")
Back to Journals » Cancer Management and Research » Volume 13
Activation of FXR Suppresses Esophageal Squamous Cell Carcinoma Through Antagonizing ERK1/2 Signaling Pathway
Authors Feng Q, Zhang H, Yao D, Zhang X, Chen WD, Wang YD
Received 22 December 2019
Accepted for publication 17 February 2021
Published 30 July 2021 Volume 2021:13 Pages 5907—5918
DOI https://doi.org/10.2147/CMAR.S243317
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Beicheng Sun
Qingqing Feng,1,* Hongli Zhang,1,* Denglin Yao,1 Xiantong Zhang,1 Wei-Dong Chen,2,3 Yan-Dong Wang1
1State Key Laboratory of Chemical Resource Engineering, College of Life Science and Technology, Beijing University of Chemical Technology, Beijing, People’s Republic of China; 2Key Laboratory of Receptors-Mediated Gene Regulation and Drug Discovery, School of Medicine, Henan University, Kaifeng, Henan, People’s Republic of China; 3Key Laboratory of Molecular Pathology, School of Basic Medical Science, Inner Mongolia Medical University, Hohhot, Inner Mongolia, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yan-Dong Wang Email [email protected]
Wei-Dong Chen Email [email protected]
Introduction: Farnesoid X receptor (FXR), a member of nuclear receptors, functionally regulates bile acid, glucose and lipid homeostasis. It is also worth noting that FXR plays a suppressor role in cancer and inflammation. However, the contribution of FXR to esophageal squamous cell carcinoma (ESCC) remains unknown.
Methods: The role of FXR activation in ESCC progression was evaluated in ESCC cell lines KYSE150 and EC109 in vitro and BALB-C nude mice in vivo. In vitro, FXR synthetic ligand GW4064 was used to detect the effects on ESCC cell proliferation, migration, apoptosis and cell cycles. To assess the effects of GW4064 on ESCC development in vivo, a xenograft tumor model was constructed. And ERK1/2 activity was evaluated by immunoblot analysis.
Results: FXR synthetic ligand GW4064 impaired esophageal squamous cell carcinoma (ESCC) proliferation and migration, induced apoptosis and cell cycle arrest in vitro, accompanied by inhibition of some inflammatory genes and promotion of pro-apoptotic genes. We then found that FXR activation decreased the phosphorylation levels of ERK1/2 induced by tumor necrosis factor-α (TNF-α) in ESCC cells. Consistent with these results, GW4064 suppressed ESCC tumorigenesis in a xenograft model and suppressed the phosphorylation of ERK1/2 in tumors.
Discussion: These findings identify that activating FXR may serve as a promising therapy or adjuvant therapeutic tool for controlling ESCC development.
Keywords: FXR, farnesoid X receptor, GW4064, ESCC, ERK1/2
Introduction
The incidence of esophageal cancer is regional, and esophageal cancer is the 8th most common cancer in the world.1 Esophageal squamous cell carcinoma (ESCC) has a high incidence rate in East Asia, Eastern and Southern Africa, and Southern Europe due to ethnicity, genetic, and dietary.2–4 For the treatment of ESCC, chemotherapy, chemoradiotherapy, and esophagectomy are the mainstays; however, the 5-year survival rate is still poor.5 Current therapeutic strategies for ESCC are limited; thus, it remains urgent to understand pathogenetic mechanisms of ESCC and develop effective strategies to control ESCC.
Mitogen-activated protein kinase (MAPK) pathway, consisting of ERK1/2, JNK1/2, p38 and ERK5, is one of the most classic signal transduction pathways and involves multiple physiological responses.6 And blocking the activation of ERK1/2 may suppress the development of colorectal cancer, pancreatic cancer and bladder cancer.7–9 Recently, Ma et al reported that triptolide displays antitumor activity in ESCC in vitro and xenograft model by inhibiting the phosphorylation of ERK1/2.10 Thus, discovering novel therapeutic drugs or targets that block conventional MAPK/ERK activation would be valuable for repressing tumor development.
Farnesoid X receptor (FXR), one member of nuclear receptor superfamily, is involved in bile acid homeostasis.11–13 In addition, FXR is shown to contribute to multiple physiological metabolisms, such as lipid, fatty acid and glucose metabolism.14 As a ligand-activated transcription factor, upon binding to the ligands, such as natural bile acids (BAs) or synthetic ligands, FXR would initiate regulation of a series of downstream genes. And GW4064 is the most commonly used synthetic ligand in research.11 Over the past decade, accumulating evidence has suggested that FXR activation has suppression function in cancer and inflammation. For example, FXR is a suppressor in hepatic inflammatory through antagonizing NF-κB signaling.15 FXR activation prevents tumor-stimulatory activities of cancer-associated fibroblasts in breast cancer.16 In a recent study, Liu et al reported GW4064 attenuated lipopolysaccharide-induced ileocolitis.17 It suggests that FXR has key roles in different cancers.
In the present research, we demonstrated that synthetic FXR ligand GW4064 dramatically suppressed ESCC cell growth, migration and cell cycle, and promoted cell apoptosis. In addition, GW4064 suppressed ESCC progression in a xenograft model. Furthermore, we reveal that GW4064 inhibits ERK1/2 signaling in ESCC cells. This study implied that activation of FXR by GW4064 is an effective treatment for ESCC.
Materials and Methods
Collection of Tissue Specimens
Human colorectal samples and stomach samples were collected from the Department of General Surgery at Affiliated Huaihe Hospital of Henan University between 2016 and 2017. These patients with Colorectal Cancer, or Gastric cancer, underwent complete surgical resection of the tumor at Affiliated Huaihe Hospital. The tumor tissues and paracancerous tissue samples were snapped frozen and stored in liquid nitrogen until subsequent analysis. Written informed consents were obtained from all subjects, and this study was approved by the Ethics Committee of Affiliated Huaihe Hospital of Henan University, Kaifeng, China.
Animals
Mice were purchased from Beijing Experimental Animal Center (License No. SCXK (Jing) 2002–0003). All animal studies were conducted with approval from the Animal Research Ethics Committee of School of Basic Medical Sciences of Henan University and followed the NIH guidelines for the care and use of laboratory animals. For in vivo tumorigenesis assay, male BALB-C nude mice (4-week old) were each injected subcutaneously into the left-forelimb armpit with EC109 cells (4×106 cells per mouse, n=6–8) in a total volume of 150 μL. GW4064 was dissolved in DMSO and configured to a concentration of 6 mg/mL. When the tumor size reached 100 mm3, GW4064 (30 mg/Kg body weight) was intraperitoneally injected once every three days. In control, mice were injected intraperitoneally with vehicle DMSO. Tumor growth was evaluated with a caliper by measuring tumor length (L) and width (W), and tumor volume was calculated according to 1/2×LW2. The mice were sacrificed after 7th measurement (30 days) and the tumors were removed and weighed.
