")
Back to Journals » Nature and Science of Sleep » Volume 11
Activation of ATM-c-IAP1 Pathway Mediates the Protective Effects of Estradiol in Human Vascular Endothelial Cells Exposed to Intermittent Hypoxia
Authors Lin YN, Lan XF , Liu ZR , Yan YR, Zhou JP, Li N, Sun XW, Li QY
Received 18 September 2019
Accepted for publication 5 November 2019
Published 28 November 2019 Volume 2019:11 Pages 357—366
DOI https://doi.org/10.2147/NSS.S231456
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Steven A Shea
Ying Ni Lin,1,2,* Xiao Fei Lan,1,3,* Zhuo Ran Liu,4,* Ya Ru Yan,1,2 Jian Ping Zhou,1,2 Ning Li,1,2 Xian Wen Sun,1,2 Qing Yun Li1,2
1Department of Respiratory and Critical Care Medicine, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, People’s Republic of China; 2Institute of Respiratory Disease, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, People’s Republic of China; 3Department of Respiratory Medicine, Shanghai Tongren Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200335, People’s Republic of China; 4Department of Thyroid and Vascular Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qing Yun Li
Department of Respiratory and Critical Care Medicine, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, People’s Republic of China
Tel +86-21-64370045
Email [email protected]
Purpose: Chronic intermittent hypoxia (CIH) contributes to the increased risk of cardiovascular diseases in obstructive sleep apnea (OSA). We previously reported the anti-apoptotic effects of estradiol (E2) on IH-exposed human umbilical vein endothelial cells (HUVECs). Herein, we employed a proteomic analysis to elucidate the mechanisms of the protective effects of E2 under IH exposure.
Methods: HUVECs were divided into three groups: control, IH, and IH+E2 group. Isobaric tags for relative and absolute quantification (iTRAQ) were performed to compare protein profiles among the groups. Some of the identified proteins were validated by Western blotting.
Results: A total of 185 proteins were differentially expressed in the IH+E2 group compared to the IH group. Bioinformatics analysis indicated that the effects of E2 may be linked to the regulation of cellular stress response. Among the differentially expressed proteins, we identified that serine-protein kinase ataxia telangiectasia mutated (ATM) and its downstream target, cellular inhibitor of apoptosis protein 1 (c-IAP1), were up-regulated by E2. We also observed that E2 decreased the level of cleaved caspase-3 and inhibited cell apoptosis in IH-exposed HUVECs. The inhibition of ATM abolished the anti-apoptotic effect of E2.
Conclusion: The ATM-c-IAP1 pathway is involved in the cardioprotective effects of E2 in HUVECs exposed to IH.
Keywords: estradiol, obstructive sleep apnea, OSA, intermittent hypoxia, IH, endothelial dysfunction, proteomics
Introduction
Obstructive sleep apnea (OSA), which is characterized by repetitive collapse of the upper airway during sleep, increases the risk for cardiovascular diseases (CVD) including hypertension, ischemic heart disease, and heart failure.1 Males are more likely to suffer from OSA and cardiovascular comorbidities than females.2,3 Moreover, the transition to menopause is independently related to an increased likelihood of developing OSA and concomitant hypertension.4,5 Estrogen replacement therapy (ERT) could reduce the CVD risk. However, ERT targets estrogen receptors in a non-selective manner, and long-term estrogen treatment could carry risks of endometrial cancer, breast cancer and thrombotic episodes.6 Therefore, revealing the molecular mechanisms of estrogen-mediated cardiovascular protection will help to explore the target(s) for the prevention and treatment of the CVD complications in OSA patients without causing the long-term side effects of estrogen.
Chronic intermittent hypoxia (CIH), which is the major pathophysiological characteristic of OSA, contributes to the development of CVD through inducing oxidative stress, aggravating systemic inflammation and increasing endothelial cell apoptosis.7 A proteomic analysis of vascular wall of male and female mice under CIH revealed a sex difference in the expressions of inflammation-related proteins, lipid metabolism-related proteins, and renin-binding protein.8 Additionally, we previously found that ovariectomized mice were more prone to the CIH-induced vascular injury than the sham-operated ones, and also observed that estradiol (E2) decreased IH-induced oxidative stress and cell apoptosis in human umbilical vein endothelial cells (HUVECs).9 However, the molecular mechanisms of the protective effects of E2 on IH-induced endothelial injury are still poorly understood. Thus, in this study, we established an IH cell model and performed an iTRAQ-based quantitative proteomics analysis to explore molecular mechanisms of the protective effects of E2 on IH-induced endothelial injury.
Materials and Methods
IH Exposure and 17β-Estradiol Treatment in HUVECs
HUVECs (ECV304) obtained from Blood Research Institute (Shanghai, China) were cultured in Dulbecco’s Modified Eagle’s medium (DMEM, Hyclone, USA) supplied with 10% fetal bovine serum (FBS, Gibco, USA) in a humidified incubator under 5% CO2 at 37°C. Cells used in the study were from the same batch. The cells were divided into the three groups: control group, IH group (cells were exposed to IH), and IH+E2 group [cells were exposed to IH with the treatment of 10−7mol/L 17β-estradiol (Sigma, USA)]. KU-55933, which is a specific inhibitor for serine-protein kinase ataxia telangiectasia mutated (ATM), was purchased from MedChemExpress (MCE). After pre-incubation with KU-55933 for 24 hrs, HUVECs were added with E2 and exposed to IH for another 16 hrs. IH exposure (1% O2 for 5 mins followed by 21% O2 for 5 mins, 6 cycles/h, and 16 hrs) was controlled by a self-designed computer program.
