory and others have now focused on modulators that bind to allosteric sites on mAChRs, allowing these compounds to display unprecedented subtype selectivity. Over the past couple of decades, the discovery of small molecules capable of selectively targeting the M1 or M4 mAChR subtypes has allowed researchers to elucidate the roles of these receptors in regulating cognitive and behavioral disturbances in preclinical animal models. Here, we provide an overview of these promising preclinical and clinical studies, which suggest that M1- and M4-selective modulators represent viable novel targets with the potential to successfully address a broad range of symptoms observed in patients with AD and SZ.
Keywords: muscarinic receptors, schizophrenia, Alzheimer's disease
Introduction
Schizophrenia (SZ) and Alzheimer’s disease (AD) are two devastating disorders of the central nervous system (CNS) that present clinically with cognitive impairments and psychotic symptoms. Psychosis is the hallmark symptom of SZ and manifests as hallucinations, disordered thought/speech, and delusions. While these psychotic symptoms are commonly associated with SZ, it has become well documented that these patients also experience cognitive and behavioral disturbances that are not adequately addressed by currently prescribed typical and atypical psychotics.1 Conversely, the most commonly associated symptoms of AD are cognitive in nature and include deficits in learning and memory. However, 50%–80% of AD patients display psychotic and behavioral disturbances that are correlated with poor social and functional outcomes.2 While these two diseases arise from separate etiologies, there is a large amount of overlap in the cognitive deficits and psychotic symptoms that are observed. Currently available therapies for these conditions fail to alleviate the broad range of symptoms experienced by patients and are often hampered by dose-limiting side effects, emphasizing the need for novel therapeutics with which to treat these patients.
Another commonality between AD and SZ is the apparent involvement of dysregulated cholinergic signaling in the brain.3,4 Acetylcholine (ACh) is a neurotransmitter that modulates neuronal function in several areas of the CNS associated with AD and/or SZ pathology, including the striatum, cortex, hippocampus, and prefrontal cortex.5 ACh mediates its actions via two families of receptors, termed the muscarinic ACh receptors (mAChRs) and the nicotinic ACh receptors (nAChRs). Here, we review the potential of mAChR modulation for the treatment of AD and SZ; however, modulation of nAChRs could also provide novel therapeutic avenues for treating these diseases (see Taly et al6 for a comprehensive review).
The mAChR family consists of five subtypes (M1–M5) that can be found throughout the CNS and periphery. These receptors are guanosine nucleotide-binding protein (G-protein)-coupled receptors and can be subdivided based on their canonical signaling pathways. M1, M3, and M5 all signal primarily via the Gαq G-protein and induce Ca2+ mobilization and inositol trisphosphate (IP3) production, while M2 and M4 signal via the Gαi G-protein to inhibit cyclic adenosine monophosphate (cAMP) production. As discussed in further detail below, treatments that broadly augment cholinergic signaling have demonstrated clinical efficacy in treating the cognitive and behavioral deficits observed in AD and SZ patients. However, the clinical utility of these treatments is curtailed by peripherally mediated side effects. The recent discovery of compounds that selectively act at the M1 or M4 receptor have suggested that these receptors may provide viable drug targets with which to safely and effectively treat AD and SZ patients.
Alzheimer’s disease
AD is the most commonly diagnosed form of dementia and currently affects approximately 35 million individuals worldwide.7 AD is a progressive neurodegenerative disease that is characterized by a host of cognitive deficits, including impairments in learning and memory. In addition to the well-documented cognitive impairments, AD patients also display behavioral disturbances, including anxiety, depression, and psychosis.8 Age is the primary risk factor for AD, and the disease usually manifests in individuals after the age of 60 years. Due to an aging population, the prevalence of AD is predicted to rise to 66 million people by the year 2030. This devastating disease burdens not only the afflicted and their families, but also generates a global financial burden, with dementias costing society approximately US$604 billion in 2010 alone.7 Given the necessity for increased attention and care, AD places a great burden and strain on the daily lives of patients, families, and caregivers.
The hallmarks of AD pathology are the accumulation of amyloid-beta (Aβ) peptide aggregates (neuritic plaques) and hyperphosphorylated tau protein (neurofibrillary tangles).9,10 The popular amyloid cascade hypothesis posits that the gradual build-up of Aβ plaques leads to neuronal inflammation, dysfunction, and, eventually, cell death. The two brain regions most critically affected by this degeneration are the cortex and hippocampus, both of which are involved in cognition, learning, and memory.11 Several lines of evidence suggest that impaired cholinergic signaling plays a key role in mediating both the cognitive and the behavioral impairments observed in AD patients.12 The basal forebrain cholinergic system is disproportionately affected in AD patients, with a robust loss of cholinergic neurons, including those innervating the hippocampus and cortex.4,13,14 In addition, administration of nonselective muscarinic antagonists can produce or exacerbate cognitive deficits in animals,15 as well as in AD patients and both young and old control subjects,16,17 suggesting that mAChRs can directly modulate cognition. The current primary treatments for AD symptoms are acetylcholinesterase inhibitors (AChEIs) such as donepezil, tacrine, galantamine, and rivastigmine, which potentiate cholinergic signaling.18,19 These treatments not only provide improvements in cognitive symptoms associated with AD,20,21 but also show efficacy in treating the psychiatric symptoms.22,23 Unfortunately, cardiovascular and gastrointestinal side effects are often observed with these treatments, effects thought to be mediated by peripherally located ACh receptors. Despite this, AChEIs remain modestly beneficial for treating AD and other forms of dementia.24 Collectively, these findings highlight the importance of the cholinergic system in mediating the cognitive and behavioral deficits observed in AD patients and highlight the need to develop cholinergic therapeutics that can provide clinical efficacy in the absence of peripherally mediated side effects.