Cell Culture
ESCC cell lines KYSE150, EC109 and TE-1 were purchased from China Infrastructure of Cell Line Resource, and were incubated in complete RPMI-1640 culture medium (with L-glutamine) supplied with 10% (vol/vol) fetal bovine serum and 1% (vol/vol) penicillin-streptomycin. Cells were pre-plated into 6-well plates and treated with GW4064 (1.5 μM, 3 μM) for 24 h. GW4064 was added to the culture medium as 1000× stock solutions in DMSO, and the vehicle (0.1% DMSO) was added as a control group as described previously.18,19 Next, cells were incubated with or without TNF-α (10 ng/mL, PeproTech) for 6 hours and then were harvested for RNA extraction. For protein assay, cells were pre-plated into 6 cm plates, and then were incubated with GW4064 (1.5 μM, 3 μM) for 24 h. Next, cells were treated with TNF-α for 3 h and total proteins were extracted.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was extracted from cells and tumors with Trizol, and the detailed procedure was carried out as described previously.20–22 Then complementary DNA strand was synthesized from 3 μg total RNA using M-MLV Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). Quantitative real-time PCR was performed using SYBR™ Select Master Mix (Applied Biosystems™) in 7500 Real Time PCR System (Applied Biosystems). Amplification of human β-actin or mouse 36B4 was used as a loading control for normalization of gene expression. Relative mRNA expression levels were described in our results. Primer sequences are shown in Table S1.
Immunoblot Analysis
Total proteins were isolated with lysis buffer from cells or tumors, and then 10% SDS/PAGE gel was used to separate proteins. The proteins were then transferred to a nitrocellulose membrane as described previously. Bands on blots were visualized and the relative density of protein expression was analyzed with a computerized digital imaging system using Tanon software. β-actin was used as an internal reference for normalization of protein levels. All the antibodies applied in the present study were purchased from Cell Signaling Technology.
Cell Proliferation and Migration Assay
Proliferation and migration assays were performed using Real Time Cellular Analysis (RTCA). For proliferation, 7000 cells/well were seeded into E-plate, and cells were incubated with GW4064 (1.5 μM, 3 μM) for RTCA. For migration, 1×105 cells with GW4064 treatment (1.5 μM, 3 μM) were cultured in the upper chamber with a serum-free medium, and the lower chamber was in a 1640 medium with 10% FBS. Then, the RTCA was used to detect cell migration.
Flow Cytometric Assay for Detection of Apoptotic Cells
Detection of apoptosis was stained with the Annexin V-FITC/PI kit according to the instructions (BD Pharmingen Annexin V: FITC Apoptosis Detection Kit I), and then quantitatively analyzed by flow cytometry. Briefly, 4×105 cells/well were pre-plated into 6-well plates, and then were treated with the indicated reagents for 24 h and 48 h, respectively. Floating cells and adherent cells were collected, and washed twice with PBS. Next, the resuspended cells were incubated with annexin V-FITC and PI in the dark room for 15 min. After staining, the cell suspension was detected with a flow cytometer.
Flow Cytometric Assay for Detection of Cell Cycle
Detection of cell cycle was performed with the DNA content quantitation assay (Solarbio) according to the instructions and then quantitatively analyzed by flow cytometry. 4×105 cells/well were pre-plated into 6-well plates, and then were treated with the indicated reagents for 24 h and 48 h, respectively. Adherent cells were collected, and washed twice with PBS. The cells were fixed with 75% pre-cooled ethanol at 4°C for 2 h, and were washed with PBS. Then, 100 μL of RNase A was used to treat cells to remove RNA interference (30 min, 37°C). Cells were incubated with PI in a dark room for 15 min, and then were detected with a flow cytometer.
Statistics
All data represent at least three independent experiments and were expressed as the mean ± SEM. The Student’s t test and a two-way analysis of variance (ANOVA), followed by Bonferroni’s post-hoc test, were performed. A P value less than 0.05 was considered to be significant.
Results
Activation of FXR Impaired Proliferation and Migration of Human ESCC Cells
Human colorectal samples and stomach samples were collected from the Department of General Surgery at Affiliated Huaihe Hospital of Henan University. FXR expression of human colon (n=8), stomach issue (n=8), EC109 cells and KYSE150 cells were quantified in Figure 1. The relative levels of FXR were lower in esophageal cancer cells (EC109 and KYSE150) than that in human colon and gastric tissues. However, the FXR agonist GW4064 still activated the FXR target genes in ESCC cells (Supplementary Fig. 1). And FXR agonist GW4064 induced FXR target genes, PLTP, BACS, and BAT expression in KYSE150 cells, SHP, STD, BACS, PLTP, BAT, and VLDLR in EC109 cells (Supplementary Fig. 1).
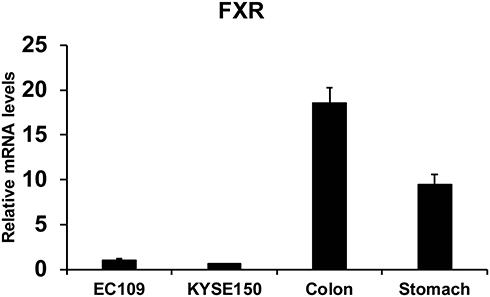 |
Figure 1 FXR levels of human colon (n=8), stomach issue (n=8), EC109 cells and KYSE150 cells were quantified. |
It is recognized that blockade of cell proliferation and migration abilities plays key roles in therapy for cancer. IC50 of GW4064 against KYSE150 and EC109 cells were 5.1 μM and 4.6 μM, respectively (Supplementary Fig. 2). MTT assay showed that the inhibitory effect of 1.5 μM of GW4064 on cell proliferation was more significant than that of GW4064 at the concentrations of 0.5 μM and 1 μM (Supplementary Fig. 3). In addition, Real Time Cellular Analysis (RTCA) was performed to detect the effect of FXR activation by GW4064 on proliferation and migration in KYSE150, EC109 cells, and TE-1. The proliferation of KYSE150 and EC109 cells was suppressed by GW4064 at the concentrations 1.5 μM and 3 μM (Figure 2A and B). Meanwhile, GW4064-treated cells showed a considerable reduction in migration potential of ESCC cells (Figure 2C and D). Consistently, the proliferation and migration of TE-1 cells were also suppressed by GW4064 at the concentrations 1.5 μM and 2 μM (Supplementary Fig. 4). We noticed that GW4064 has a concentration-dependent inhibitory effect on the proliferation and migration of ESCC cells. These results suggest that FXR activation by GW4064 impaired ESCC cell proliferation and migration.