Protein Digestion and iTRAQ Labeling
HUVECs were washed with PBS, harvested by SDT lysis buffer. The concentration of total proteins was measured by the bicinchoninic acid (BCA) kit (Thermo Scientific, USA). iTRAQ labeling was performed according to the kit protocol. For protein digestion, 330 μg of protein of each sample was treated with 100 mM dithiothreitol (DTT), and then the cysteine residues were blocked by adding 100 μl 0.05 M iodoacetamide (IAA) for 30 mins at room temperate under dark conditions. Then, the proteins were digested with trypsin at 37°C for 16 hrs. The resultant peptides mixture samples (50 μg for each sample) were labeled using an iTRAQ Reagent-8plex Multiplex Kit from Applied Biosystems (Thermo Fisher Scientific Inc., DE, USA). The iTRAQ-labeled peptide mixtures were then pooled together and dried.
SCX Fractionation and LC-MS/MS Analysis
The iTRAQ-labeled mixed peptides were fractionated using strong cation exchange chromatography (SCX) using the AKTA Purifier system (GE Healthcare) on a polysulfoethyl column (PolyLCInc, Maryland, USA). The collected 36 fractions were pooled to give a final total of four fractions, and desalted on C18 Cartridges (Sigma). The fractions were then analyzed using a Q Exactive MS (Thermo Finnigan) equipped with Easy nLC (Thermo Fisher Scientific Inc.). The peptide mixture was reconstituted in Buffer A (0.1% formic acid), and was loaded onto a C18 capillary column (2 cm x 100 μm, 5 μm, Dionex, Thermo Fisher Scientific Inc., DE, USA), and separated in a C18 capillary column (75 μm x 100 mm, 3 μm, Dionex, Thermo Fisher Scientific Inc., DE, USA) with a linear gradient of Buffer B (80% acetonitrile and 0.1% formic acid) at a flow rate of 250 nL/min. The gradient was begun with 0–35% B for 100 mins, increased to 100% B in the following 8 mins, and held on 100% B from 108 to 120 mins. MS data acquisition was performed using nano-LC-MS/MS with a quadrupole-Orbitrap mass spectrometer (Q-Exactive; Thermo Fisher Scientific Inc.). Protein identification and quantification were performed using Mascot (version 2.2) and Proteome Discoverer Version 1.4 software (Thermo Fisher Scientific Inc.) against the UniProt human database. For protein identification, the Mascot search parameters were set as previously described:10,11 type of search: MS/MS Ion search; enzyme: trypsin; mass values: monoisotopic; max missed cleavages: 2; fixed modifications: carbamidomethyl (C), iTRAQ8plex (N-term), iTRAQ8plex (K); variable modification: oxidation (M), iTRAQ8plex (Y); peptide mass tolerance: ± 20 ppm; fragment mass tolerance: 0.1Da; protein mass: unrestricted; peptide false discovery rate (FDR) ≤1%. The average of the reporter ion intensities for the two biological replicates was considered for relative quantification. To identify differentially expressed proteins, we performed Student’s t-test comparisons between the iTRAQ reporter ions’ intensities. The protein was considered to be differentially expressed if a p-value was less than 0.01 [the iTRAQ ratio (IH/control or IH+E2/IH ) was <0.89 or >1.12].10,11
Bioinformatic Analysis
Gene Ontology (GO) term analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the differentially expressed proteins were performed using the Annotation, Visualization, and Integrated Discovery (DAVID) online tool (version 6.8).12,13 The protein–protein interaction network of the proteins was generated using Cytoscape (version 3.6.1) based on the String database (version 10.5).14,15
Preparation of Cell Lysates and Western Blotting
Cells were washed with PBS twice and were harvested with whole-cell lysis buffer containing 10% SDS. The total protein concentrations of the lysates were measured by a Pierce BCA protein assay kit (Thermo Fisher Scientific). Cell lysates were boiled for 5 mins in 2 × Laemmli buffer, separated on an 8% or 12% SDS-polyacrylamide gel, and transferred to a polyvinylidene fluoride membrane (Immobilon-P, Millipore). Membranes were washed in Tris-buffered saline containing 0.05% Tween 20 (TBS-T) and blocked for 1 hr at room temperature in TBS-T with 5% skim milk or 5% bovine serum albumin (BSA). The membranes were then incubated overnight at 4°C with antibodies against ATM (1:1000, Cell signaling technology, 2873), c-IAP1 (1:1000, Cell signaling technology, 7065), caspase 3 (1:1000, Cell signaling technology, 9662S) and β-tubulin (1:1000, Abcam, ab179513). The membranes were washed with TBS-T and incubated with secondary goat anti-rabbit IgG (1:2000, Merck Millipore, 401315) antibody for 1 hr at room temperature. The bound antibodies were detected with an Immobilon Western kit (Millipore, Billerica, USA), and the signals were visualized using a chemiluminescence detection system (LAS-4000, FUJIFILM).
Measurement of Cell Apoptosis
The apoptosis rate of HUVECs was analyzed by flow cytometer (BD, USA) using Annexin V-FITC Apoptosis Detection Kit (BD, USA) according to the manufacturer’s protocol.
