")
Back to Journals » Journal of Inflammation Research » Volume 14
Activation and Inhibition of the NLRP3 Inflammasome by RNA Viruses
Authors Choudhury SM , Ma X , Abdullah SW , Zheng H
Received 7 December 2020
Accepted for publication 27 February 2021
Published 26 March 2021 Volume 2021:14 Pages 1145—1163
DOI https://doi.org/10.2147/JIR.S295706
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
SK Mohiuddin Choudhury,* XuSheng Ma,* Sahibzada Waheed Abdullah, HaiXue Zheng
State Key Laboratory of Veterinary Etiological Biology, National Foot and Mouth Disease Reference Laboratory, Key Laboratory of Animal Virology of Ministry of Agriculture, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou, 730046, Gansu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: HaiXue Zheng
State Key Laboratory of Veterinary Etiological Biology, National Foot and Mouth Disease Reference Laboratory, Key Laboratory of Animal Virology of Ministry of Agriculture, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou, 730046, Gansu, People’s Republic of China
Tel +86-931-8342086
Email [email protected]
Abstract: Inflammation refers to the response of the immune system to viral, bacterial, and fungal infections, or other foreign particles in the body, which can involve the production of a wide array of soluble inflammatory mediators. It is important for the development of many RNA virus-infected diseases. The primary factors through which the infection becomes inflammation involve inflammasome. Inflammasomes are proteins complex that the activation is responsive to specific pathogens, host cell damage, and other environmental stimuli. Inflammasomes bring about the maturation of various pro-inflammatory cytokines such as IL-18 and IL-1β in order to mediate the innate immune defense mechanisms. Many RNA viruses and their components, such as encephalomyocarditis virus (EMCV) 2B viroporin, the viral RNA of hepatitis C virus, the influenza virus M2 viroporin, the respiratory syncytial virus (RSV) small hydrophobic (SH) viroporin, and the human rhinovirus (HRV) 2B viroporin can activate the Nod-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) inflammasome to influence the inflammatory response. On the other hand, several viruses use virus-encoded proteins to suppress inflammation activation, such as the influenza virus NS1 protein and the measles virus (MV) V protein. In this review, we summarize how RNA virus infection leads to the activation or inhibition of the NLRP3 inflammasome.
Keywords: inflammation, NLRP3 inflammasome, RNA viruses, activation and inhibition of NLRP3, viral protein and genome
Overview and Consequence of Inflammasomes Modulation
Innate immunity discriminates between host components and microorganisms and it is the immune system’s first line of defense against infection. The innate immune system involves germline-encoded receptors in cells, which are called pattern recognition receptors (PRRs).1,2 PRRs are categorized into two groups according to their localization: membrane-bound and cytoplasmic PRRs.3 Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) are membrane-bound PRRs, while Nod-like receptors (NLRs) and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) are cytoplasmic PRRs.3 PRRs can initiate multiple cellular responses such as transcription of various genes which result in the exclusion of the pathogens. They can also co-operate with each other for the optimum clearance of the pathogens. PRRs and inflammasomes cooperate to initiate a signal of infection and tissue damage, resulting in an innate immune response until the body returns to a normal state.4 PRRs and inflammasomes mediated inflammation play two critical roles. One, it builds a strong barrier against infection. Two, when overreaction occurs, it increases the production of cytokines and inflammatory storm to reduce excessive tissue damage and to minimize the risk.5 Membrane-bound PRRs like TLRs and CLRs localize at the plasma membrane or endosomes whether cytoplasmic PRRs like RLRs and NLRs localize at the cytoplasm.6 TLRs are a type of membrane-bound PRR that are classified as type-1 integral membrane glycoproteins.7,8 TLRs recognize the broad range of proteins, nucleic acids, glycans, etc, and they activate MAP kinase, NF-κβ, IRF pathways such as TLR4 recognizes lipopolysaccharide (LPS) of a gram (-) bacterial cell wall.9 CLRs recognize glycans from the wall of fungi, some bacteria, and they activate kinase syk, CARD9/MALT1/Bcl-10 adapter complex such as Dectin-1/CLEC7A can recognize β-1,3-glucans of the cell wall of various fungus.10–12 On the other hand, RLRs depend on mitochondrial adapter protein MAVS and they trigger the antiviral responses like type-1 interferon production.13 Additionally, NLRs recognize bacterial, viral, parasitic and fungal PAMPs.14 Absent In Melanoma 2 (AIM-2) detects viral and bacterial DNA.15 They form multiprotein signalling complexes knows as inflammasome and secret interleukins-1β, interleukins-18 activating caspase-1 such as NOD-1 and NOD-2 recognize bacterial peptidoglycan.16
PRRs are activated in response to pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).17 They induce IFN signaling or other inflammatory pathways and thereby eliminate invading pathogens and repair damaged tissues. PAMPs are related to bacteria, viruses, or other microorganisms that cause disease, while DAMPs involve various host cell components such as tumor cells, dead or dying cells, or products released from cells. DAMPs are released during cell death or damage. Usually, they do not need pathogenic infection.18,19 As they evolved before the other parts of the immune system (including adaptive immunity), they are also known as primitive PRRs. DAMP molecule such as S100A9 is required for efficient replication of Influenza A virus and modulating pro-inflammatory responses.20 There are five main PRRs that act as cytosolic PAMP and DAMP sensors and form protein complexes known as inflammasomes. To date, five PRRs (NLRP1, NLRP3, NLRC4, Pyrin and AIM2) have been shown to form inflammasomes.21,22 Emerging evidence indicates that several other members of the NLR family and the PYHIN family, including NLRP6, NLRP7, NLRP12 and IFI16, can also form inflammasomes, but their composition remains obscure.22 Three of these PRRs are NLRP1, NLRP3 and NLRC4, which are all NLRs. The other two PRRs are AIM2-like receptors (ALRs) and pyrin.21,23 All five PRRs can recruit apoptosis-associated speck-like protein containing a CARD (ASC). Inactive pro-caspase-1 then interacts with oligomerized ASC. This induces self-cleavage of procaspase-1 to form bioactive caspase-1, which contains two heterodimers of p20 and p10. Following this, caspase-1 cleaves pro-IL-1β and pro-IL-18 and the active cytokines are released from the cells, triggering inflammation. The release of various inflammatory cytokines and the antigen-specific adaptive immune response of the host are connected through PRRs.3,24,25
This review discusses the roles and mechanisms of NLRP3 inflammasome activation and inhibition during RNA virus infections.