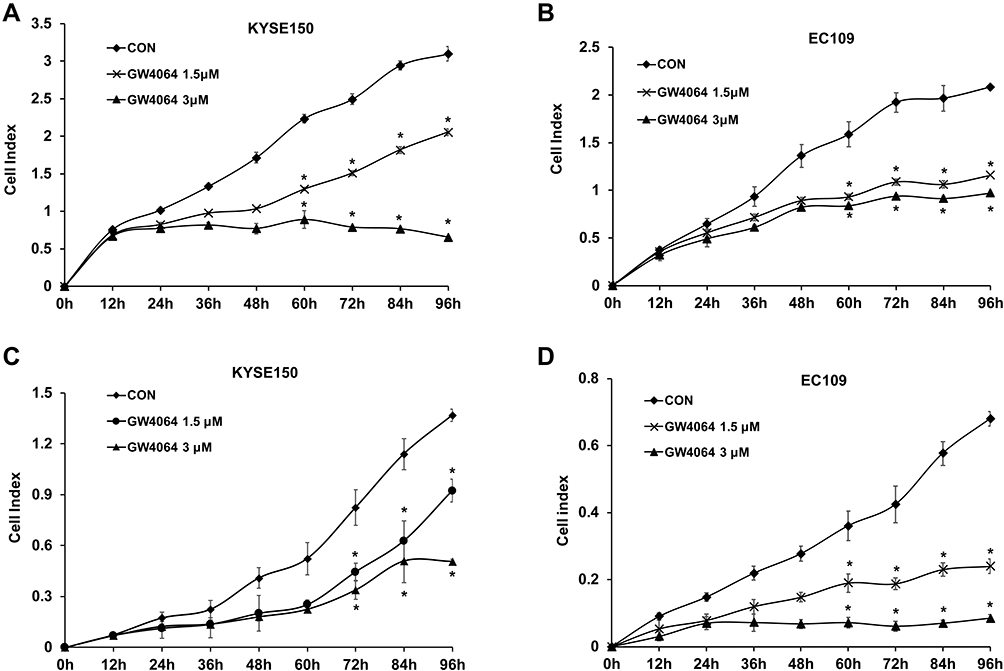 |
Figure 2 FXR agonist GW4064 impairs proliferation and migration of ESCC cells. (A) GW4064 inhibited proliferation of KYSE150 cells. Proliferation assay was performed using Real Time Cellular Analysis (RTCA). (n=5) (B) GW4064 inhibited proliferation of EC109 cells (n=5) (C) Real Time Cellular Analysis (RTCA) assay confirmed that GW4064 inhibited KYSE150 cell migration. (n=5) (D) GW4064 impaired EC109 cell migration. *P < 0.05 versus the control groups. CON, DMSO-treated group; GW4064 1.5μM, 1.5μM GW4064-treated group GW4064 3μM, 3μM GW4064-treated group. The impedance of the electron flow caused by adherent cells is reported using a unitless parameter called the cell index, where cell index = (time point n impedance - impedance in the absence of cells)/nominal impedance value). |
FXR Activation Induced Apoptosis of Human ESCC Cells
To investigate the effect of FXR activation on ESCC cell apoptosis, flow cytometry assay was employed. And KYSE150 and EC109 cells were treated with GW4064 (3 μM) for 24 h and 48 h. It was observed that there was a significant increase in the proportion of apoptotic cells in the treated group compared to the control group (Figure 3A and B).
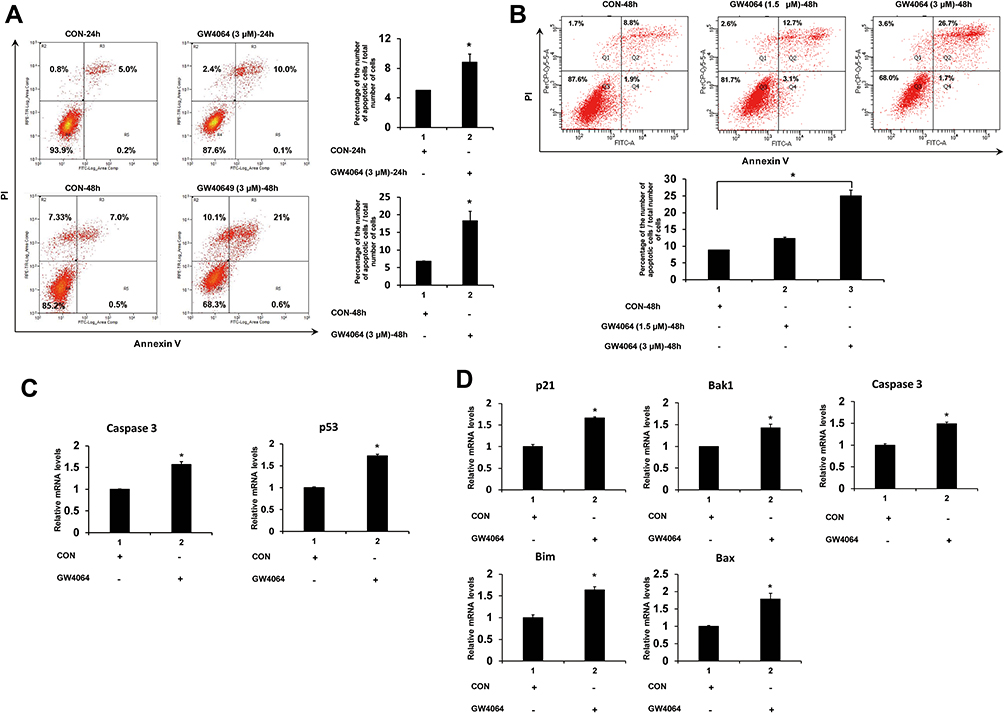 |
Figure 3 FXR activation induced apoptosis of human ESCC cells. (A) Representative fluorescence-activated cell sorting analysis of apoptosis of KYSE150 cells treated by GW4064. KYSE150 cells were treated with DMSO (control groups), GW4064 (3μM) for 24h and 48 h, and then harvested and stained with the apoptotic kit. Con, control. *P < 0.05, (n=3) (B) Representative fluorescence-activated cell sorting analysis of apoptosis of EC109 cells treated by GW4064. EC109 cells were treated with DMSO as control groups (Con-48h), 1.5μM of GW4064 (GW4064 (1.5μM)-48h) or 3μM of GW4064 (GW4064 (3μM)-48h) for 48 h. And then cells were harvested for analysis. *P<0.05 (n=3) (C) Relative mRNA levels of pro-apoptotic genes were increased in the GW4064-induced KYSE150 cells. *P < 0.05 versus the control groups. (n=3) (D) GW4064 induced the expression of pro-apoptotic genes in EC109 cells. *P < 0.05 versus the control groups. (n=3). |
In detail, we observed that FXR activation by GW4064 (3 μM) in KYSE150 cells resulted in 2-fold higher of the percentage of apoptotic cells than that of the control group after treatment for 24 h and resulted in 3-fold higher of the percentage after treatment for 48 h (Figure 3A). A considerable difference of EC109 apoptotic cell percentage can be observed among the control group (8.8%), GW4064 1.5 μM group (12.3%) and GW4064 3 μM group (25.1%) after treatment for 48 h (Figure 3B). Collectively, FXR activation with GW4064 induced cell apoptosis. Next, we determined the transcription levels of apoptosis-related genes. We found that GW4064 treatment (1.5 μM) in KYSE150 cells increased the expression levels of pro-apoptosis genes p53 and caspase 3 (Figure 3C). And the transcription levels of p21, Bak1, Bim, Bax and caspase 3 were increased in GW4064-treated EC109 cells (Figure 3D). In addition, FXR activation with GW4064 increased the expression levels of pro-apoptosis proteins Cleaved-PARP, p53 and p21 in KYSE150 cells and EC109 cells (Supplementary Fig. 5). Overall, these data indicate that GW4064 can induce apoptosis in ESCC cells.