Schizophrenia
SZ is a severe and debilitating psychiatric disease that affects approximately 1% of the population.25 It is characterized by multiple symptom clusters, including positive symptoms, negative symptoms, and cognitive impairments. The onset of SZ symptoms usually occurs early in life (before 30 years of age), between adolescence and young adulthood. There is marked variability in the symptomatology between individual SZ patients, and the neurobiology of the disease is complex. Accordingly, it has been hypothesized that SZ symptoms can arise as a result of numerous underlying etiologies that are poorly understood.26 The hallmark psychotic symptoms of SZ are the positive cluster and include auditory hallucinations, delusional beliefs, and disorganized thoughts and speech. SZ patients also exhibit negative symptoms, including anhedonia, dysfunctional social interactions, and poverty of thoughts and speech, as well as cognitive disturbances affecting several behavioral domains, including working memory, attention, and executive function. Furthermore, the above-mentioned symptom groups are commonly accompanied by disruptions in mood and substance abuse, which affect 40%–80% of SZ patients.27,28 Given the vast spectrum of symptoms observed in SZ patients, it is important to develop therapeutic treatments that can provide efficacy in treating positive, negative, and cognitive deficits.
Although the causes of SZ remain largely unknown, research over the past couple of decades has focused on the dysregulation of signaling by monoamines such as dopamine and serotonin. The prevailing dopaminergic hypothesis attributes positive symptoms to hyperdopaminergic activity in striatal and mesolimbic pathways, while negative symptoms are ascribed to hypodopaminergic activity in the medial prefrontal cortical and mesocortical pathways.29 Current treatments include both typical (eg, haloperidol and chlorpromazine) and atypical (eg, risperidone and clozapine) antipsychotics, which act on the dopaminergic system and D2 dopamine receptors in particular. These treatments show partial efficacy in reducing psychotic or positive symptoms;30 however, they demonstrate little to no efficacy in addressing negative symptoms and cognitive impairments, which can prevent patients from participating fully and productively in society.31,32 Despite the beneficial effects of treating positive symptoms in SZ patients, up to 74% of patients on atypical antipsychotics discontinue use after 18 months due to adverse parkinsonian-like and metabolic syndrome side effects.1 Accumulating evidence suggests that the three clusters of SZ symptoms cannot be ascribed solely to alterations in monoaminergic signaling as dysregulation of glutamatergic, γ-aminobutyric acid (GABA)-ergic, and cholinergic systems have also been reported.3,33–35 The therapeutic efficacy of AChEIs in ameliorating cognitive deficits in AD has led to the hypothesis that these same drugs could be effective as an adjunct medication in SZ patients. Unfortunately, the results from clinical trials with AChEIs in SZ patients have been disappointing,36 likely owing to dose-limiting effects caused by activation of peripheral receptors. Given the shortcomings of these current therapies, it is imperative that novel approaches are developed to provide more comprehensive clinical efficacy with reduced adverse side effects.
Targeting muscarinic receptors for treatment of AD and SZ
The efficacy of AChEIs observed in patients with AD highlights the potential of cholinergic modulation in treating both cognitive- and psychosis-related behavioral disturbances. Furthermore, administration of nonselective muscarinic antagonists can induce cognitive deficits and psychosis in humans,16,37 indicating that mAChR activation may provide pro-cognitive and antipsychotic efficacy. Accordingly, several mAChR agonists have been developed and have entered clinical testing with the goal of ameliorating the behavioral and cognitive deficits observed in numerous psychiatric diseases. Of these, the M1/M4-preferring agonist xanomeline was the only one to progress to a phase III clinical trial, where it was assessed for efficacy in ameliorating cognitive deficits observed in AD patients. While xanomeline showed a trend toward improving cognitive function in these patients, this effect did not reach statistical significance. However, this agonist did produce surprisingly robust and dose-dependent reductions in hallucinations, delusions, vocal outbursts, and other behavioral disturbances in these patients.38,39 The efficacy of xanomeline in treating psychotic and behavioral disturbances in AD patients led to a more recent double-blinded, placebo-controlled outcome trial to determine if similar efficacy could be observed in patients with SZ. This study reported that xanomeline treatment produced robust improvements in both the positive and the negative symptoms of patients with SZ.40 Both the magnitude and the time-course of xanomeline efficacy were superior to those previously reported with atypical antipsychotics, with statistically significant effects observed after only 1 week of treatment. In addition, xanomeline produced statistically significant improvements in verbal learning and short-term memory, indicating efficacy in treating cognitive symptoms.40 Unfortunately, gastrointestinal side effects were observed, and dose limitations have removed it from consideration for long-term clinical use. However, these two seminal studies provide strong clinical validation of mAChRs as targets for the treatment of both psychotic and cognitive disturbances in AD and SZ.