Models of NLRP3 Activation
The NLRP3 inflammasome is the most well-studied inflammasome. Many families of viruses can activate it. For full NLRP3 inflammasome activation, two signals are required.26 As we see in Figure 1, Signal 1 (the primary, first, or priming signal) can be initiated by TLRs and RLRs or by a protein receptor, which leads to the upregulation of pro-caspase-1 and pro-IL-1β and pro-IL-18.27 Signal 2 for NLRP3 inflammasome activation, involving pro-caspase-1 recruitment to NLRP3 and the subsequent production of mature caspase-1 and IL-1β and IL-18, involves stress signals associated with tissue damage or infection.28 The mechanisms of NLRP3 inflammasome activation are still not fully understood.
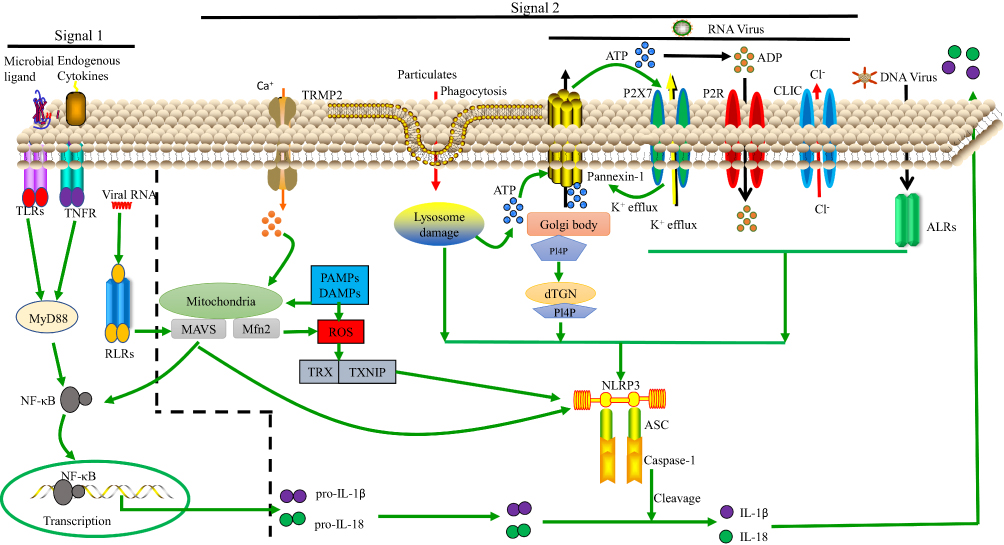 |
Figure 1 Model of NLRP3 activation. Signals 1 and 2 are both essential for NLRP3 activation. Regarding signal 1, the NF-κB pathway can be activated by TLRs, RLRs and TNF-α signaling, leading to pro-IL-1β and pro-IL-18 upregulation. Regarding signal 2, various PAMPs and DAMPs induce ASC assembly and caspase-1 maturation, which involves NLRP3 activation. In some cases, this is related to the presence of a high concentration of extracellular ATP and subsequent K+, Cl− efflux via P2X purinoceptor 7 (P2RX7) channels, pannexin-1 or CLIC channels. Additionally, nigericin, asbestos, and other particulates can induce lysosomal damage, releasing cathepsin B into the cytosol. Moreover, ROS can activate NLRP3 by inducing Ca2+ influx through transient receptor potential melastatin 2 (TRPM2) channels. The ALRs can interact with NLRP3 leading to pro-IL-1β and pro-IL-18 upregulation. Lastly, NLRP3 can bind to the dispersed trans-Golgi network (dTGN) via PI4P. Downstream of these processes, IL-1β and IL-18 are released. |
Signal 1, Involving TLRs or RLRs, Primes NLRP3 Inflammasome Activation
TLR signaling can be important for priming regarding NLRP3 inflammasome activation. Activation of TLR7, TLR3, and TLR2 primes NLRP3 inflammasome activation, while TLR-knockout macrophages did not induce caspase-1 maturation after stimulation with ATP.27
To better understand signal 1 induced by viral infections, innate immune signaling during influenza virus infection was investigated.29 Influenza virus genomic RNA in endosomes can be recognized by TLR7.30,31 The lack of IL-1β release from TLR7-deficient bone marrow-derived DCs infected with influenza virus revealed that TLR7 signaling was needed for pro-IL-1β transcription and the release of mature IL-1β.29
Cytosolic double-stranded RNA (dsRNA) can activate the NLRP3 inflammasome by interacting with TLR.32 To determine whether TLRs are needed for NLRP3 inflammasome activation, wildtype and MyD88/TRIF-deficient macrophages were transfected with a synthetic analog of dsRNA, poly(I: C), to assess caspase-1 activation after poly(I:C) stimulation. Cytosolic poly(I:C) but not extracellular poly(I:C) was able to activate caspase-1, and this process was independent of TLR signaling. These results confirmed that cytosolic poly(I:C) is able to activate NLRP3 inflammasomes without TLR signaling.32
To determine the molecular mechanisms underlying NLRP3 expression, NLRP3 mRNA and protein levels were assessed after treatment of murine serosal macrophages with TLR agonists.33 In particular, to determine whether NF-κB is required for NLRP3 expression, pretreatment with JSH-23 (a specific inhibitor of NF-κB transcriptional activity) was performed. JSH-23 significantly attenuated the lipopolysaccharide (LPS)- or peptidoglycan (PGN)-induced NLRP3 mRNA and protein expression. Similarly, after JSH-23 pretreatment, LPS- or PGN-induced expression of NLRP3 firm state mRNA and protein were both comprehensively inhibited. These results indicate that TLR-induced NF-κB activity is needed for NLRP3 activation in macrophages.33
TLR signaling is thought to initiate NLRP3 activation in human monocytes.27,34,35 Additionally, TLR-initiated NLRP3 activation in macrophages/monocytes is thought to be a useful model for molecular research on NLRP3 expression.33 TLR signaling in macrophages activates NF-κB and AP-1.36 TLR-induced NF-κB activation allows NF-κB to bind to the NF-κB binding sites in the NLRP3 promoter and thereby regulate NLRP3 transcription.33
TLRs can detect extracellular as well as intracellular (vacuolar) stimuli, whereas NLRs seem to respond only to cytosolic stimuli. It has been shown that, together, TLRs and NLRs can efficiently regulate innate immune responses.37 For example, TLR and NLR signaling are both essential for the secretion of IL-1β. TLRs can induce 35-kDa pro-IL-1β expression,24 whereas NLRs are responsible for IL-1β maturation and secretion.38–41
RLRs are cytoplasmic sensors that recognize viral RNA, which induces signal 1 transduction and triggers intracellular innate immune defenses and inflammatory responses. RIG-I can lead to pro-IL-1β and pro-IL-18 transcription by interacting with mitochondrial antiviral-signaling protein (MAVS) and a complex of the adaptors CARD-containing protein 9 (CARD9) and Bcl-10, which activates NF-κβ. RIG-I can also bind to ASC, triggering mature caspase-1 production, which leads to inflammasome activation.42 The 5ʹ-triphosphate dsRNA of certain negative-stranded RNA viruses (such as influenza A and MV) can activate cytosolic RIG-I which involves in NF-κB activation and transcription of pro-IL-1β and pro-IL-18 by initiating signal 1.43,44 The interaction of RIG-I with MAVS can induce recruitment of various molecules, leading to NF-κB activation. RIG-I can also bind to ASC, triggering mature caspase-1 production, which leads to inflammasome activation and IL-1β secretion.45 These findings show that RIG-I has an important role in signal 1 of the inflammasome activation process. In summary, TLRs and RIG-I can both induce the transcription of antiviral genes such as pro-IL-1β and pro-IL-18.