FXR Activation Induced Cell Cycle Arrest
As the cell cycle arrest contributed to the anticancer effect, we next sought to investigate the cell cycle distribution by flow cytometry in KYSE150 and EC109 cells with or without GW4064 treatment. We observed an increase in the percentage of cells in the G0/G1 phase after 1.5 μM GW4064 treatment (76.3%) and 3 μM GW4064 treatment (76.4%) compared to the control group (60.8%) at 24 h, suggesting that FXR activation induced KYSE150 cell cycle arrest in the G0/G1 phase (Figure 4A). Similarly, G0/G1 phase arrest also was seen in EC109 cells after GW4064 treatment for 24 h (Figure 4B). The G0/G1 phase distribution was increased from 51.6% in control group to 66.8% in 1.5 µM GW4064-treated group and 72.4% in 3 µM GW4064-treated group in EC109 cells. Together, these data indicated that FXR activation by GW4064 contributed to cell cycle G0/G1 arrest of ESCC cells and demonstrated one conceivable mechanism by which FXR activation suppresses ESCC process.
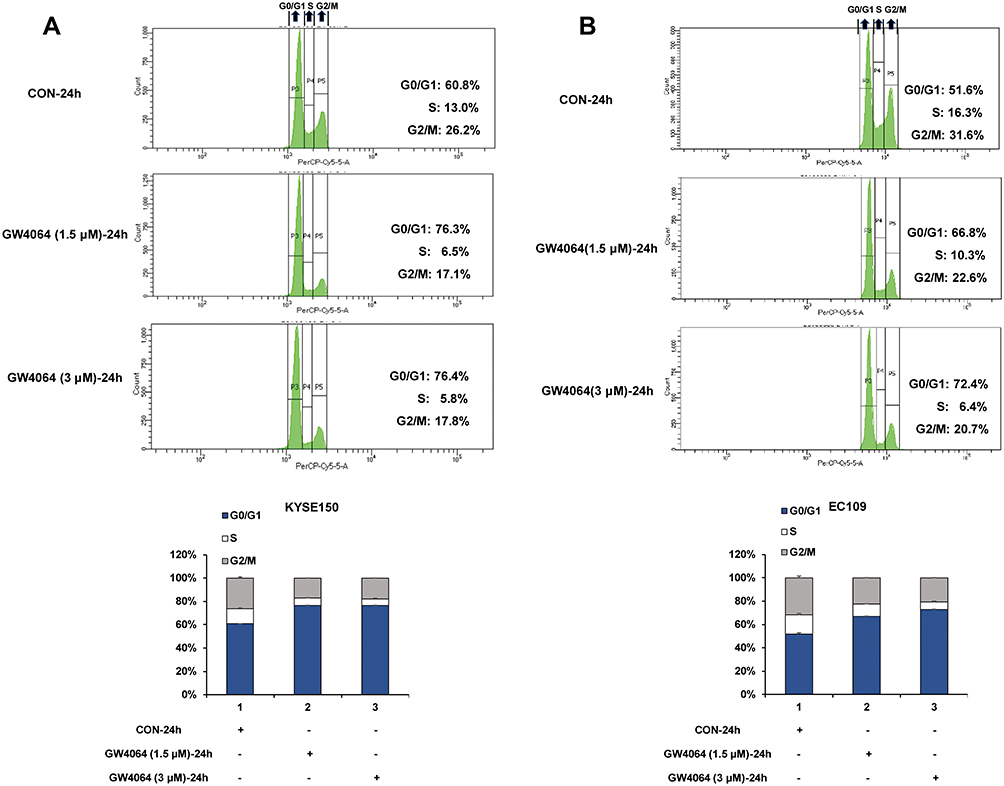 |
Figure 4 FXR activation induced cell cycle arrest. (A) Representative histograms of KYSE150 cells treated for 24 h in the absence or presence of GW4064 (1.5μM or 3μM) and the cell cycle distribution after GW4064 treatment for 24 h. (B) Representative histograms of EC109 cells treated for 24 h in the absence or presence of GW4064 (3μM) and the cell cycle distribution after GW4064 treatment. (n=3). |
GW4064 Regulated ESCC Cell Inflammatory Response
Next, we determined the transcription levels of proliferation and migration-associated inflammatory factors and proinflammatory genes. We found that GW4064 treatment (1.5 μM) in KYSE150 cells increased the expression levels of FXR target genes SHP and BSEP and decreased gene expression levels of c-fos, CyclinD1, IL-6 and MMP7 (Figure 5A). And the transcription levels of c-fos, MMP7, MMP12 and cdc25 were lessened in GW4064-treated EC109 cells (Figure 5B). Furthermore, we observed FXR agonist GW4064 downregulated TNF-α-induced proinflammatory genes levels in KYSE150 cells (Figure 5C). And GW4064 downregulated proinflammatory genes IP-10 and TNF-α levels induced by TNF-α in EC109 cells (Figure 5D). These findings support the notion that FXR agonist GW4064 can inhibit ESCC cell inflammatory response.