Statistical Analysis
Quantitative data are presented as mean ± SD. Comparison among multiple groups was performed using one-way ANOVA, followed by Tukey’s multiple comparison test using GraphPad Prism 6.0 (GraphPad software, La Jolla, CA, USA). A p-value less than 0.05 was considered statistically significant.
Results
Identification of Differentially Expressed Proteins Between IH and IH+E2
We first identified 145 differentially expressed proteins between control and IH group, 67 of which were up-regulated and 78 were down-regulated in the IH group (Table S1). The differentially expressed proteins were cataloged according to biological process (BP), cellular components (CC), and molecular functions (MF) by Gene Ontology (GO) enrichment analysis (Figure 1). In biological process, the enriched items included RNA splicing (GO: 0008380), regulation of signal transduction by p53 class mediator (GO: 1901796), cellular response to vitamin D (GO: 0071305), regulation of catalytic activity (GO: 0050790), and regulation of cellular response to heat (GO: 1900034).
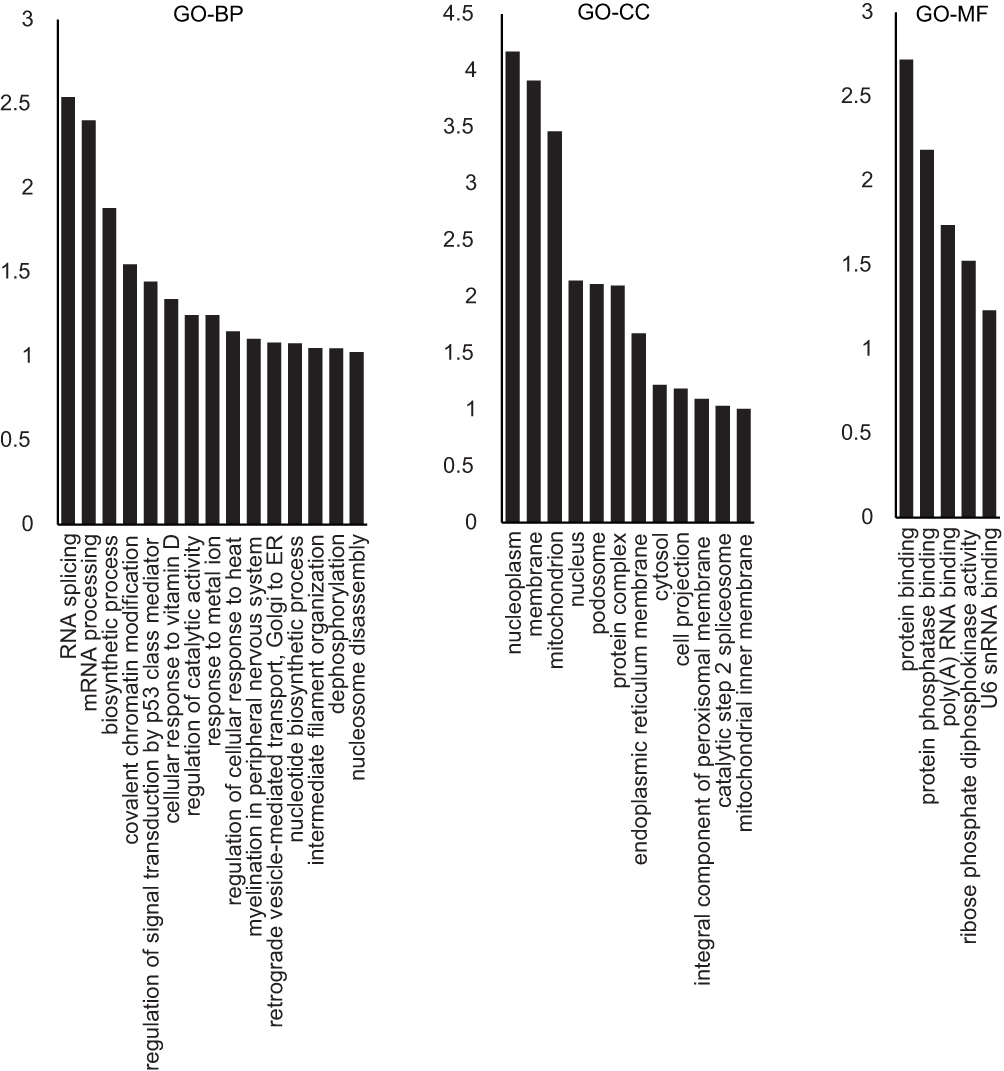 |
Figure 1 GO analysis of the differentially expressed proteins between IH-exposed HUVECs and the control group. The distribution bar charts of the biological processes (GO-BP), cellular components (GO-CC), and molecular functions (GO-MF) are shown. |
Then, we did the comparison between IH and IH+E2 group. A total of 185 differentially expressed proteins were identified between IH and IH+E2 group, with 107 up-regulated and 78 down-regulated in the IH+E2 group (Figure 2 and Table S2). The GO enrichment analysis showed that in biological process, regulation of signal transduction by p53 class mediator (GO: 1901796), DNA methylation (GO: 0006306), regulation of cellular response to heat (GO: 1900034), DNA damage checkpoint (GO: 0000077), cell-cycle arrest (GO: 0007050) and negative regulation of apoptotic process (GO: 0043066) were within the enriched items (Figure 3A). For cellular components, the items of mitochondrion (GO: 0005829) accounts for the largest portion of proteins followed by cytosol (GO: 0005829) and nucleoplasm (GO: 0005654). For molecular function, protein binding (GO: 0005515), poly (A) RNA binding (GO: 0044822) and protein phosphatase binding (GO: 0019903) are the top three significantly enriched items (Figure 3A).