Allosteric modulation of muscarinic receptors
The efficacy of AChEIs in treating AD patients, in conjunction with the efficacy of xanomeline in improving both the cognitive and the behavioral disturbances observed in patients with either AD or SZ, highlight the potential of cholinergic modulation in treating these diseases. However, the poorly tolerated gastrointestinal side effects of AChEIs and xanomeline limit the clinical utility of these compounds.40,41 The adverse side effects observed with nonselective modulation of the cholinergic system are thought to be primarily mediated by peripherally located M2 and M3 receptors.42 Accordingly, it has been hypothesized that selective modulation of the M1, M4, or M5 subtype could maintain the clinical efficacy observed with nonselective cholinergic treatments without the adverse-effect liability. A critical obstacle in these efforts has been the high conservation of the orthosteric ACh-binding site across the five mAChR subtypes, making it difficult to develop subtype-selective ACh-site ligands. However, an alternative strategy of targeting allosteric sites that are distinct from the ACh-binding site has been used with success at numerous G-protein-coupled receptors (for review, see Conn et al).43 By targeting less conserved sites, it has been possible to develop compounds with unprecedented subtype selectivity. Allosteric ligands can modulate receptor signaling via multiple mechanisms. Allosteric agonists bind to an allosteric site and directly cause receptor activation. Alternatively, positive allosteric modulators (PAMs) bind to allosteric sites where they have no effect alone, but increase the affinity and/or efficacy of endogenous agonists. Because allosteric modulators do not directly activate the receptor, but instead potentiate activation by orthosteric ligands, they maintain the temporal and spatial signaling of cholinergic circuits. In some instances, molecules can act as both an allosteric agonist and an allosteric potentiator, indicating that these mechanisms of receptor regulation are not exclusive in nature.44 As discussed below, the discovery of subtype-selective M1 and M4 agonists and modulators have greatly advanced our understanding of the importance of these receptors and emphasize the potential utility of targeting M1 and M4 in the treatment of AD and SZ.
Targeting M1 muscarinic receptors for cognitive symptoms observed with AD and SZ
The M1 mAChR subtype is the most predominantly expressed mAChR subtype in the CNS and is expressed in several brain regions implicated in the regulation of cognitive processes, including the striatum, prefrontal cortex, and hippocampus.45,46 Many of the studies examining the role of the M1 receptor in the CNS have utilized M1 knockout (KO) mice that do not express the M1 receptor. Interestingly, these M1-deficient mice display increased amphetamine-induced hyperlocomotion and dopamine neurotransmission,47 indicating that M1 modulation may have antipsychotic potential. However, the majority of studies have focused on the role of M1 in regulating cognitive processes. N-methyl-D-aspartate (NMDA) receptors play a critical role in regulating synaptic plasticity, and disrupted NMDA-receptor neurotransmission is thought to underlie the cognitive deficits observed in numerous psychiatric diseases. M1 mAChRs have been demonstrated to potentiate NMDA-receptor signaling in the hippocampus and cortex,48,49 brain areas intimately associated with learning and memory. In addition, M1 KO mice displayed reduced hippocampal long-term potentiation, a mechanism heavily implicated in learning and memory. Behaviorally, M1 KO animals display deficits in several medial prefrontal cortex-dependent cognitive tasks, including non-matching-to-sample, win-shift radial arm maze, and social discrimination tasks.50 Finally, studies in mice exhibiting AD-like Aβ plaque pathologies found that deletion of M1 increased amyloidogenic processes, suggesting that M1 may play a role in regulating AD disease progression.51 Collectively, these studies provided key rationale to pursue compounds targeting the M1 mAChR for the treatment of cognitive symptoms observed in neuropsychiatric diseases.