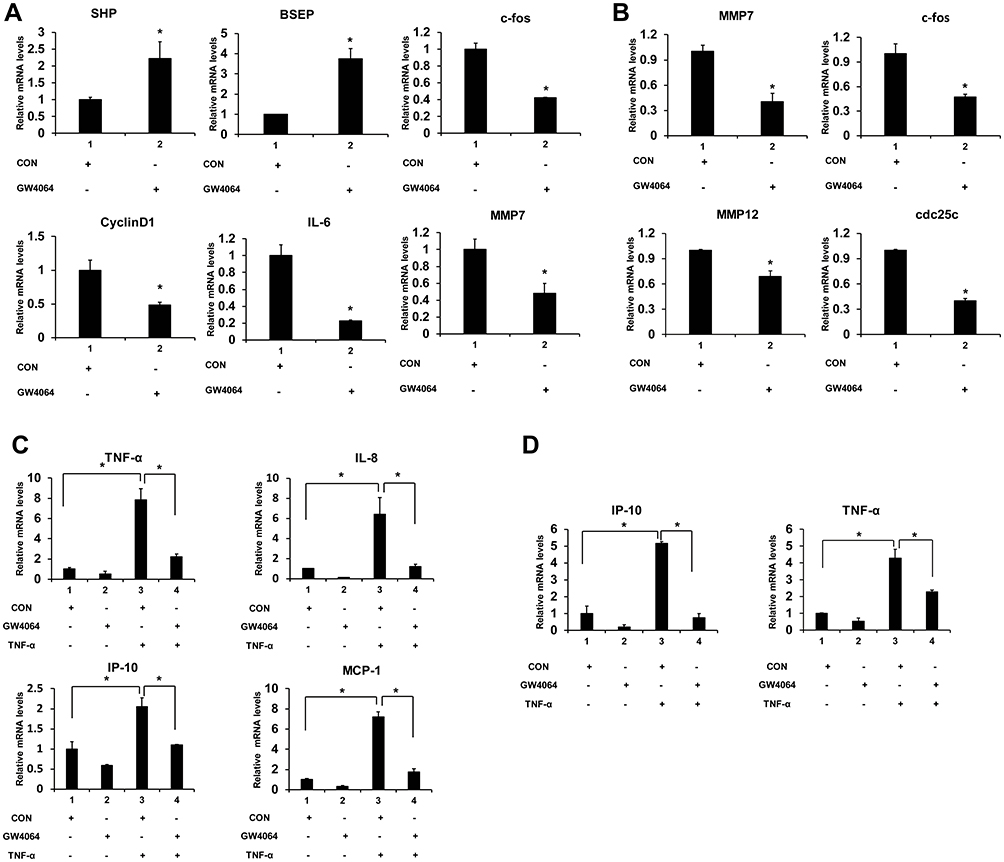 |
Figure 5 FXR agonist GW4064 decreased the mRNA levels of proinflammatory genes in ESCC cells. (A) GW4064 influenced FXR target genes and suppressed c-fos, CyclinD1, IL-6 and MMP7 expression in KYSE150 cells. (B) GW4064 downregulated MMP7, c-fos, MMP12 and cdc25 gene expression in EC109 cells. (C) The mRNA levels of inflammatory cytokines induced by TNF-α were reduced upon GW4064 in KYSE150 cells. (D) GW4064 decreased the inflammatory gene expression induced by TNF-α in EC109 cells. *P < 0.05. (n=3). |
GW4064 Suppressed the Tumorigenesis in vivo
To further confirm the effects of GW4064 on ESCC development in vivo, a xenograft tumor model was constructed. The xenograft experiments showed smaller tumor sizes in the GW4064-treated group than those in the control group (Figure 6A–C). Moreover, we detected the tumor mass in each group and the results showed that the tumor weight was significantly reduced in the GW4064-treated groups (Figure 6D). GW4064 suppressed tumor sizes in a dose-dependent manner (Supplementary Fig. 6). Thus, these results suggest that GW4064 suppressed ESCC progression in vivo.
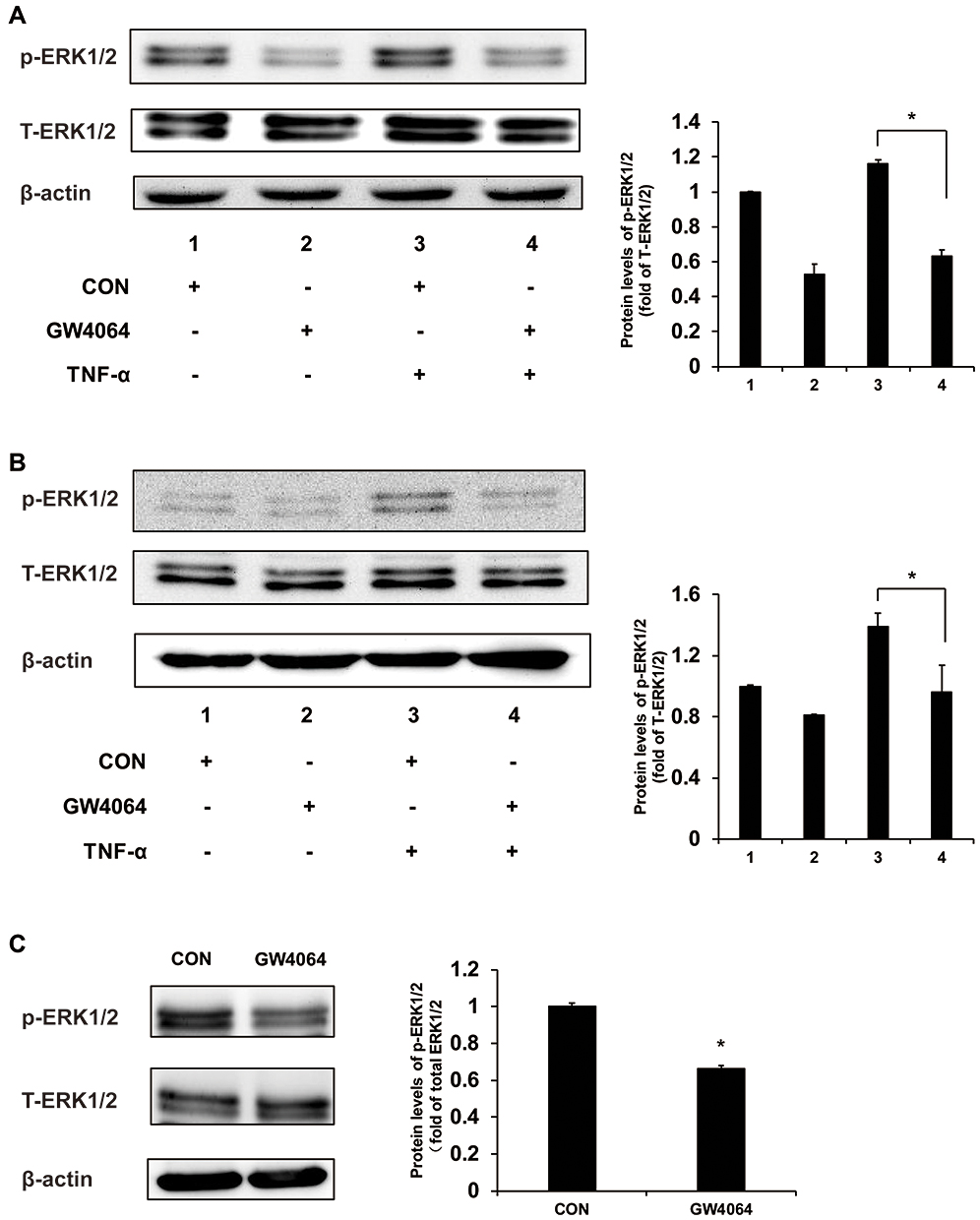 |
Figure 6 FXR agonist GW4064 suppressed ERK1/2 cell signaling pathway. (A) GW4064 suppressed the ERK1/2 pathway induced by TNF-α in KYSE150 cells. (B) Phospho-ERK1/2 levels were decreased upon TNF-α treatment (10 ng/mL) in EC109 cells. (C) Phospho-ERK1/2 protein levels were downregulated in GW4064-treated xenograft tumors. *P < 0.05. (n = 3). |
FXR Activation Reduced ERK1/2 Activity in vivo and in vitro
We have found that the expression levels of some proinflammatory genes, such as c-fos, cdc25 and Bim, were downregulated in the GW4064-treated ESCC cells, and these genes may be mediated by MAPK/ERK1/2 signaling. To understand the mechanism by which FXR activation regulates ESCC development, we performed immunoblot analysis to examine the phosphorylation levels of ERK1/2 in ESCC cells and tumors. We observed that FXR agonist GW4064 suppressed the phosphorylation of ERK1/2 induced by TNF-α by about 45% and 33% in KYSE150 and EC109 cells, respectively (Figure 7A and B). Consistently, the phosphorylation levels of ERK1/2 in the tumors treated by GW4064 were lower than that in the control group (Figure 7C). These results suggest that FXR activation by GW4064 suppressed ERK1/2 phosphorylation in vivo and in vitro.