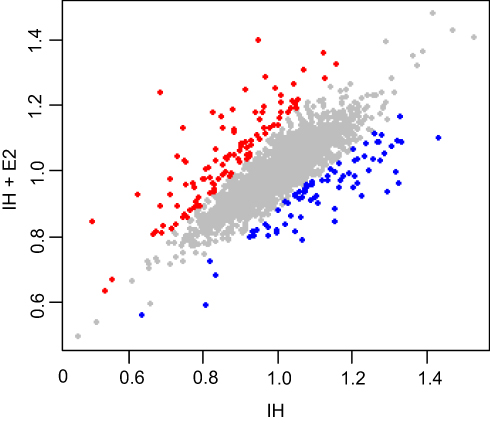 |
Figure 2 Differentially expressed proteins between IH-exposed HUVECs treated with or without estradiol (E2). Proteins up-regulated (107 proteins) and down-regulated (78 proteins) in IH-exposed HUVECs treated with estradiol (IH+E2) with p < 0.01 are denoted by red and blue colored dots, respectively, while proteins do not change significantly between IH and IH+E2 group are represented by grey dots. |
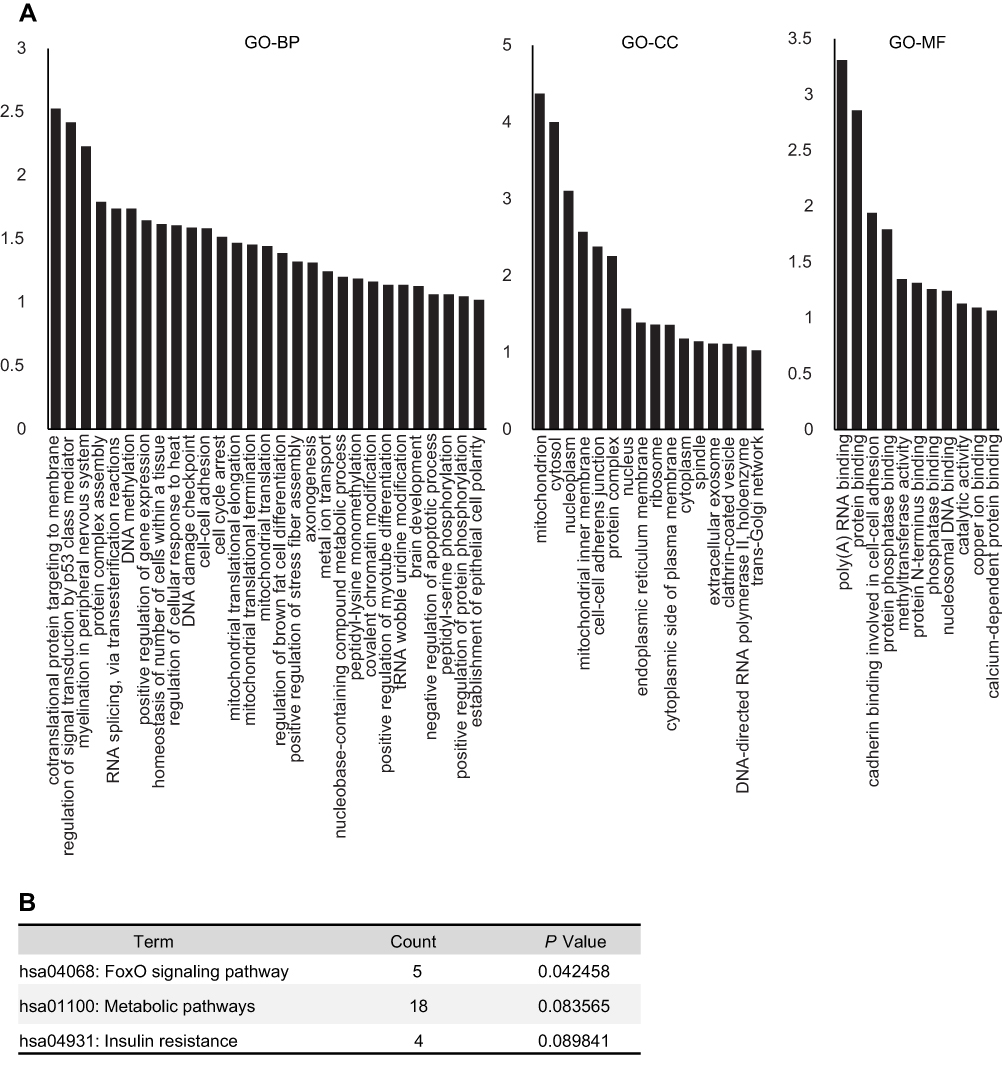 |
Figure 3 Functional annotation analysis of differentially expressed proteins between IH-exposed HUVECs treated with or without estradiol (E2). (A) GO analysis of the differentially expressed proteins. The distribution bar charts of the biological processes (GO-BP), cellular components (GO-CC), and molecular functions (GO-MF) are shown. (B) KEGG pathway analysis of the differentially expressed proteins. |
The differentially expressed proteins were mapped to KEGG pathways by DAVID. The FOXO signaling pathway, which is related to oxidative stress, DNA repair and stress resistance,16 was identified (Figure 3B). Five covered proteins [serine-protein kinase ataxia telangiectasia mutated (ATM), 5ʹ-AMP-activated protein kinase (AMPK) subunit b-1, serine/threonine-protein kinase 4, SMAD family member 2, and p38 MAPK] in FOXO signaling pathway changed significantly in IH+E2 group compared to IH group. Additionally, 18 proteins and four proteins were enriched in the metabolic pathway and insulin resistance pathway, respectively (Figure 3B).