Numerous M1-selective compounds have been discovered and subsequently tested in preclinical animal models of cognition (see Table 1 for complete list). A breakthrough came with the discovery of the first-generation M1 mAChR allosteric agonist AC-42. This compound was found to bind to an allosteric site and displayed M1-selective functional activity when assessed at muscarinic subtypes in vitro.52 However, this compound does not possess the physiochemical properties necessary for in vivo use.53 Subsequent optimization produced two analogs of AC-42 (AC-260584 and 77-LH-28-1), which maintained M1 selectivity and possessed properties suitable for use in animal models. Both AC-260584 and 77-LH-28-1 displayed antipsychotic and cognition-enhancing efficacy in pre-clinical models.54–56 Unfortunately, the efficacy of AC-260584 was confounded by nonselective effects on dopaminergic, adrenergic, and serotonergic receptors.57 Another early allosteric agonist, TBPB (1-(1’-(2-methylbenzyl)-1,4’-bipiperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-1), also exhibited impressive selectivity for M1 mAChRs and potentiated NMDA receptor currents in CA1 hippocampal cells.58 Moreover, additional pre-clinical studies with TBPB demonstrated efficacy in reducing antipsychotic-like behaviors and in reversing scopolamine-impaired acquisition of contextual fear.59 Studies in cell lines also demonstrated that TBPB promoted a non-amyloidogenic pathway and decreased Aβ production, indicating that M1 modulation may have efficacy in the treatment of both symptomatic and pathologic features of AD.58 More recently, the M1-selective allosteric agonist VU0357017 was discovered, which displayed improved potency via binding to a novel allosteric site on the M1 mAChR. VU0357017 significantly blocked scopolamine-impaired contextual fear conditioning and enhanced spatial and contextual fear learning.60,61 Interestingly, recent studies suggest that many M1 allosteric agonists, including 77-LH-28-1 and VU0357017, may act in a ‘bitopic’ manner, simultaneously binding at both allosteric and orthosteric sites.62–66 In addition to the confounding issue of bitopic binding, some M1 allosteric agonists display ‘signal bias’ or context-dependent pharmacology and differentially activate various downstream signaling pathways such as Ca2+ mobilization and β-arrestin activation.60 A recent clinical study utilizing the M1-selective allosteric agonist GSK1034702 demonstrated pro-cognitive efficacy in a nicotine abstinence model of episodic memory impairment in smokers,67 providing exciting evidence that M1-selective activation can provide pro-cognitive benefits in humans. Collectively, these preclinical and clinical findings with allosteric agonists highlight the potential utility of M1-selective activation in treating cognitive deficits observed with numerous CNS disorders.
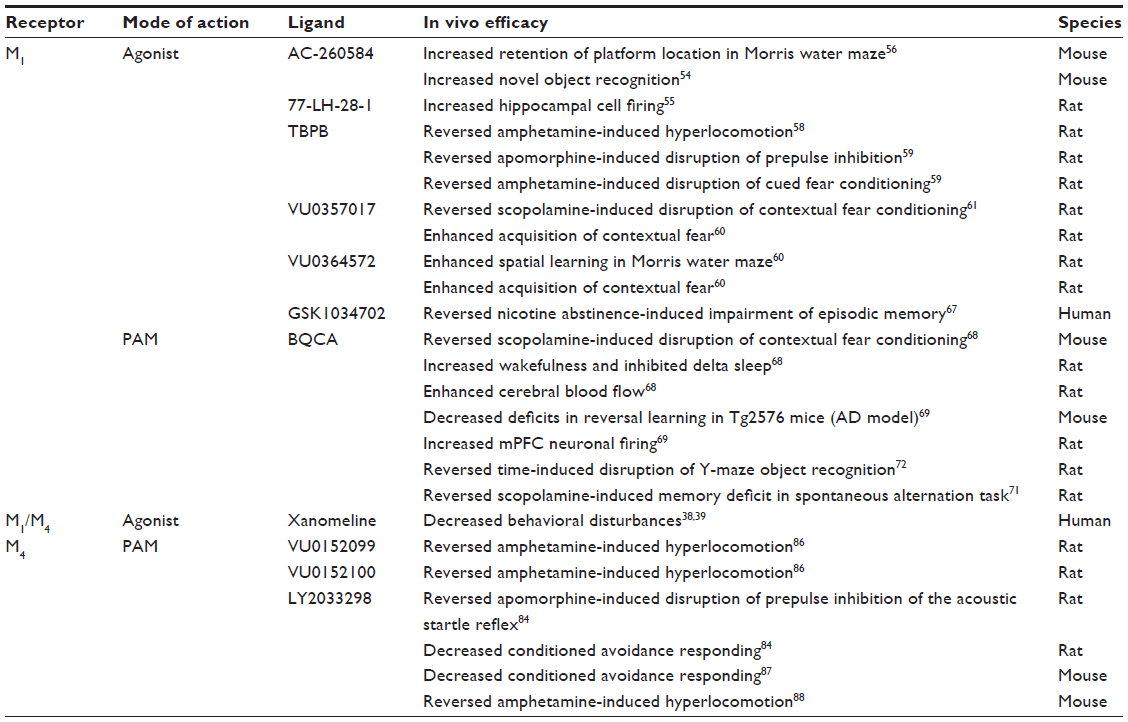 | Table 1 Antipsychotic and pro-cognitive effects of M1 and M4 selective modulators |
Another approach extensively studied in recent years involves the development of PAM compounds that bind to allosteric sites on the M1 mAChR and indirectly promote activity by enhancing the affinity and/or efficacy of the endogenous ligand ACh. The first subtype-selective M1 PAM to be characterized was benzyl quinolone carboxylic acid (BQCA);68 BQCA exhibited high selectivity with no activity at mAChR subtypes M2–M5 and induced up to a 129-fold leftward shift in ACh potency at the M1 mAChR.68,69 In brain slice electrophysiology studies, BQCA enhanced excitatory postsynaptic currents in medial prefrontal cortical neurons,69 an area critical for higher cognitive, learning, and memory functions.70 In pre-clinical animal studies, BQCA reversed scopolamine-impaired contextual fear conditioning and rescued medial prefrontal cortex-dependent discrimination reversal learning deficits in a transgenic mouse model of AD.68,69 Additionally, recent studies demonstrated that BQCA was effective in reversing memory deficits in Y-maze object recognition and spontaneous alternation tasks in rats.71,72 The discovery and characterization of BQCA exemplified the promising potential for allosteric modulators to selectively target M1 mAChRs, and has since provided a basis for further development of similar M1 PAMs.73 Recent drug discovery efforts in our group have yielded novel M1-selective PAMs VU0405652 (ML169) and VU0456940, both of which potentiate M1-mediated non-amyloidogenic amyloid precursor protein (APPsα) processing, suggesting disease-modifying potential in AD.74,75 Collectively, these studies provide evidence supporting the therapeutic potential of selectively targeting M1 mAChRs in the treatment of AD and SZ. However, continued development and characterization of M1-selective compounds is needed to fully elucidate the potential of M1-modulation in mediating symptomatic and disease-modifying efficacy in AD and SZ.