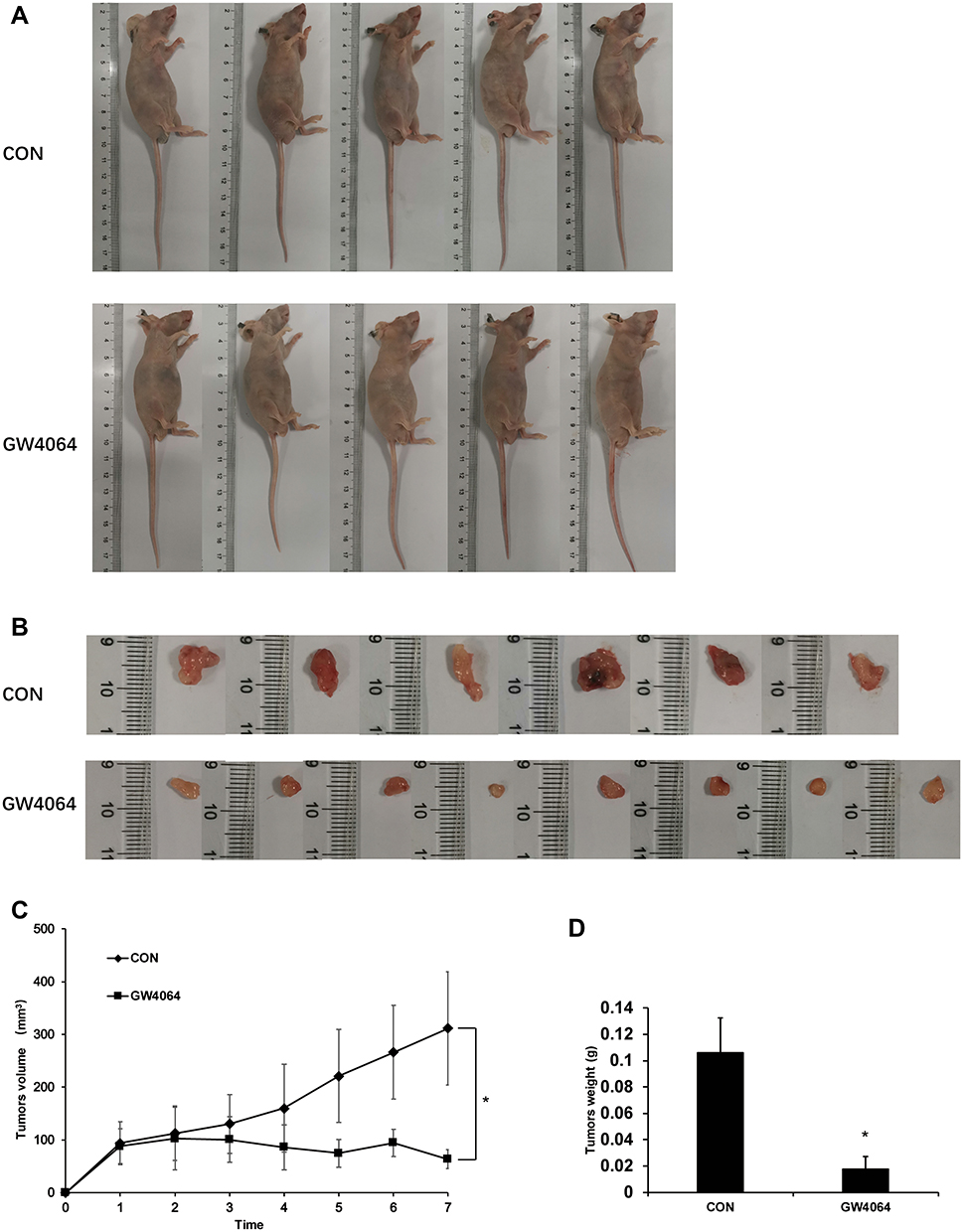 |
Figure 7 GW4064 suppressed EC109 cells-induced tumorigenesis in vivo. (A) Representative nude mice with xenograft tumors were shown. EC109 cells were injected subcutaneously into the nude mice. GW4064 (30 mg/Kg body weight) was administered by intraperitoneally injected every 3 days for 7 times. (n=6–8). (B) The tumors were excised and photographed. (C) Growth kinetics of tumor was analyzed. (D) Tumor weight was measured and analyzed. *P < 0.05. (n=6–8). |
Discussion
FXR, as a bile acid nuclear receptor, controls target gene expression by binding FXR response elements (FXREs) after ligand challenge. FXR regulates SHP, CYP7A1, CYP8B1, BACS and BAT expressions involved in bile acid metabolism, and regulates lipoprotein metabolism-related genes MDR3, PLTP, SDC1, and VLDLR. Then, multiple signaling pathways were triggered to regulate lipid and glucose metabolism, and energy homeostasis.11,23 In the recent years, FXR has been shown to be involved in various disease therapies, including liver carcinogenesis, colon cancer, endometriosis, cholangiocarcinoma, pulmonary hypertension and liver fibrosis.24–31 Guan et al reported that inhibition of FXR suppressed progression of esophageal cancer32 and De Gottardi et al showed that FXR was significantly overexpressed in Barrett’s esophagus and guggulsterone, an antagonist of bile acid receptors, induced cell apoptosis in a Barrett’s esophagus-derived cell line.33 FXR, as a nuclear receptor, is normally activated by its ligands. However, the function of FXR activation by its ligands in ESCC development is still unclear. In our current study, we reveal an inhibitory function of FXR on ESCC after activated by the agonist GW4064.
Although the relative expression levels of FXR were lower in esophageal cancer cells (EC109 and KYSE150) than that in human colon and gastric tissues (Figure 1), FXR agonist GW4064 still activated the FXR target genes in ESCC cells (Supplementary Fig. 2). We further compared the target gene responses of WT and FXR−/- mice to GW4064 treatment. GW4064 changed the gene expression levels of SHP, CYP7A1, CYP8B1 and MDR3 in WT, but not FXR−/- mouse livers (Supplementary Fig. 7), which indicated that GW4064 specifically activated FXR in vivo. In transplanted tumors, GW4064 did not increase the protein level of FXR (Supplementary Fig. 8). However, we noted that GW4064 not only activated FXR in tumor tissue but also in mouse livers in the xenograft mouse model (Supplementary Fig. 8), indicating that GW4064 has antitumor activity in the whole body in mice. This result indicates that GW4064 stimulated the nuclear transcription of FXR, but did not promote the protein expression levels of FXR. GW4064 is administered by intraperitoneal injection, and the drug reaches the whole body through blood circulation. The efficacy and safety of drugs in vivo is influenced by a variety of factors, including cell- and tissue-specific targeting, and drug stability.34 Thus, it will be interesting to explore effective and special drug delivery systems for GW4064, such as nanomaterials, in order to improve drug targeting tumor cells and alleviate the side effects of GW4064 in normal tissues.