Models of Signal 2 in NLRP3 Inflammasome Activation
Regarding signal 2, the following three models are widely supported: 1) ion channel model; 2) lysosomal rupture model; 3) reactive oxygen species (ROS) model; and as we see in Figure 1.
Ion Channel Model
This model involves a high concentration of extracellular ATP. In response to ATP binding, P2X purinoceptor 7 (P2RX7) ion channels open, causing K+ efflux, and pannexin-1 channels (which are permeable to small molecules) in the cell membrane are expressed. This helps PAMPs and DAMPs to enter into the cytosol,46 directly activating NLRP3.47 Lower concentration of cytosolic K+ induces NLRP3 activation48 whether high concentration of K+ blocks the activation of NLRP3 inflammasomes.49,50 But this mechanism is still not well understood. K+ is considered as a upstream signalling for NLRP3 activation.49 In presence of lipopolysaccharide (LPS), murine macrophages were treated with potassium ionophores such as nigericin that results in the maturation and secretion of IL-1β which is the diaphanous evidence for activation of NLRP3 inflammasomes.51 In an other study, it has been shown that the propagation of cytopathogenic viral RNA, such as VSV or EMCV, triggered lytic cell death to proceed the K+ efflux, a typical trigger for inflammatory NLRP3 activation.52
Previous studies have shown that the Ca2+ signaling plays an exigent role in NLRP3 activation.53–55 Nigericin, ATP, silica, alum, etc, induce Ca+ mobilization causing NLRP3 activation.54 It has been shown that NLRP3 activation seized while the Ca+ signaling was inhibited.54 BAPTA-AM (Ca+ chelator) blocks the NLRP3 inflammasomes activation by inhibiting IL-1β secretion.56–58 As an essential intracellular Ca2+ tank, the Endoplasmic reticulum(ER) appears critically important because the Ca2+ release chain of the ER (IP3R) reduces movement and NLRP3 activation due to the pharmacological or small hairpin RNA knockdown (shRNA) on the inositol 1,4,5-trisphosphate reception (IP3R).53 IP3, a product of phospholipase C (PLC), activates the IP3R. However, inhibition of PLC seizes the activation of NLRP3, but the activation of PLC induces IL-1β secretion in absence of exogenous stimuli.53 Another study proves that CASR and GPRC6A are only necessary to activate the extracellular Ca2+ NLRP3 activation and not to activate ATP-induced NLRP3.55 Additionally, a new research showed that the Ca2+ signaling is autonomous of NLRP3 inflammasome activation.59 Interestingly, this analysis also showed that BAPTA prevents NLRP3 activation regardless of its anticipated Ca2+ signaling inhibitory effects.59 The effect of Ca2+ signaling is therefore contentious in NLRP3 activation.
It was reported that when the extracellular Cl− concentration decreased from 130mM to 9 mM, it increased ATP-induced IL-1β secretion.60 Cl− channel blockers such as mefenamic acid, flufenamic acid, benzoic acid, etc, can inhibit the NLRP3 activation.61–63 Cation flows and a decline in intracellular Cl− concentration were detected throughout NLRP3 inflammatory activation. Moreover, chloride intracellular channel (CLIC) protein family and volume-regulated anion channel (VRAC) are crucial for NLRP3 inflammasome activation.62,63 All of these data prove that Cl− efflux is essential for NLRP3 inflammasome activation.
Like K+, Ca2+ and Cl−, Na+ are involved in NLRP3 activation.48 Mechanically, the NA+ inflow modulated the activation of NLRP3 partly by minimizing the decrease in intracellular K+.64 Thus, Na+ influx can regulate NLRP3 inflammasome activation by decreasing intracellular K+. Prolonged Zn2+ reduction has been reported to induce NLRP3 activation in macrophages.65 While Zn2+ depleted sensors are currently undisclosed, Zn2+ depletion-induced lysosome membrane destabilization is an essential activity in Zn2+-induced IL-1β secretion.65
Dispersed trans-Golgi network (dTGN) model is the consequence of ion channel model that is recently described and in the dTGN model, the binding of NLRP3 to negatively charged phosphatidylinositol-4-phosphate (PI4P) on the dTGN is essential for NLRP3 aggregation and activation.66 Diverse stimuli of PI4P lead to disassembly of the TGN. A small number of NLRP3 proteins are activated upon stimulation of PI4P, and they associate with dTGN and form large aggregates. In a recent study, extracts of activator cells (stimulated 293 NLRP3 cells) were mixed with recipient cells (293 ASC-casp 1 cells) that had been permeabilized with perfringolysin O.66 After treatment of HEK 293 cells with nigericin (which acts as a stimulus for NLRP3), caspase-1 cleavage occurred. In this study, wildtype bone marrow-derived macrophages (BMDMs) were also stimulated using various stimuli (nigericin, silica, ATP, etc), resulting in considerable TGN disassembly that induced caspase-1 and IL-1β production and after that, endogenous NLRP3 formed puncta.66 When cells with mutations leading to constitutively active NLRP367,68 were treated with nigericin, NLRP3 aggregated, creating multiple small puncta before forming large specks due to the induction of ASC oligomerization, suggesting that NLRP3 aggregation is necessary for NLRP3 activation. Thus, the formation of dTGN, which is downstream signal dependent and leads to NLRP3 recruitment, initiates NLRP3 inflammasome activation. Oligomerization of ASC PYD is initiated at dTGN-localized NLRP3 puncta, which indicates that recruitment to the dTGN activates the NLRP3 inflammasome. The presence of dTGN is necessary for the involvement and activation of NLRP3. The highly conserved KKKK motif is found in all NLRP3 orthologs, and there are at least three positively charged residues in each ortholog. Along with the KKKK motif, the additional conserved polybasic regions have a vital function in recruiting NLRP3 to the dTGN, which is critical for its later activation.66 The negatively charged phospholipids PI(3,4,5)P3 and PI(4,5)P2 on the plasma membrane surface bind to various Rab GTPases via polybasic regions, and the recruitment of NLRP3 is reported to involve a similar mechanism.69 Thus, NLRP3 is recruited to PI4P-enriched microdomains on the TGN. TGN protein, 38-kDa (TGN38) sac-1 notably decreased NLRP3 activation via its catalytic activity and for NLRP3 recruitment to the dTGN and its later activation; PI4P binding plays a vital role that is compatible with the observation that PI4P is enriched on the TGN.70 Inhibiting the K+ efflux does not prevent the disassembly of the TGN, but K+ efflux can play an important role in activating NLRP3.48 Again, K+ efflux alone is not sufficient for NLRP3 activation because K+ efflux cannot induce dTGN.66 PI4P involvement in the dTGN model is critical for K+ efflux-independent NLRP3 activation.71,72
The disassembly of the TGN into dTGN is a general cellular signal influenced by various NLRP3 stimuli, and NLRP3 is recruited through ionic bonding between its polybasic region and PI4P on the dTGN. Thereafter, the dTGN serves as a scaffold for NLRP3 to cluster and interact with ASC, activating the downstream signaling cascade. By binding to the dTGN, NLRP3 indirectly mediates responses to a great variety of molecules linked to various pathogens and dangers.66
Lysosomal Rupture Model
NLRP3 stimuli (eg, nigericin, asbestos, silica, alum, and amyloid β) primarily form aggregates. These aggregates are phagocytosed and then damage lysosomes, causing the release of cathepsin B, which directly activates NLRP3.73 It has been proved that cathepsin B inhibitor CA-074-Me seizes the activation of NLRP3 inflammasome.73–75 In such way, the non-target influence or redundancy in the family of cathepsin members may be due to the inhibition of NLRP3 activation by Cathepsin B inhibitors. A recent study has suggested that inhibition of multiple cathepsins by CA-074-Me can promote NLRP3 activation.76 However, with perhaps the exception of K+ efflux, the function of those events remains an open question in NLRP3 activation.