Targeting M4 muscarinic receptors for psychotic symptoms observed with AD and SZ
Recent evidence suggests that modulation of the M4 receptor may provide a novel avenue for the development of anti-psychotic drugs. The psychotic symptoms associated with SZ are thought to be intimately associated with hyperactive dopaminergic signaling in striatal and mesocortical pathways. Clinically prescribed typical and atypical psychotics show efficacy in reducing psychosis and exert their effects primarily via antagonizing the D2 dopamine receptor. Nonselective mAChR agonists can reduce striatal dopamine release,76 while administration of nonselective mAChR antagonists can induce psychosis in humans37 and disrupt sensorimotor gating in the preclinical prepulse inhibition rodent model.77 Conversely, the nonselective mAChR agonist BuTAC ([5R-(exo)]-6-[4-butylthio-1,2,5-thiadiazol-3-yl]-1-azabicyclo-[3.2.1]-octane) shows an antipsychotic profile when tested in numerous preclinical animal models. Administration of BuTAC reduces apomorphine-induced climbing and apomorphine-induced disruptions of prepulse inhibition78,79 and reduces conditioned avoidance responding in wild-type, but not M4 KO mice.80 Collectively, these results suggest that activation of mAChRs, and M4 in particular, may provide a novel strategy for treating psychotic symptoms in AD and SZ.
As mentioned above, clinical trials for xanomeline yielded the striking finding that administration of the M1/M4-preferring agonist significantly reduced psychosis-related symptoms in both AD and SZ patients.38–40 The M4 receptor is highly expressed in the striatum, hippocampus, and neocortex,45,46 suggesting that this mAChR subtype is ideally located to modulate dopaminergic signaling. In support of this hypothesis, M4 KO mice exhibit a hyperdopaminergic phenotype that is resistant to mAChR agonist-induced attenuation of dopamine levels.76,81 Selective deletion of M4 mAChRs on D1 dopamine receptor-expressing neurons resulted in increased locomotor activity and behavioral sensitization to psychostimulants.82 Additionally, the antipsychotic efficacy of xanomeline in preclinical animal models is attenuated in animals in which this subpopulation of M4 receptors is deleted.83 These findings support the hypothesis that M4 mAChRs represent a viable novel drug target for the treatment of psychosis in SZ, AD, and other neurological disorders.
Two novel M4-selective compounds, VU10010 and LY2033298, represented a breakthrough when they were described in 2008.84,85 VU10010 is a potent M4-selective PAM that increases affinity/efficacy of ACh to promote M4 mAChR activation. In brain slices, VU10010 selectively potentiated mAChR-mediated reductions in glutamatergic, but not GABAergic, signaling in hippocampal neurons, indicating a key role for M4 in regulating hippocampal function, and possibly in modulating cognition. Though these findings were major advances in validating the concept of selective M4 PAMs at a cellular and molecular level, VU10010 does not possess physiochemical properties suitable for in vivo dosing (displaying a high log P-value of 4.5), limiting the utility of this tool compound.85 Subsequent optimization of VU10010 led to the discovery of VU0152100 and VU0152099, both of which possessed improved chemical and pharmacokinetic characteristics making them suitable for use in rodent models. Both compounds possess log P-values a full order of magnitude less lipophilic than VU10010, resulting in improved solubility and affording homogeneous dosing solutions in multiple vehicles acceptable for in vivo studies.86 Additionally, VU0152100 and VU0152099 exhibited substantial systemic absorption and brain penetration following intraperitoneal administration.86 Both VU0152100 and VU0152099 effectively reversed amphetamine-induced hyperlocomotion, demonstrating antipsychotic-like activity in preclinical models. LY2033298, a structurally distinct M4-selective PAM, was similarly efficacious in several preclinical models of psychosis, including conditioned avoidance responding and apomorphine-impaired prepulse inhibition.84,87,88 Together, these preliminary efforts suggest that the antipsychotic effects of xanomeline may be primarily due to M4 mAChR activation. The development of M4-selective PAMs has proven to be very fruitful in substantiating the efficacy in targeting M4 mAChRs for the potential treatment of the positive symptoms of SZ. In addition, preliminary studies suggest that selective activation may enhance cognitive domains of learning and memory. However, further in vivo characterization of M4 is required to elucidate the therapeutic potential of M4 modulation in treating the cognitive impairments and psychotic symptoms associated with SZ and AD.