The protein–protein interaction networks of differentially expressed proteins were performed using the STRING application (version 1.3.2) in Cytoscape software. The network exhibited hubs containing proteins related to cellular stress response including ATM, DNA topoisomerase IIb (TOP2B), mammalian target of rapamycin (mTOR) and histone-lysine N-methyltransferase (EHMT1)17–20(Figure 4).
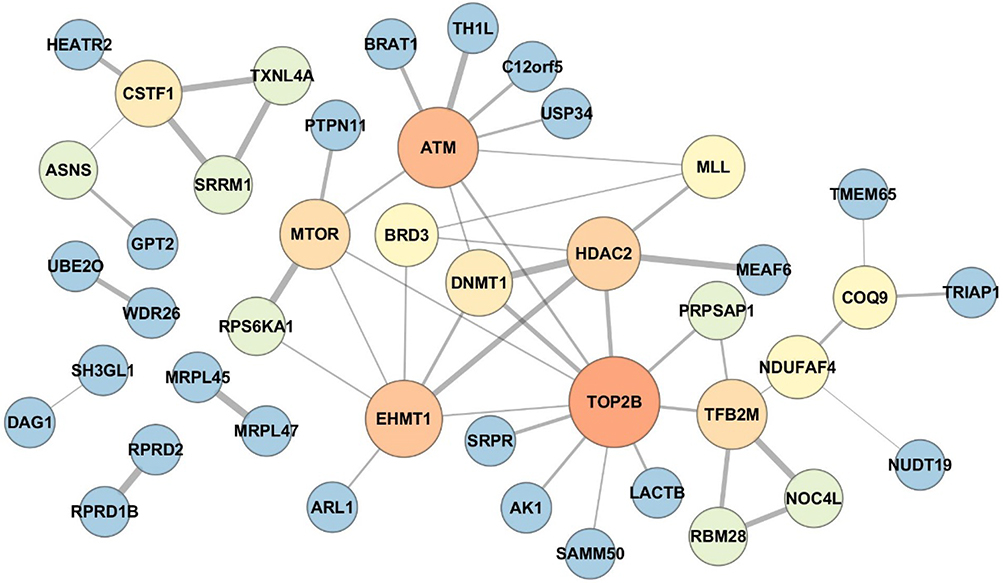 |
Figure 4 Protein–protein interaction network of the differentially expressed proteins between IH-exposed HUVECs treated with or without estradiol (E2). The protein–protein interaction network exhibited hubs containing proteins related to cellular stress response including ataxia telangiectasia mutated (ATM), DNA topoisomerase IIb (TOP2B), mammalian target of rapamycin (mTOR) and histone-lysine N-methyltransferase (EHMT1). The color and size of the nodes were set proportionally to the node degree by Cytoscape. The higher was the node degree, the larger was the size and brighter was the color of the node. |
Identification of ATM and c-IAP1 as Targets of Estradiol Under IH Exposure
We previously found that E2 suppressed oxidative stress and decreased cell apoptosis in IH-exposed HUVECs.9 In this study, a group of differentially expressed proteins between the IH and IH+E2 group were involved in cellular stress response (such as DNA damage and heat response) based on functional analysis. Among those proteins, ATM, which has functions of redox sensing17 and regulating DNA damage repair pathway,21 was up-regulated in IH+E2 compared to the IH group, which was validated by Western blotting (Figure 5). Moreover, its downstream target cellular inhibitor of apoptosis proteins, c-IAP1,22 which is encoded by BIRC2, was also increased by E2, which was verified by Western blotting (Figure 5). The previous study showed that IAPs inhibit the activity of the terminal effector cell death protease, caspases-3.23 We also observed that E2 decreased the level of cleaved caspase-3 in IH-exposed HUVECs (Figure 5). KU-55933, which is a specific inhibitor for ATM, decreased the level of ATM, increased the level the cleaved caspase-3, and promoted cell apoptosis (Figure 6A and B). E2 suppressed cell apoptosis in IH-exposed HUVECs, which was abolished by KU-55933 (Figure 6C). These results indicated that E2 targets the ATM-c-IAP pathway to suppress IH-induced cell apoptosis in HUVECs.