ROS Model
The ROS model71 represents a common pathway underlying NLRP3 inflammasome activation.77 Mitochondria are the main intracellular organelles that produce ROS.78 Nigericin, asbestos, silica, and alum induce ROS production, and ROS is generated by NADPH oxidase.79 ROS leads to K+ fluxes, activating the NLRP3 inflammasome.75,80 However, ROS inhibitors manipulate the priming modulation of NLRP3 as used in large concentration.81 Again, mitochondrial DNA release can occur downstream activation of NLRP3.82 Another study reported that mitochondria-associated adaptor MAVS can activatedNLRP3 inflammasome in presence of soluble stimuli such as ATP, nigericin but not particulate matter such as alum or monosodium urate.83 However, some other studies show that mitochondrial MAVS activate NLRP3 inflammasome in presence of RNA viruses but not non-viral stimuli such as ATP or nigericin.32,84–86 RNA virus such as murine norovirus (MNV) leads to Gasdermin D (GSDSD) dependent pyroptosis resulting NLRP3 activation in STAT-1 deficient macrophages displayed increased MAVS mediated IL-1β secretion.87 Additionally, Mitofusin-2, an outer membrane protein of mitochondria-responsible for mitochondrial fusion is required for NLRP3 activation after infection with RNA viruses such as influenza, measles or encephalomyocarditis virus (EMCV).88 Still there is a need for deep research to unveil the exact mechanism of mitochondrial effect in NLRP3 activation.
Inflammation During RNA Virus Infections
Cytokines in the human body can be induced by viral or bacterial infections and by various other kinds of health conditions.89 Excessive cytokine and chemokine production in the body can cause morbidity and even mortality in various infectious diseases. Few effective treatments to reduce severe inflammation have been developed. A major cause of this issue is the poor understanding of the relationships between cell biology and inflammation. Some immune cells can produce soluble immune mediators, and these cells have a prominent role in combating viral and bacterial infections.90 The cytokines tumor necrosis factor α (TNF-α) and interleukin 1 (IL-1) play vital roles in many acute and chronic disorders. Many patients with numerous diseases (eg Shock, osteoporosis, trauma, damage of organs, rheumatoid arthritis) caused by inflammatory cytokines and chemokines have been ill for several decades.91 Despite these long-term issues, there are few effective therapeutics to solve these problems.
RNA viruses are a diverse class of viruses that are capable of inducing the production of inflammatory factors in various species of animals, which can cause fatal diseases, including in humans. Respiratory viruses such as coronavirus (CoV) and influenza virus can cause a massive rise in cytokine production, which can have devastating effects on the host, even leading to death in some cases.92,93 CoV replicates its genome through the type I interferon (IFN) signaling pathway and builds up a base for a secondary attack, causing massive respiratory inflammation related to cytokine and chemokine secretion.92 The cytokine amphiregulin mediates tissue repair and neutralizes inflammation in the host body.93 Ebola virus can cause viral hemorrhagic fever (VHF), which is induced by massive inflammation and has resulted in over 11,000 deaths in West Africa and has caused a great deal of concern when cases were diagnosed in the USA. In humans and animals, VHF can involve intravascular coagulation, hemorrhagic fever, internal bleeding, and bleeding from the mouth, eyes, and other body orifices. VHF viruses mainly target dendritic cells (DCs), macrophages, and monocytes, which are the principal sources of inflammatory cytokines.94
Dengue virus infection can cause dengue hemorrhagic fever (DHF) and multiorgan failure.95,96 The virus is endemic in many areas across the globe, especially in tropical and subtropical regions. It needs mosquitoes to act as a vector to complete its primary infection cycle and transmission to humans. DENV NS1-mediated activation of the cathepsin L/heparanase pathway during DHF can cause multiorgan failure.97 Several cytokines (TNF-α, CCL2, CXCL10, IL-1, 6, 8, and 10) underlie the clinical manifestations.89 However, further developments are required to fully determine the precise mechanisms underlying DHF, as the current animal models are limited because the virus does not naturally infect non-human species.
At present, researchers are exploring the molecular and cellular contributors to “inflammation” that causes various health conditions. Based on this research, new vaccines and drugs that target inflammatory cascades may be discovered, which will be helpful for the treatment of these health conditions.