Summary
The recent discovery of novel allosteric agonists and modulators of M1 and M4 mAChRs have further validated the approach of targeting these muscarinic subtypes in the treatment of cognitive and behavioral impairments present in AD and SZ. Despite the fact that most discovery efforts are still in the preclinical phase of development, there are now several tool compounds that continue to provide important findings, furthering our fundamental understanding of the role of mAChRs and these debilitating neuropsychiatric diseases. Data from clinical trials demonstrating the efficacy of xanomeline in AD and SZ patients have created intense interest in the pursuit of highly selective M1 and M4 activators to ultimately provide novel therapeutic options with minimal adverse side effects.
Disclosure
Dr Conn receives research funding and salary support from Bristol-Meyers Squibb and AstraZeneca. Dr Conn is an inventor on patents that protect different classes of mAChR allosteric modulators. The authors have no other conflicts of interest in this work.
References
Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–1223. | |
Borroni B, Costanzi C, Padovani A. Genetic susceptibility to behavioural and psychological symptoms in Alzheimer disease. Curr Alzheimer Res. 2010;7(2):158–164. | |
Sarter M, Lustig C, Taylor SF. Cholinergic contributions to the cognitive symptoms of schizophrenia and the viability of cholinergic treatments. Neuropharmacology. 2012;62(3):1544–1553. | |
Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2(8000):1403. | |
Mesulam MM, Mufson EJ, Wainer BH, Levey AI. Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1–Ch6). Neuroscience. 1983;10(4):1185–1201. | |
Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8(9):733–750. | |
Batsch NL, Mittelman MS. World Alzheimer Report 2012: Overcoming the Stigma of Dementia. London: Alzheimer’s Disease International; 2012. Available from: http://www.alz.co.uk/research/WorldAlzheim erReport2012.pdf. Accessed. January 6, 2013. | |
Grossberg GT. Diagnosis and treatment of Alzheimer’s disease. J Clin Psychiatry. 2003;64 Suppl 9:3–6. | |
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. | |
Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6(4):487–498. | |
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. | |
Woolf NJ. The critical role of cholinergic basal forebrain neurons in morphological change and memory encoding: a hypothesis. Neurobiol Learn Mem. 1996;66(3):258–266. | |
Bartus RT, Dean RL 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217(4558):408–414. | |
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215(4537):1237–1239. | |
Pazzagli A, Pepeu G. Amnesic properties of scopolamine and brain acetylcholine in the rat. Int J Neuropharmacol. 1965;4(5):291–299. | |
Rusted JM, Warburton DM. The effects of scopolamine on working memory in healthy young volunteers. Psychopharmacology (Berl). 1988;96(2):145–152. | |
Sunderland T, Tariot PN, Cohen RM, Weingartner H, Mueller EA 3rd, Murphy DL. Anticholinergic sensitivity in patients with dementia of the Alzheimer type and age-matched controls. A dose-response study. Arch Gen Psychiatry. 1987;44(5):418–426. | |
Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006;(1):CD005593. | |
Muñoz-Torrero D. Acetylcholinesterase inhibitors as disease-modifying therapies for Alzheimer’s disease. Curr Med Chem. 2008; 15(24):2433–2455. | |
Delrieu J, Piau A, Caillaud C, Voisin T, Vellas B. Managing cognitive dysfunction through the continuum of Alzheimer’s disease: role of pharmacotherapy. CNS Drugs. 2011;25(3):213–226. | |
Feldman H. Treating Alzheimer’s disease with cholinesterase inhibitors: what have we learned so far? Int Psychogeriatr. 2002; 14 Suppl 1:3–5. | |
Kaufer D. Beyond the cholinergic hypothesis: the effect of metrifonate and other cholinesterase inhibitors on neuropsychiatric symptoms in Alzheimer’s disease. Dement Geriatr Cogn Disord. 1998;9 Suppl 2:8–14. | |
Rösler M. The efficacy of cholinesterase inhibitors in treating the behavioural symptoms of dementia. Int J Clin Pract Suppl. 2002;(127):20–36. | |
Barten DM, Albright CF. Therapeutic strategies for Alzheimer’s disease. Mol Neurobiol. 2008;37(2–3):171–186. | |
Bhugra D. The global prevalence of schizophrenia. PLoS Med. 2005;2(5):e151; quiz e175. | |
Andreasen NC, Carpenter WT Jr. Diagnosis and classification of schizophrenia. Schizophr Bull. 1993;19(2):199–214. | |
Tollefson GD, Sanger TM, Lu Y, Thieme ME. Depressive signs and symptoms in schizophrenia: a prospective blinded trial of olanzapine and haloperidol. Arch Gen Psychiatry. 1998;55(3):250–258. | |
Westermeyer J. Comorbid schizophrenia and substance abuse: a review of epidemiology and course. Am J Addict. 2006;15(5):345–355. | |
Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148(11):1474–1486. | |