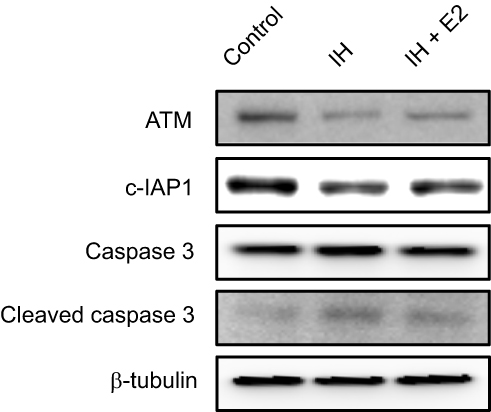 |
Figure 5 Verification of protein expression levels of ATM and c-IAP1 in IH-exposed HUVECs treated with or without estradiol (E2). E2 increased the levels of ATM and c-IAP1, and decreased the level of cleaved caspase 3 in IH-exposed HUVECs. Protein expression levels of ATM, c-IAP1 and downstream target, caspase 3, in IH-exposed HUVECs treated with or without E2 were verified by Western blotting. β-tubulin was used as an internal control. |
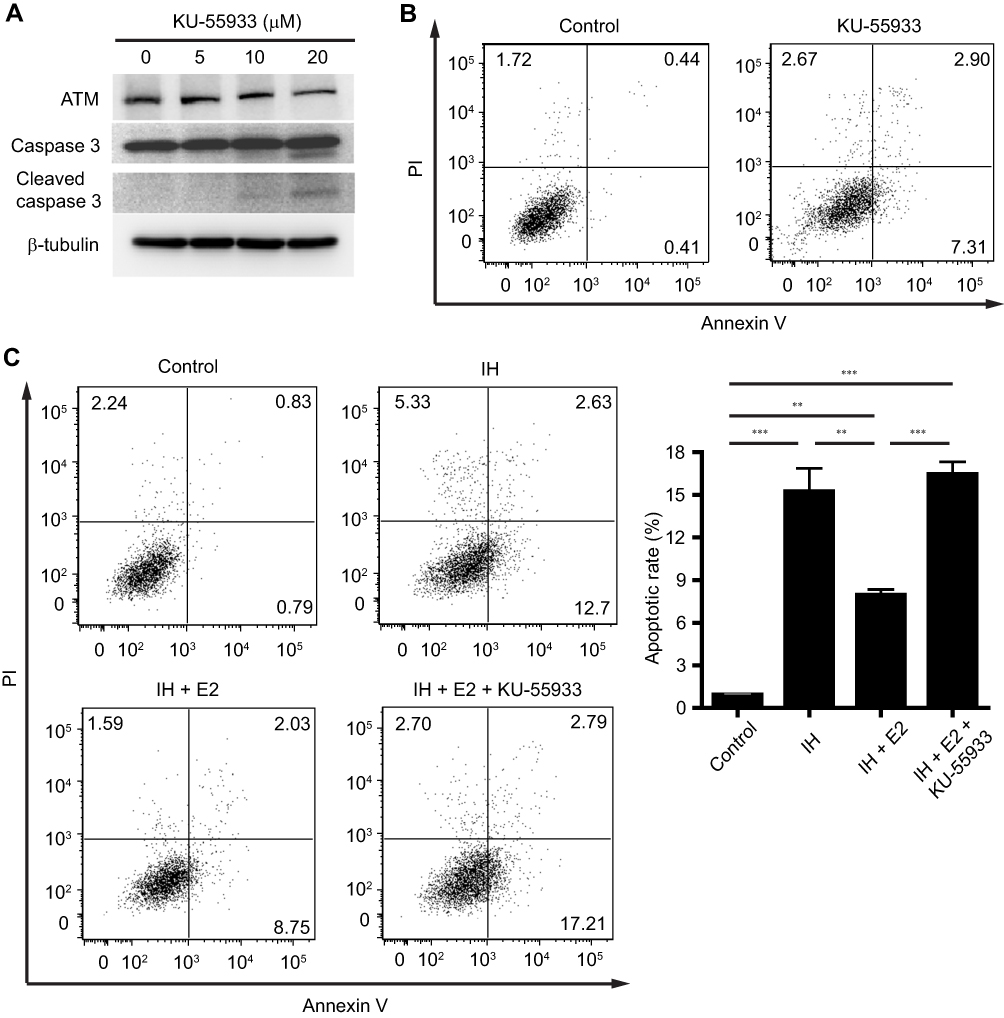 |
Figure 6 The anti-apoptotic effect of estradiol (E2) was abolished by inhibition of ATM. (A) HUVECs were treated with the indicated concentrations of KU-55933 for 24 hrs, which is a specific inhibitor for ATM. KU-55933 decreased the level of ATM, and increased the level of cleaved caspase 3 in HUVECs. Protein expressions of ATM, caspase 3, and cleaved caspase 3 were analyzed by Western blotting. β-tubulin was used as an internal control. (B) KU-55933 (20 µM) induced cell apoptosis in HUVECs. Representative flow cytometric analysis of cell apoptosis in HUVECs was shown. (C) E2 suppressed cell apoptosis in IH-exposed HUVECs, which was cancelled by KU-55933. The left panel shows the representative flow cytometric analysis of cell apoptosis in HUVECs, and the right panel shows the quantification of cell apoptotic rate. **P < 0.01 and ***P < 0.001 by one-way ANOVA followed by Tukey’s multiple comparison test. |
Discussion
In order to understand the molecular mechanisms of the protective effects of E2 on IH-induced endothelial injury, we compared the iTRAQ data between IH-exposed HUVECs supplemented with and without E2. A total of 185 differentially expressed proteins were identified. Functional analysis of the differentially expressed proteins indicated that the vascular protective effects of E2 under IH exposure may be linked to the regulation of cellular stress response including DNA damage response.