RNA Viruses That Activate NLRP3 Inflammasomes
As we see in Figure 2, various RNA viruses from various families can activate the NLRP3 inflammasome. Such as Encephalomyocarditis virus (EMCV), Hepatitis C virus (HCV), Sendai virus, Human immunodeficiency virus-1 (HIV1), Respiratory syncytial virus (RSV), Human rhinovirus (HRV), Zika virus (ZIKV), Influenza virus (IAV), Dengue virus (DENV), Rift valley virus (RVFV), Measles virus (MV), Sendai virus (SeV), Enterovirus 71 (EV-71), Coronavirus (SARS-CoV) have already demonstrated that activate the NLRP3 inflammasome. An in vitro study of rotavirus supplied the first evidence, and then the genomes of Sendai virus (SeV) and influenza A virus were observed to activate caspase-1 in murine macrophages.47
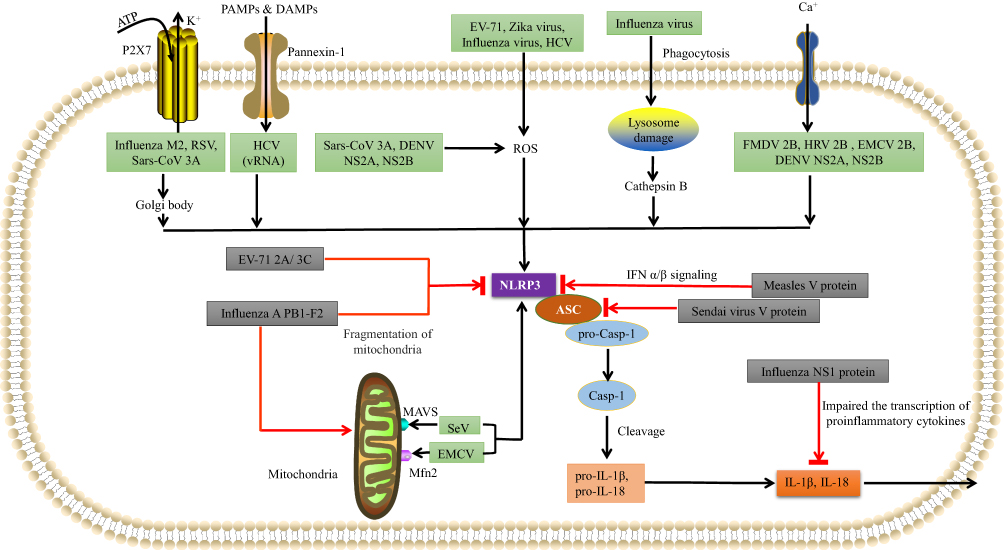 |
Figure 2 Various RNA viruses can activate NLRP3 inflammasomes. EV-71, Zika virus (ZIKV), influenza virus, and hepatitis C virus (HCV), DENV NS2A, NS2B use the ROS-dependent pathway to form the NLRP3 inflammasome complex. Influenza virus can also rupture lysosomes and then activate NLRP3 inflammasomes via a cathepsin B-dependent process. SeV uses MAVS, and EMCV uses mitofusin-2 to activate NLRP3. FMDV 2B, HRV 2B, and EMCV 2B proteins activate NLRP3 via the Ca2+ model, while HCV viral RNA, RSV viral RNA, Sars-CoV 3A and influenza M2 use the K+ model involving pannexin channels. Influenza M2 and RSV also use the Golgi network to activate NLRP3. Various RNA viruses can inhibit NLRP3 inflammasomes using virus-encoded proteins. The influenza NS1 protein prevents caspase-1 activation and impairs the transcription of pro-inflammatory cytokines to prevent IL-1β and IL-18 release. The influenza A PB1-F2 protein impairs mitochondria in order to inhibit NLRP3 activation. The measles virus (MV) V protein prevents the transcription of IFN-α/β. The SeV V protein prevents NLRP3-mediated ASC oligomerization. The EV-71 2A and 3C proteases directly cleave NLRP3. |
Coronaviridae Family (+)ssRNA Virus
Severe acute respiratory syndrome coronavirus (Sars-CoV) is a (+)ssRNA virus that belongs to the Coronaviridae family.98 The WHO (World Health Organization) declared COVID-19 a pandemic, and as of 16 December 2020, approx. Globally, 73.5million cases with 1.64 million deaths have been reported.99,100 IL-1β, IL-18, and LDH (lactate dehydrogenase) upsurges have been reported in sera in patients with COVID-19, indicating inflammasome network involvement.101–103 Similarly, the activation of NLRP3 inflammasome was discovered in a recent report on COVID-19 patients.104 The microscopic analysis combined with luminescent assays in one study showed the formation of NLRP3 and ASC puncta, activation of caspase-1, and secretion of IL-1β in COVID-19 patients with PBMCs (peripheral blood mononuclear cells) during the disease and in postmortem lung tissues.104 Moreover, the level of Casp-1p20 and IL-18 in patients with COVID-19 has been an important marker for determining the disease’s severity.104 It was reported that Sars-CoV 3A viroporin could induce the NLRP3 inflammasome activation and IL-1β production via mitochondrial ROS and ion channel.105 Recent research reveals that the SARS-CoV E protein, composed of just 76 amino acids, forms permeable Ca2+ ion channels and stimulates the NLRP3 inflammasome.106 IL-1β secretion was significantly higher in LPS-primed BMDMs co-transfected with Sars-Cov 3A viroporin resulting in that Sars-CoV 3A viroporin induces IL-1β secretion.105 Increasing extracellular K+ concentration blocks the 3A viroporin mediated IL-1β secretion in BMDMs. It proves that Sars-Cov 3A viroporin activates NLRP3 inflammasome by K+ channel. However, Mito-TEMPO treated BMDMs shows a lower secretion of 3A viroporin mediated IL-1β, indicating that mitochondrial ROS is essential for Sars-Cov 3A viroporin mediated NLRP3 activation.105 All of these data prove that Sars-Cov 3A viroporin can activate NLRP3 inflammasome via mitochondrial ROS and K+ channel.