Sawa A, Snyder SH. Schizophrenia: neural mechanisms for novel therapies. Mol Med. 2003;9(1–2):3–9. | |
Hill SK, Bishop JR, Palumbo D, Sweeney JA. Effect of second- generation antipsychotics on cognition: current issues and future challenges. Expert Rev Neurother. 2010;10(1):43–57. | |
Murphy BP, Chung YC, Park TW, McGorry PD. Pharmacological treatment of primary negative symptoms in schizophrenia: a systematic review. Schizophr Res. 2006;88(1–3):5–25. | |
Lisman JE, Coyle JT, Green RW, et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31(5):234–242. | |
Marin O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13(2):107–120. | |
Noetzel MJ, Jones CK, Conn PJ. Emerging approaches for treatment of schizophrenia: modulation of glutamatergic signaling. Discov Med. 2012;14(78):335–343. | |
Thakurathi N, Vincenzi B, Henderson DC. Assessing the prospect of donepezil in improving cognitive impairment in patients with schizophrenia. Expert Opin Investig Drugs. 2013;22(2):259–265. | |
Osterholm RK, Camoriano JK. Transdermal scopolamine psychosis. JAMA. 1982;247(22):3081. | |
Bodick NC, Offen WW, Levey AI, et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol. 1997;54(4):465–473. | |
Bodick NC, Offen WW, Shannon HE, et al. The selective muscarinic agonist xanomeline improves both the cognitive deficits and behavioral symptoms of Alzheimer disease. Alzheimer Dis Assoc Disord. 1997;11 Suppl 4:S16–S22. | |
Shekhar A, Potter WZ, Lightfoot J, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165(8):1033–1039. | |
Conn PJ, Jones CK, Lindsley CW. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci. 2009;30(3):148–155. | |
Bymaster FP, Carter PA, Yamada M, et al. Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur J Neurosci. 2003;17(7):1403–1410. | |
Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8(1):41–54. | |
Noetzel MJ, Rook JM, Vinson PN, et al. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol. 2012;81(2):120–133. | |
Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11(10):3218–3226. | |
Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14(5 Pt 2):3351–3363. | |
Gerber DJ, Sotnikova TD, Gainetdinov RR, Huang SY, Caron MG, Tonegawa S. Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. Proc Natl Acad Sci U S A. 2001;98(26):15312–15317. | |
Marino MJ, Conn PJ. Direct and indirect modulation of the N-methyl D-aspartate receptor. Curr Drug Targets CNS Neurol Disord. 2002;1(1):1–16. | |
Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 1998;95(19):11465–11470. | |
Anagnostaras SG, Murphy GG, Hamilton SE, et al. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci. 2003;6(1):51–58. | |
Davis AA, Fritz JJ, Wess J, Lah JJ, Levey AI. Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J Neurosci. 2010;30(12):4190–4196. | |
Spalding TA, Trotter C, Skjaerbaek N, et al. Discovery of an ectopic activation site on the M(1) muscarinic receptor. Mol Pharmacol. 2002;61(6):1297–1302. | |
Langmead CJ, Fry VA, Forbes IT, et al. Probing the molecular mechanism of interaction between 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1- butyl]-piperidine (AC-42) and the muscarinic M(1) receptor: direct pharmacological evidence that AC-42 is an allosteric agonist. Mol Pharmacol. 2006;69(1):236–246. | |
Bradley SR, Lameh J, Ohrmund L, et al. AC-260584, an orally bioavailable M(1) muscarinic receptor allosteric agonist, improves cognitive performance in an animal model. Neuropharmacology. 2010;58(2):365–373. | |
Langmead CJ, Austin NE, Branch CL, et al. Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist, 77-LH-28-1. Br J Pharmacol. 2008;154(5):1104–1115. | |
Vanover KE, Veinbergs I, Davis RE. Antipsychotic-like behavioral effects and cognitive enhancement by a potent and selective muscarinic M-sub-1 receptor agonist, AC-260584. Behav Neurosci. 2008;122(3):570–575. | |
Heinrich JN, Butera JA, Carrick T, et al. Pharmacological comparison of muscarinic ligands: historical versus more recent muscarinic M1-preferring receptor agonists. Eur J Pharmacol. 2009;605(1–3):53–56. | |
Jones CK, Brady AE, Davis AA, et al. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J Neurosci. 2008;28(41):10422–10433. | |
Kane A. The In Vivo Characterization of TBPB, a Novel Allosteric Agonist of M1 Muscarinic Receptors: Implications for the Role of the M1 Muscarinic Receptor in Treatment of Schizophrenia [undergraduate thesis]. Nashville (TN): Vanderbilt University; 2008. | |
Digby GJ, Noetzel MJ, Bubser M, et al. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J Neurosci. 2012;32(25):8532–8544. | |
Lebois EP, Bridges TM, Lewis LM, et al. Discovery and characterization of novel subtype-selective allosteric agonists for the investigation of M(1) receptor function in the central nervous system. ACS Chem Neurosci. 2010;1(2):104–121. | |