CIH induces oxidative stress,9,24 and consequently causes oxidative DNA damage and cytotoxicity, leading to endothelial cell apoptosis. In the study, we exposed HUVECs to 1% O2 for 5 mins followed by 5 mins at 21% O2 in the gas phase (equivalent to 6 cycles/hr). Due to the diffusion time to equilibrate gas concentrations across the whole culture medium, it was unable to subject the cells to the pathophysiological IH frequency in severe sleep apnea,25 which could be a limitation of the settings for in vitro IH exposure. Although the IH cycle rate was below moderate-to-severe sleep apnea, we observed an increase in malondialdehyde (MDA) level, a decrease in superoxide dismutase (SOD) activity, and an increase in the apoptotic rate in IH-exposed HUVECs in a previous study,9 indicating that our cell model may mimic sleep apnea to some extent. Consistently, we found that regulation of signal transduction by p53 class mediator, which regulates cell apoptotic process, was among the most enriched biological process of the differential expressed proteins between IH-exposed HUVECs and control group, indicating that the cell apoptotic process was altered in IH-exposed HUVECs.
E2 supplementation could suppress IH-induced oxidative stress, decrease endothelial cell apoptosis, and thereby prevent the vascular injury of CIH.9,26,27 In this study, we noticed that the kinase ATM, which performs a redox-sensing function, was upregulated by E2 treatment in IH-exposed HUVECs. A previous study showed that in cerebral endothelial cells subjected to oxygen-glucose deprivation, knock-down of ATM accentuates mitochondrial DNA damage, and enhances death of cerebral endothelial cells.22 Deficiency of one or two alleles of ATM accelerates atherosclerosis in apoE−/− mice, indicating a protective role of ATM in vascular diseases.28 ATM regulates the reactive oxygen species (ROS) level through activation of antioxidant enzymes including catalase, thioredoxin, SOD, and heme oxygenase-1 (HO-1),29,30 whereas the absence of functional ATM leads to increased oxidative stress and makes cells more susceptible to oxidative damage.31 Moreover, as part of the stress response, ATM regulates the expression of IAPs through alteration of nuclear factor-kB (NF-kB) activity. Knock-down of ATM reduces c-IAP2 expression, increases the activities of caspase-3 and -8, and accentuates cell death of cerebrovascular endothelial cells.22 In the study, we observed that E2 increased c-IAP1 expression, decreased the level of cleaved caspase-3 and suppressed IH-induced cell apoptosis in HUVECs. The inhibition of ATM cancelled the anti-apoptotic effect of E2. It can be assumed that upon the stress condition induced by IH, E2 may exert anti-oxidant and anti-apoptotic effects through increasing the expressions of ATM and c-IAP1, and thereby show a protective effect on IH-induced endothelial cell injury. The findings provide clues that targeting the ATM-c-IAP1 pathway may protect against endothelial cell injury in OSA. Both chloroquine and resveratrol are able to promote ATM autophosphorylation and activate ATM.28,32 Treatment of ATM+/+ apoE−/- mice with low-dose chloroquine decreases blood pressure and attenuates atherosclerosis in an ATM-dependent manner.28 Whether chloroquine or resveratrol could treat endothelial cell injury in OSA by activating the ATM-c-IAP1 pathway will need further investigation in IH-exposed cell and animal models.
Conclusion
In summary, we identified 185 differentially expressed proteins between IH-exposed HUVECs treated with and without E2. Functional analysis of the differentially expressed proteins implied many aspects of the effects of E2, especially on the regulation of cellular stress response including DNA damage response, during IH exposure. Further, we identified the ATM-c-IAP1 pathway as the potential target of E2. Thus, E2 may protect against oxidative stress and endothelial apoptosis through promoting the ATM-c-IAP1 pathway. The findings facilitate the understanding of the molecular mechanisms of E2 and provide valuable clues for the exploration of the target(s) for the prevention and treatment of the CVD complication in OSA patients.
Acknowledgments
This work was funded by the National Natural Science Foundation of China (81700084, 81070068, 81770084, 81570082, 81700085) and Shanghai Key Discipline for Respiratory Disease (2017ZZ02014), Key Research Program of Shanghai Science and Technology Commission (18140903600).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Golbin JM, Somers VK, Caples SM. Obstructive sleep apnea, cardiovascular disease, and pulmonary hypertension. Proc Am Thorac Soc. 2008;5(2):200–206. doi:10.1513/pats.200708-143MG
2. Hedner J, Bengtsson-Bostrom K, Peker Y, Grote L, Rastam L, Lindblad U. Hypertension prevalence in obstructive sleep apnoea and sex: a population-based case-control study. Eur Respir J. 2006;27(3):564–570. doi:10.1183/09031936.06.00042105
3. Sanchez-de-la-Torre A, Abad J, Duran-Cantolla J, et al. Effect of patient sex on the severity of coronary artery disease in patients with newly diagnosis of obstructive sleep apnoea admitted by an acute coronary syndrome. PLoS One. 2016;11:7. doi:10.1371/journal.pone.0159207
4. Young T, Finn L, Austin D, Peterson A. Menopausal status and sleep-disordered breathing in the Wisconsin Sleep Cohort Study. Am J Respir Crit Care Med. 2003;167(9):1181–1185. doi:10.1164/rccm.200209-1055OC
5. Xu HJ, Lan XF, Li QY, et al. Factors affecting blood pressure profile in pre and postmenopausal women with obstructive sleep apnea hypopnea syndrome. Sleep Breath. 2015;19(1):169–174. doi:10.1007/s11325-014-0983-z
6. Anagnostis P, Paschou SA, Katsiki N, Krikidis D, Lambrinoudaki I, Goulis DG. Menopausal hormone therapy and cardiovascular risk. Where are we now? Curr Vasc Pharmacol. 2018;8(91576):1570161116666180709095348.