Orthomyxoviridae Family (-)ssRNA Virus
Influenza virus is an (-)ssRNA virus that belongs to the Orthomyxoviridae family, which includes seven genera.107 Influenza virus is the most studied RNA virus and it is capable of activating NLRP3 inflammasomes.108 Recent research has shown that one of the mechanisms underlying NLRP3 activation during influenza virus infection involves influenza virus activating signal 1 (the priming signal). Signal 1 can involve activating TLR-7 signaling in macrophages and DCs, which leads to the synthesis of various pro-inflammatory cytokines such as pro-IL-18 and pro-IL-1β.109 The presence of the influenza virus M2 protein in the TGN then initiates signal 2 to activate the NLRP3 inflammasome. For signal 2, the M2 proteins act as ion channels, exporting H+ from the acidified Golgi body. This creates an ionic imbalance, leading to K+ efflux combined with Na+ efflux, and ROS production, which activates the NLRP3 inflammasome.109
Moreover, during infection of BMDMs with wildtype influenza virus, the BMDMs secreted pro-inflammatory cytokines such as IL-18 and IL-1β. However, IL-18 and IL-1β were not secreted during infection with mutated influenza virus expressing non-functional M2. In contrast, after LPS-stimulated BMDMs and bone marrow-derived DCs were transduced with recombinant lentivirus expressing M2, the NLRP3 inflammasome was activated. The function of the M2 proteins as ion channels has been suggested as a vital step for NLRP3 inflammasome activation.109 Additionally, ATP release from dying influenza-infected cells amplified NLRP3 activation.29
Phenuiviridae Family (-)ssRNA Virus
Rift Valley fever virus (RVFV) is an (-)ssRNA virus that belongs to the family Phenuiviridae and the genus Phlebovirus.110 RVFV activates NLRP3 inflammasomes via MAVS.26 RVFV-infected cells secrete IL-1β, which involves the NLRP3 inflammasome, ASC oligomerization, and caspase-1 maturation.85 Conventional DCs from the bone marrow of wildtype mice were infected with RVFV, which led to IL-1β release. To assess whether NLRP3 and ASC are required for IL-1β release during RVFV infection, NLRP3- and ASC-deficient cells were also infected, resulting in negligible amounts of IL-1β secretion. It was also observed that viral replication was vital for the processing of IL-1β.85 The mitochondrial protein MAVS directly interacts with NLRP3 and also influences IL-1β secretion in response to various NLRP3 activators (eg SeV V protein).83 The molecular interaction of NLRP3 and MAVS in DCs infected with RVFV was observed by confocal microscopy.85 Thus, RVFV brings about the release of IL-1β by activating the NLRP3 inflammasome.
Pneumoviridae Family (-)ssRNA Virus
Respiratory syncytial virus (RSV) is an (-)ssRNA virus that belongs to the family Pneumoviridae and the genus Orthopneumovirus.111 For RSV, NLRP3 inflammasome activation is dependent on K+ efflux and ROS production, and caspase-1-mediated IL-1β secretion subsequently occurs.112 293T cells were infected with RSV to observe the function of caspase-1 regarding IL-1β secretion. In RSV-infected wildtype 293T cells expressing caspase-1, IL-1β secretion was higher when the cells were transfected with pcDNA 3.1 empty vector, pro-caspase-1, and pro-IL-1β. In contrast, in RSV-infected caspase-1-knockout 293T cells, the level of IL-β in the supernatant was dramatically reduced compared to after the infection of wildtype cells. These results show that caspase-1 is essential for IL-1β production during RSV infection.112 Additionally, the NLRP3 inflammasome was found to be necessary for IL-1β secretion by RSV-infected NLRP3-expressing cells. Moreover, ASC plays a critical role as a significant element of the NLRP3 inflammasome,112 as infection of HEK-293T cells with RSV resulted in a higher level of IL-1β secretion. This indicated that, for IL-1β secretion, ASC plays a critical role during RSV infection.112 Furthermore, RSV activates NF-κB during infection.113 After using BAY 11–7082 (an NF-κB inhibitor) in RSV-infected cells, IL-1β production was lower. This indicated that NF-κB signaling is essential for NLRP3 expression during RSV infection.112
MyD88 plays a crucial role as an adaptor protein during TLR-mediated signal transduction. After TLR2 and MyD88 knockout in BMDMs, RSV infection dramatically reduced the IL-1β level in the supernatant in comparison to the level released by infected wildtype and infected TLR4-knockout BMDMs.2 This indicated that the TLR2/MyD88 pathway is essential for the production and release of IL-1β.2 In other studies, it was shown that intracellular ROS production occurs during RSV infection.85,114
Paramyxoviridae Family (-)ssRNA Viruses
Measles virus (MV) is an (-)ssRNA virus that belongs to the family Paramyxoviridae and the genus Morbillivirus.115 It is a non-segmented RNA virus that encodes phosphoprotein (P), protein V, and protein C.116 MV activates NLRP3 inflammasomes, which leads to caspase-1-mediated production of mature IL-1β.2 Phorbol 12-myristate 13-acetate (PMA)-stimulated THP-1 cells infected with MV secrete IL-1β. However, THP-1 cells that were infected with UV-treated MV only released a low level of IL-1β. These findings indicate that MV can induce IL-1β release by THP-1 cells.2 IL- 1β secretion was assessed in THP-1 cells after using short hairpin RNA to target human NLRP3, which indicated that MV activates NLRP3 inflammasomes. When HEK 293T cells were transfected with the MV V protein, co-precipitation with NLRP3 was observed, indicating that the V protein interacts with NLRP3.117 Thus, MV infection can activate NLRP3 and cause IL-1β secretion.
Sendai virus (SeV) is an (-)ssRNA virus that belongs to the family Paramyxoviridae and the genus Respirovirus.118 When SeV infected 293T cells and mouse embryonic fibroblasts, it induced the formation of huge MAVS signaling complexes composed of functional prison-like MAVS aggregates.119 Infecting NLRP3-transfected cells with SeV led to NLRP3-mediated caspase-1 activation, but this did not occur in the absence of NLRP3. These results indicate that SeV infection leads to NLRP3-dependent caspase-1 activation.86 SeV infection of THP-1 cells caused caspase-1 activation and IL-1β secretion, while SeV infection of MAVS-knockdown THP-1 cells significantly decreased the formation of active caspase-1 and the mature form of IL-1β (p17). These findings indicate that MAVS is crucial for caspase-1-related NLRP3 activation.86
Newcastle disease virus (NDV) is an (-)ssRNA virus that belongs to the family Paramyxoviridae and the new genus Avulavirus.120 It can induce the cleavage of pro-IL-1β by caspase-1 and the subsequent release of IL-1β, which involves activating NLRP3 inflammasomes.114 This IL-1β release, which is mediated by caspase-1, was demonstrated in THP-1 monocytic cells infected with the highly virulent Herts/33 strain of NDV.114 NDV-induced activation of NLRP3 was confirmed by infecting BMDMs from wildtype and NLRP3-deficient mice. NDV-induced activation of NLRP3 was confirmed in THP-1 cells expressing short hairpin RNA targeting human NLRP3.114 These findings confirm that NDV activates NLRP3 inflammasomes and thereby induces IL-1β secretion.
Retroviridae Family (+)ssRNA Virus
Human immunodeficiency virus (HIV) is an (+)ssRNA virus that belongs to the family Retroviridae and the genus Lentivirus.121 HIV infection activates NLRP3 inflammasomes, resulting in IL-1β release.122 Monocyte-derived DCs were isolated from healthy individuals and HIV-1-positive patients and infected with inactivated (aldrithiol-2-treated) HIV-1.122 NLRP3 mRNA expression was higher in the DCs from healthy individuals, and this was followed by increases in CASP-1 and IL-1β mRNA levels and IL-1β secretion, while stimulation with inactivated HIV-1 failed to activate inflammasome and cytokine production in HIV-DC.122 These findings strongly suggest that the whole genome of HIV can activate NLRP3 inflammasomes. However, the precise NLRP3 activation mechanism and host–virus immunological associations remain unknown. The mechanisms underlying NLRP3 inflammasome activation related to HIV infection need further investigation.