Avlani VA, Langmead CJ, Guida E, et al. Orthosteric and allosteric modes of interaction of novel selective agonists of the M1 muscarinic acetylcholine receptor. Mol Pharmacol. 2010;78(1):94–104. | |
Digby GJ, Utley TJ, Lamsal A, et al. Chemical modification of the M(1) agonist VU0364572 reveals molecular switches in pharmacology and a bitopic binding mode. ACS Chem Neurosci. 2012;3(12):1025–1036. | |
Gregory KJ, Sexton PM, Christopoulos A. Allosteric modulation of muscarinic acetylcholine receptors. Curr Neuropharmacol. 2007;5(3):157–167. | |
Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov. 2013;12(8):630–644. | |
Davie BJ, Christopoulos A, Scammells PJ. Development of M1 mAChR allosteric and bitopic ligands: prospective therapeutics for the treatment of cognitive deficits. ACS Chem Neurosci. 2013; 4(7):1026–1048. | |
Nathan PJ, Watson J, Lund J, et al. The potent M(1) receptor allosteric agonist GSK1034702 improves episodic memory in humans in the nicotine abstinence model of cognitive dysfunction. Int J Neuropsychopharmacol. 2013;16(4):721–731. | |
Ma L, Seager MA, Wittmann M, et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci U S A. 2009;106(37):15950–15955. | |
Shirey JK, Brady AE, Jones PJ, et al. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J Neurosci. 2009;29(45):14271–14286. | |
Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. | |
Chambon C, Jatzke C, Wegener N, Gravius A, Danysz W. Using cholinergic M1 receptor positive allosteric modulators to improve memory via enhancement of brain cholinergic communication. Eur J Pharmacol. 2012;697(1–3):73–80. | |
Chambon C, Wegener N, Gravius A, Danysz W. A new automated method to assess the rat recognition memory: validation of the method. Behav Brain Res. 2011;222(1):151–157. | |
Kuduk SD, Beshore DC. Novel M(1) allosteric ligands: a patent review. Expert Opin Ther Pat. 2012;22(12):1385–1398. | |
Reid PR, Bridges TM, Sheffler DJ, et al. Discovery and optimization of a novel, selective and brain penetrant M1 positive allosteric modulator (PAM): the development of ML169, an MLPCN probe. Bioorg Med Chem Lett. 2011;21(9):2697–2701. | |
Tarr JC, Turlington ML, Reid PR, et al. Targeting selective activation of M(1) for the treatment of Alzheimer’s disease: further chemical optimization and pharmacological characterization of the M(1) positive allosteric modulator ML169. ACS Chem Neurosci. 2012;3(11):884–895. | |
Threlfell S, Clements MA, Khodai T, et al. Striatal muscarinic receptors promote activity dependence of dopamine transmission via distinct receptor subtypes on cholinergic interneurons in ventral versus dorsal striatum. J Neurosci. 2010;30(9):3398–3408. | |
Ukai M, Okuda A, Mamiya T. Effects of anticholinergic drugs selective for muscarinic receptor subtypes on prepulse inhibition in mice. Eur J Pharmacol. 2004;492(2–3):183–187. | |
Jones CK, Eberle EL, Shaw DB, McKinzie DL, Shannon HE. Pharmacologic interactions between the muscarinic cholinergic and dopaminergic systems in the modulation of prepulse inhibition in rats. J Pharmacol Exp Ther. 2005;312(3):1055–1063. | |
Rasmussen T, Fink-Jensen A, Sauerberg P, et al. The muscarinic receptor agonist BuTAC, a novel potential antipsychotic, does not impair learning and memory in mouse passive avoidance. Schizophr Res. 2001;49(1–2):193–201. | |
Watt ML, Rorick-Kehn L, Shaw DB, et al. The muscarinic acetylcholine receptor agonist BuTAC mediates antipsychotic-like effects via the M4 subtype. Neuropsychopharmacology. Epub August 2, 2013. | |
Gomeza J, Zhang L, Kostenis E, et al. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M(4) muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96(18):10483–10488. | |
Jeon J, Dencker D, Wörtwein G, et al. A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors. J Neurosci. 2010;30(6):2396–2405. | |
Dencker D, Wörtwein G, Weikop P, et al. Involvement of a subpopulation of neuronal M4 muscarinic acetylcholine receptors in the antipsychotic-like effects of the M1/M4 preferring muscarinic receptor agonist xanomeline. J Neurosci. 2011;31(16):5905–5908. | |
Chan WY, McKinzie DL, Bose S, et al. Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc Natl Acad Sci U S A. 2008;105(31):10978–10983. | |
Shirey JK, Xiang Z, Orton D, et al. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat Chem Biol. 2008;4(1):42–50. | |
Brady AE, Jones CK, Bridges TM, et al. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J Pharmacol Exp Ther. 2008;327(3):941–953. | |
Leach K, Loiacono RE, Felder CC, et al. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology. 2010;35(4):855–869. | |
Suratman S, Leach K, Sexton P, Felder C, Loiacono R, Christopoulos A. Impact of species variability and ‘probe-dependence’ on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br J Pharmacol. 2011;162(7):1659–1670. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.