7. Castellon X, Bogdanova V. Chronic inflammatory diseases and endothelial dysfunction. Aging Dis. 2016;7(1):81–89. doi:10.14336/AD.2015.0803
8. Li QY, Feng Y, Lin YN, et al. Gender difference in protein expression of vascular wall in mice exposed to chronic intermittent hypoxia: a preliminary study. Genet Mol Res. 2014;13(4):8489–8501. doi:10.4238/2014.October.20.25
9. Lan XF, Zhang XJ, Lin YN, et al. Estradiol regulates txnip and prevents intermittent hypoxia-induced vascular injury. Sci Rep. 2017;7(1):017–10442. doi:10.1038/s41598-017-10442-7
10. Li Q, Li J, Liu S, et al. A comparative proteomic analysis of the buds and the young expanding leaves of the tea plant (Camellia sinensis L.). Int J Mol Sci. 2015;16(6):14007–14038. doi:10.3390/ijms160614007
11. Cai T, Wu B, Tang X, et al. iTRAQ-based proteomic analysis reveals possible target-related proteins and signal networks in human osteoblasts overexpressing FGFR2. Proteome Sci. 2018;16(12):018–0140. doi:10.1186/s12953-018-0140-x
12. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi:10.1038/nprot.2008.211
13. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi:10.1093/nar/gkn923
14. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
15. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi:10.1093/nar/gkw937
16. Furukawa-Hibi Y, Kobayashi Y, Chen C, Motoyama N. FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid Redox Signal. 2005;7(5–6):752–760. doi:10.1089/ars.2005.7.752
17. Zhang Y, Lee JH, Paull TT, et al. Mitochondrial redox sensing by the kinase ATM maintains cellular antioxidant capacity. Sci Signal. 2018;11:538. doi:10.1126/scisignal.aaq0702
18. Zhang A, Lyu YL, Lin CP, et al. A protease pathway for the repair of topoisomerase II-DNA covalent complexes. J Biol Chem. 2006;281(47):35997–36003. doi:10.1074/jbc.M604149200
19. Aramburu J, Ortells MC, Tejedor S, Buxade M, Lopez-Rodriguez C. Transcriptional regulation of the stress response by mTOR. Sci Signal. 2014;7(332):2005326. doi:10.1126/scisignal.2005326
20. Watanabe S, Iimori M, Chan DV, Hara E, Kitao H, Maehara Y. MDC1 methylation mediated by lysine methyltransferases EHMT1 and EHMT2 regulates active ATM accumulation flanking DNA damage sites. Sci Rep. 2018;8(1):018–29239. doi:10.1038/s41598-018-29239-3
21. Tichy A, Vavrova J, Pejchal J, Rezacova M. Ataxia-telangiectasia mutated kinase (ATM) as a central regulator of radiation-induced DNA damage response. Acta Medica Cordoba. 2010;53(1):13–17.
22. Yin KJ, Chen SD, Lee JM, Xu J, Hsu CY. ATM gene regulates oxygen-glucose deprivation-induced nuclear factor-kappaB DNA-binding activity and downstream apoptotic cascade in mouse cerebrovascular endothelial cells. Stroke. 2002;33(10):2471–2477. doi:10.1161/01.STR.0000030316.79601.03
23. Deveraux QL, Reed JC. IAP family proteins–suppressors of apoptosis. Genes Dev. 1999;13(3):239–252. doi:10.1101/gad.13.3.239
24. Badran M, Golbidi S, Devlin A, Ayas N, Laher I. Chronic intermittent hypoxia causes endothelial dysfunction in a mouse model of diet-induced obesity. Sleep Med. 2014;15(5):596–602. doi:10.1016/j.sleep.2014.01.013
25. Farré R, Almendros I, Montserrat JM, Gozal D, Navajas D. Gas partial pressure in cultured cells: patho-physiological importance and methodological approaches. Front Physiol. 2018;9:1803. doi:10.3389/fphys.2018.01803
26. Ribon-Demars A, Pialoux V, Boreau A, et al. Protective roles of estradiol against vascular oxidative stress in ovariectomized female rats exposed to normoxia or intermittent hypoxia. Acta Physiol. 2018;27(10):13159.
27. Sudoh N, Toba K, Akishita M, et al. Estrogen prevents oxidative stress-induced endothelial cell apoptosis in rats. Circulation. 2001;103(5):724–729. doi:10.1161/01.CIR.103.5.724
28. Schneider JG, Finck BN, Ren J, et al. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006;4(5):377–389. doi:10.1016/j.cmet.2006.10.002
29. Barzilai A, Rotman G, Shiloh Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair (Amst). 2002;1(1):3–25. doi:10.1016/S1568-7864(01)00007-6
30. Yu JH, Cho SO, Lim JW, Kim N, Kim H. Ataxia telangiectasia mutated inhibits oxidative stress-induced apoptosis by regulating heme oxygenase-1 expression. Int J Biochem Cell Biol. 2015;60:147–156. doi:10.1016/j.biocel.2015.01.002
31. Kamsler A, Daily D, Hochman A, et al. Increased oxidative stress in ataxia telangiectasia evidenced by alterations in redox state of brains from Atm-deficient mice. Cancer Res. 2001;61(5):1849–1854.
32. Lee J-H, Guo Z, Myler LR, Zheng S, Paull TT. Direct activation of ATM by resveratrol under oxidizing conditions. PLoS One. 2014;9(6):e97969–e97969. doi:10.1371/journal.pone.0097969
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.