Flaviviridae Family (+)ssRNA Virus
Zika virus (ZIKV) is an (+)ssRNA virus that belongs to the family Flaviviridae and the genus Flavivirus.123 ZIKV encodes an idiosyncratic polypeptide that contains several non-structural proteins, including the viral RNA-dependent RNA polymerase (RdRp) NS5.124 NS5 can activate the NLRP3 inflammasome by binding to leucine-rich repeat (LRR) and NACHT domains, but not the pyrin domain (PYD).125 Via a de novo mechanism, this RdRp initiates the synthesis of viral genomic RNA,126 and it also suppresses signal transducer and activator of transcription 2 (STAT2) and thus inhibits type 1 IFN signaling.127 It localizes to the nucleus.127 NLRP3, ASC, and caspase-1 are diffusely located in the cytoplasm and NS5 interacts with NLRP3 via its LRR and NACHT domains and allows the assembly of the NLRP3 inflammasome complex. An NS5–NLRP3–ASC spherelike structure is formed, with NS5 located in the interior, NLRP3 interacting with NS5, and ASC on the periphery.125 ZIKV infection induces IL-1β secretion, and this was explored by the knockdown of NLRP3 inflammasome constituents in THP-1 macrophages,2 which indicated that the NLRP3 inflammasome is crucial for ZIKV-induced IL-1β secretion. The inflammasome complex has a ring-like structure128 and the formation of specks of oligomerized ASC indicates the formation of the inflammasome complex,28 with ASC oligomerization providing strong evidence of the induction of NLRP3 inflammasome activation.129 ZIKV increases the formation of specks of oligomerized ASC, which directly indicates that inflammasomes, involving caspase-1, are crucial for ZIKV-induced IL-1β secretion.125
Dengue virus (DENV) is a mosquito-borne (+)ssRNA virus that belongs to the family Flaviviridae and the genus Flavivirus having four serotypes, DENV 1–4.130 It contains three structural proteins and seven nonstructural proteins.131 It was reported that DENV could activate NLRP3 inflammasome and IL-1β production in human blood macrophages and platelets.132,133 However, the Nonstructural proteins 2A, 2B, and structural protein M activate the NLRP3 inflammasome.134,135 M protein of DENV is responsible for endothelial dysfunction, and vascular leakage resulting in IL-1β mediated NLRP3 activation.134 DENV stimulates inflammatory NLRP3 assembly in platelets. By controlling IL-1β, platelets may increase vascular permeability.132 DENV M mediated vascular permeability and leakage was confirmed by infecting BMDMs with lentivirus-M isolated from NLRP3+/+ and NLRP3−/- mice and treated with LPS plus ATP or LPS plus nigericin. NLRP3 was activated by ATP or nigericin in lentivirus-M-treated BMDMs of NLRP3+/+ mice. IL-1β production was significantly higher in ATP or nigericin in lentivirus-M-treated BMDMs of NLRP3+/+ mice, resulting in NLRP3 involvement M-mediated IL-1β production and secretion.134 Another study showed that NS2A and NS2B of DENV-2 induce IL-1β secretion and activation of NLRP3 inflammasome through Ca++ ion channel and mitochondrial ROS model.135 THP-1 and HMEC-1 cells were infected with DENV-2 or treated with LPS plus ATP. Detection of IL-1β secretion from supernatant or Caspase-1 and ASC oligomerization indicates that DENV-2 can induce NLRP3 inflammasome. Detection of a higher level of NLRP3 expression, ASC oligomerization, and Caspase-1 activation from LPS-treated HMEC-1 cells transfected with NS2A or NS2B indicates that NS2A and NS2B can activate IL-1β mediated NLRP3 inflammasome.135 Above all, results suggest that DENV M, NS2A, and NS2B proteins induce NLRP3 inflammasome activation.
Hepatitis C virus (HCV) is an (+)ssRNA virus that belongs to the family Flaviviridae and the genus Flavivirus.136 It was reported that HCV could induce the activation of NLRP3 inflammasome and the production of IL-1β by intrahepatic macrophages that delegates liver inflammation.137 HCV infects hepatocytes causing liver inflammation and tissue damage, leading to fibrosis and cirrhosis. These mechanisms underlie liver disease and are believed to affect the initiation of liver cancer.138,139 RNA sequencing from liver biopsies was done in control and Chronic HCV-infected patient group to assess the host response with the liver disease during HCV infection. Higher expression of IL-1β from the HCV-infected patient group indicates that HCV can induce IL-1β production and secretion by liver macrophages in chronic HCV-infected patients. HCV infected MyD88-knockdown THP-1 showed higher expression of IL-1β whether HCV infected MAVS-knockdown THP-1 had no significant IL-1β expression. Again HCV infected TLR7-knockdown THP-1 showed significant IL-1β mRNA expression reduction resulting from that HCV stimulated IL-1β secretion is MyD88 dependent through TLR7 signalling.137 Another study showed that HCV genomic RNA is responsible for NLRP3 inflammasome activation in human myeloid cells.140 HCV infected THP-1 was treated with glyben (potassium channel inhibitor) in a dose-dependent manner resulting in the blockage of IL-1β maturation, indicating HCV-induced NLRP3 activation is potassium efflux dependent.137 Transfection of HCV RNA into diphenyl iodonium (ROS inhibitor) pretreated THP-1 cells causing reduced IL-1β secretion, which means that HCV RNA-induced NLRP3 activation is ROS dependent.140 Above all data prove that HCV induced NLRP3 inflammasome activation in potassium efflux and ROS-dependent manner.