MAPK/ERK pathway is involved in tumor development, including regulation of cell proliferation, migration, and apoptosis.35 Blocking ERK1/2 pathway can alleviate and even prevent tumorigenesis and deterioration.7–9 ERK1/2 inhibitor FR180204 has been reported as a suppressor to impair mesothelioma cell proliferation and diminish colorectal cancer cell viability.36,37 Furthermore, Ma et al found triptolide impaired ESCC cell proliferation and migration, and arrested cell cycle and promoted apoptosis via ERK1/2 pathway.10 Here we show that FXR agonist GW4064 suppressed ESCC development and also inhibited ERK1/2 pathway. We found that GW4064 impaired ESCC cell proliferation, migration, and promoted apoptosis in vitro. And we observed GW4064 inhibited ESCC tumor growth in vivo. As shown in Figure 8, these data show FXR activation may regulate ERK1/2 to suppress ESCC development.
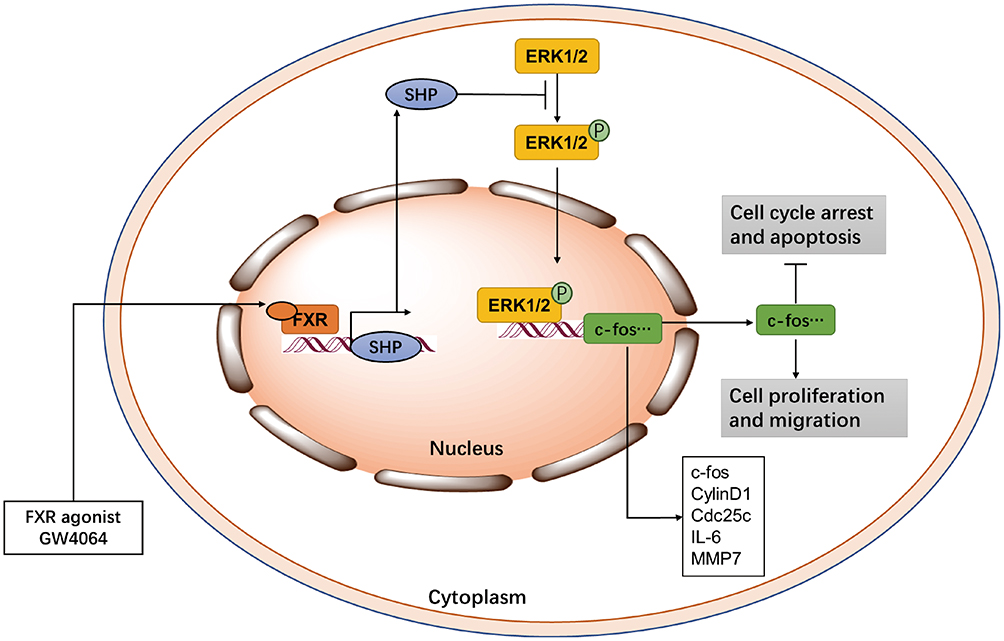 |
Figure 8 GW4064 affects the development of ESCC by activating FXR. |




Several publications proposed an important function for MMP7 in accelerating cell proliferation and cancer development.38–40 Previous reports found activating FXR inhibited MMP7 expression in gastric tissue and colon cancer cells.26,41 And Peng et al clarified that the FXR interacted with a novel non-traditional negative FXREs in the MMP7 5ʹ promoter and then inhibited MMP7 transcription.26 In our study, FXR activation significantly inhibited MMP7 mRNA levels in ESCC cells. We noted that FXR activation also suppressed other proto-oncogene, inflammatory gene expression and promoted some apoptosis-related gene expression in ESCC cells. These results indicate FXR activation may regulate these functional genes to suppress ESCC development.
In summary, the current study defined that FXR activation plays an antitumor role in ESCC development. Briefly, FXR activation inhibited ESCC cell growth, induced apoptosis and cell cycle arrest, and suppressed ERK1/2 pathway. In addition, FXR ligand GW4064 inhibited the ESCC development in the mouse xenograft model. These results indicate that FXR agonist ligands have the potential to be possible anti-ESCC therapeutic drugs.
Funding
This work is supported by the National Natural Science Foundation of China (Grant No. 81672433, No. 81970551 and No. 81370537), the Fundamental Research Funds for the Central Universities and Research projects on biomedical transformation of China-Japan Friendship Hospital (Grant No. PYBZ1803), the Fundamental Research Funds for the Central Universities (Grant No. PT2001) to Y.-D.W., the National Natural Science Foundation of China (Grant No. 81970726 and No. 81472232), Henan Provincial Natural Science Foundation (Grant No.182300410323), Program for Science & Technology Innovation Talents in Universities of Henan Province (HASTIT, Grant No. 13HASTIT024) and Plan for Scientific Innovation Talent of Henan Province to W.-D.C.
Disclosure
The authors have declared that no conflicts of interest exist.
References
1. Huang FL, Yu SJ. Esophageal cancer: risk factors, genetic association, and treatment. Asian J Surg. 2016;41(3):S1015958416302019. doi:10.1016/j.asjsur.2016.10.005
2. Cristina B, Fabio L, Jacques F, et al. Trends in oesophageal cancer incidence and mortality in Europe. Int J Cancer. 2010;122(5):1118–1129.
3. Reid BJ, Barrett MT, Galipeau PC, et al. Barrett’s esophagus: ordering the events that lead to cancer. Eur J Cancer Prevent. 1996;5 Suppl 2(Supplement2):57. doi:10.1097/00008469-199612002-00009
4. Prateek S, Kenneth MQ, John D, et al. A critical review of the diagnosis and management of Barrett’s esophagus: the AGA Chicago Workshop. Gastroenterology. 2004;127(1):310–330. doi:10.1053/j.gastro.2004.04.010
5. Hiroyuki K, Masanobu N. Treatments for esophageal cancer: a review. Gen Thorac Cardiovasc Surg. 2013;61(6):330–335. doi:10.1007/s11748-013-0246-0
6. Marie C, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83.
7. Cicenas J, Zalyte E, Rimkus A, Dapkus D, Noreika R, Urbonavicius S. JNK, p38, ERK, and SGK1 Inhibitors in Cancer. Cancers. 2017;10(1). doi:10.3390/cancers10010001
8. Qin Y, Hu Q, Ji S. et al. Homeodomain-interacting protein kinase 2 suppresses proliferation and aerobic glycolysis via ERK/cMyc axis in pancreatic cancer. Cell Prolif;2019. e12603. doi: 10.1111/cpr.12603
9. Yin H, Yang X, Gu W, et al. HMGB1-mediated autophagy attenuates gemcitabine-induced apoptosis in bladder cancer cells involving JNK and ERK activation. Oncotarget. 2017;8(42):71642–71656. doi:10.18632/oncotarget.17796
10. Yanchun M, Yi W, Lu W, et al. Triptolide prevents proliferation and migration of Esophageal Squamous Cell Cancer via MAPK/ERK signaling pathway. Eur J Pharmacol. 2019;851:43–51. doi:10.1016/j.ejphar.2019.02.030
11. Wang YD, Chen WD, Moore DD, Huang W. FXR: a metabolic regulator and cell protector. Cell Res. 2008;18(11):1087–1095.
12. Wang YD, Yang F, Chen WD, et al. Farnesoid X receptor protects liver cells from apoptosis induced by serum deprivation in vitro and fasting in vivo. Mol Endocrinol. 2008;22(7):1622–1632. doi:10.1210/me.2007-0527
13. Wang YD, Chen WD, Li C, et al. Farnesoid X receptor antagonizes JNK signaling pathway in liver carcinogenesis by activating SOD3. Mol Endocrinol. 2015;29(2):322–331. doi:10.1210/me.2014-1225
14. Wang YD, Chen WD, Huang W. FXR, a target for different diseases. Histol Histopathol. 2008;23(5):621–627. doi:10.14670/HH-23.621
15. Wang Y, Chen W, M, Yu D, Forman B, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008;48(5):1632–1643. doi:10.1002/hep.22519
16. Barone I, Vircillo V, Giordano C, et al. Activation of Farnesoid X Receptor impairs the tumor-promoting function of breast cancer-associated fibroblasts. Cancer Lett. 2018;437:89–99. doi:10.1016/j.canlet.2018.08.026
17. Liu HM, Liao JF, Lee TY. Farnesoid X receptor agonist GW4064 ameliorates lipopolysaccharide-induced ileocolitis through TLR4/MyD88 pathway related mitochondrial dysfunction in mice. Biochem Biophys Res Commun. 2017;490(3):841–848. doi:10.1016/j.bbrc.2017.06.129
18. Liu Y, Binz J, Numerick MJ, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J Clin Invest. 2003;112(11):1678–1687. doi:10.1172/JCI18945
19. Guo F, Xu Z, Zhang Y, et al. FXR induces SOCS3 and suppresses hepatocellular carcinoma. Oncotarget. 2015;6(33):34606–34616. doi:10.18632/oncotarget.5314
20. Zhang Q, Su J, Wang Z, et al. MicroRNA-149* suppresses hepatic inflammatory response through antagonizing STAT3 signaling pathway. Oncotarget. 2017;8(39):65397–65406. doi:10.18632/oncotarget.18541
21. Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology (Baltimore, Md). 2011;54(4):1421–1432. doi:10.1002/hep.24525
22. Guo C, Qi H, Yu Y, et al. The G-Protein-Coupled Bile Acid Receptor Gpbar1 (TGR5) Inhibits Gastric Inflammation Through Antagonizing NF-kappaB Signaling Pathway. Front Pharmacol. 2015;6:287. doi:10.3389/fphar.2015.00287
23. Pathak P, Xie C, Nichols RG, et al. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology (Baltimore, Md). 2018;68(4):1574–1588. doi:10.1002/hep.29857
24. Jiang Y, Iakova P, Jin J, et al. Farnesoid X receptor inhibits gankyrin in mouse livers and prevents development of liver cancer. Hepatology. 2013;57(3):1098–1106. doi:10.1002/hep.26146
25. Huang XF, Zhao WY, Huang WD. FXR and liver carcinogenesis. Acta Pharmacol Sin. 2015;36(1):37–43. doi:10.1038/aps.2014.117
26. Peng Z, Chen J, Drachenberg CB, Raufman JP, Xie G. Farnesoid X receptor represses matrix metalloproteinase 7 expression, revealing this regulatory axis as a promising therapeutic target in colon cancer. J Biol Chem. 2019. doi:10.1074/jbc.RA118.004361
27. Gadaleta RM, Garcia-Irigoyen O, Moschetta A. Bile acids and colon cancer: is FXR the solution of the conundrum? Mol Aspects Med. 2017;56:66–74. doi:10.1016/j.mam.2017.04.002
28. Wu PL, Zeng C, Zhou YF, Yin L, Yu XL, Xue Q. Farnesoid X Receptor Agonist GW4064 Inhibits Aromatase and ERbeta Expression in Human Endometriotic Stromal Cells. Reprod Sci. 2018. doi:10.1177/1933719118808912
29. Di Matteo S, Nevi L, Costantini D, et al. The FXR agonist obeticholic acid inhibits the cancerogenic potential of human cholangiocarcinoma. PLoS One. 2019;14(1):e0210077. doi:10.1371/journal.pone.0210077
30. Comeglio P, Filippi S, Sarchielli E, et al. Therapeutic effects of the selective farnesoid X receptor agonist obeticholic acid in a monocrotaline-induced pulmonary hypertension rat model. J Endocrinol Invest. 2019.
31. Ferrell JM, Pathak P, Boehme S, Gilliland T, Chiang JYL. Deficiency of both farnesoid X receptor and Takeda G protein-coupled receptor 5 exacerbated liver fibrosis in mice. Hepatology (Baltimore, Md). 2019. doi:10.1021/jacs.5b09974
32. Guan B, Li H, Yang Z, Hoque A, Xu X. Inhibition of farnesoid X receptor controls esophageal cancer cell growth in vitro and in nude mouse xenografts. Cancer. 2013;119(7):1321–1329.
33. De Gottardi A, Dumonceau JM, Bruttin F, et al. Expression of the bile acid receptor FXR in Barrett’s esophagus and enhancement of apoptosis by guggulsterone in vitro. Mol Cancer. 2006;5:48. doi:10.1186/1476-4598-5-48
34. Tibbitt MW, Dahlman JE, Langer R. Emerging Frontiers in Drug Delivery. J Am Chem Soc. 2016;138(3):704–717.
35. Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015;35(6):600–604. doi:10.3109/10799893.2015.1030412
36. Honda M, Kanno T, Fujita Y, Gotoh A, Nakano T, Nishizaki T. Mesothelioma cell proliferation through autocrine activation of PDGF-betabeta receptor. Cell Physiol Biochem. 2012;29(5–6):667–674. doi:10.1159/000176386
37. Ragusa M, Statello L, Maugeri M, et al. Specific alterations of the microRNA transcriptome and global network structure in colorectal cancer after treatment with MAPK/ERK inhibitors. J Mol Med. 2012;90(12):1421–1438. doi:10.1007/s00109-012-0918-8
38. Said AH, Raufman JP, Xie G. The role of matrix metalloproteinases in colorectal cancer. Cancers. 2014;6(1):366. doi:10.3390/cancers6010366
39. Lu L, Ma GQ, Liu XD, et al. Correlation between GDF15, MMP7 and gastric cancer and its prognosis. Eur Rev Med Pharmacol Sci. 2017;21(3):535–541.
40. Zhang Q, Liu S, Parajuli KR, et al. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene. 2017;36(5):687–699. doi:10.1038/onc.2016.240
41. Lian F, Xing X, Yuan G, et al. Farnesoid X receptor protects human and murine gastric epithelial cells against inflammation-induced damage. Biochem J. 2011;438(2):315–323. doi:10.1042/BJ20102096
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.