Picornaviridae Family (+)ssRNA Viruses
Encephalomyocarditis virus (EMCV) is an (+)ssRNA virus that belongs to the family Picornaviridae and the genus Cardiovirus.141 Recently, a study demonstrated the relationship between EMCV and NLRP3 activation. The EMCV 2B viroporin interacted with and activated the NLRP3 inflammasome, which was originally diffusely distributed in the cytosol; in contrast, the viral RNA was unable to activate the NLRP3 inflammasome.142 Moreover, the 2B viroporin increased the [Ca2+]cyt in LPS-primed mouse BMDMs infected with EMCV, which induced IL-1β secretion.142 Thus, EMCV can induce IL-1β secretion in an NLRP3-dependent manner.143 The 2B viroporin localizes to the endoplasmic reticulum (ER) and Golgi body, which reduces the [Ca2+] in these organelles.144 To assess the role of the 2B viroporin in changes in [Ca2+]cyt, 2B was transfected into cells and the calcium kinetics were assessed using calcium-sensitive fluorescent probes, which indicated that 2B has a significant role in activating the NLRP3 inflammasome in a mechanism involving the ER and Golgi body network. After EMCV infection, there was a large increase in the fluorescence intensity.142 This indicated that EMCV infection increases the [Ca2+]cyt via fluxes (involving the stores in the ER and Golgi body) that, in turn, activate the NLRP3 inflammasome in a calpain-independent manner. Another study showed that mitochondrial ROS can induce NLRP3 inflammasome activation.145 The kinetic changes in mitochondria produced by ROS indicated that NLRP3 activation was dependent on mitochondrial ROS. The level of mitochondrial ROS was higher at 3 h after EMCV infection but decreased at 24 h after infection.142 This indicates that EMCV can activate NLRP3 inflammasomes by increasing mitochondrial ROS.
Enterovirus 71 (EV-71) is an (+)ssRNA virus that belongs to the family Picornaviridae and the genus Enterovirus.146 EV-71 can activate NLRP3 inflammasomes, resulting in the release of IL-1β.147 THP-1 cells were infected with EV-71 to explore whether EV-71 activates NLRP3, and IL-1β secretion was detected in the supernatant. It was also confirmed that heat- or UV- inactivated EV-71 was unable to induce IL-1β secretion by THP-1 cells.147 These findings indicate that EV-71 can induce the release of IL-1β. Again, caspase-1 activation and ASC oligomerization are the firm references for inflammasome activation.129 After EV-71 infection of THP-1 cells, mature IL-1β as well as caspase-1 was detected in the supernatant and ASC was oligomerized. Additionally, Ac-YVAD-CHO (a caspase-1 inhibitor) prevented EV-71-induced caspase-1-mediated IL-1β secretion.147 These results show that EV-71 can activate NLRP3 and induce IL-1β.
Human rhinovirus (HRV) is an (+)ssRNA virus that belongs to the family Picornaviridae and the genus Enterovirus.148 There are three species (A–C). The HRV 2B viroporin can activate both the NLRP3 and NLRC5 inflammasomes.149 After bronchial epithelial cells were infected with HRV or transfected with the 2B viroporin, 2B localized to the Golgi body, as did NLRC5, NLRP3, and ASC. After treatment with BAPTA-AM (a Ca2+ chelator) or verapamil (a Ca2+ channel inhibitor), 2B activated the NLRP3 inflammasome by increasing [Ca2+]cyt, which indicated that the HRV 2B viroporin activates NLRP3 inflammasomes in a process that involves Ca2+ channels.150
Foot and mouth disease virus (FMDV) is an (+)ssRNA picornavirus that belongs to the genus Aphthovirus.151 The 2B protein of FMDV, a non-structural protein, can induce NLRP3 activation and IL-1β secretion.152 It has been reported that signal 2 for NLRP3 activation in response to FMDV infection involves K+ efflux and Ca2+ influx but is ROS and cathepsin B independent. The 2B protein can change the membrane permeability, causing an imbalance regarding [Ca2+]cyt in the host cell.153 In particular, amino acids 140–145 of the 2B protein are responsible for IL-1β release after NLRP3 activation as they are involved in pore formation in the cell membrane, which allows ion efflux. FMDV RNA also activates NF-κβ, which is involved in signal 1 and regulates the transcription of pro-IL-1β and pro-IL-18. After FMDV infection, activated NLRP3 induces IL-1β secretion, independent of the RIG-1 inflammasome. FMDV-infected RAW264.7 cells secreted IL-1β after NLRP3 inflammasome activation by the induction of NF-κβ p65, which is involved in signal 1. Mice and porcine macrophages infected with FMDV produced ASC, caspase-1, and IL-1β levels that varied over time and with different infection doses. The FMDV 2B protein activates NLRP3 inflammasomes by decreasing [K+]cyt and increasing [Ca2+]cyt, causing the release of IL-1β.152 This evidence shows that the FMDV 2B protein can activate NLRP3 inflammasomes via ion channel formation in a ROS-independent manner.
These findings show that the proteins of certain RNA viruses activate NLRP3 inflammasomes, including by binding to specific sites on NLRP3. However, the precise mechanisms underlying NLRP3 activation via these proteins are not yet known. Therefore, further investigation is needed to understand the complete mechanisms underlying NLRP3 activation by these proteins.
RNA Viruses That Inhibit NLRP3 Inflammasomes Using Virus-Encoded Proteins
It has been reported that many viral proteins can inhibit inflammasome activation as we see in Figure 2.154
Orthomyxoviridae Family (-)ssRNA Virus
Mutant influenza viruses without NS1 or with NS1 with deletion of the N-terminal RNA-binding domain (but not the C-terminal region) can induce the secretion of IL-1β and IL-18 after the production of mature caspase-1. This process involves the N-terminal domain of NS1.155 Additionally, the PB1-F2 protein of influenza virus reduces the mitochondrial membrane potential, which inhibits NLRP3 activation.156 Again, PB1-F2 protein of highly pathogenic influenza A (H7N9) seized the IL-1β secretion from IAV-infected macrophages resulting the inhibition of RNA-induced NLRP3 inflammasome activation.157 However, this protein is the subject of debate concerning its pro-inflammatory properties. Another study has shown that extracellular protein aggregate PB1-F2 phagocytose is known for its macrophages contributing to NLRP3 overactivity and increased IL-1β and IL-18 secretion.158 It is still unknown how extracellular and intracellular fabricate PB1-F2 contributes to viral pathogenesis.157 PYD-containing viral proteins, such as the Shope fibroma virus (SFV)-encoded PYD-only protein/macromolecule (vPOP) and the M13L protein of poxvirus, co-localize and associate with ASC via PYD–PYD interactions and then prevent caspase-1 activation and IL-1β production.159,160 The NS1 protein of influenza A virus can inhibit the NLRP3 inflammasome,161 with NS1 proteins from both extremely infective and low-pathogenicity strains dramatically reducing IL-1β and IL-18 secretion by LPS- and ATP-treated THP-1 cells. NS1 overexpression inhibits the transactivation mediated by the transcription factor NF-κβ, reducing the transcription of pro-inflammatory cytokines. The interaction of NS1 with NLRP3 in NS1-expressing THP-1 cells prevented NLRP3 inflammasome activation.161 These results show that the influenza virus NS1 protein can inhibit NLRP3 inflammasomes; this involves reducing the transactivation mediated by NF-κβ, thus preventing caspase-1 maturation and IL-1β and IL-18 secretion.
Paramyxoviridae Family (-)ssRNA Viruses
The MV V protein can interact with both NLRP3 and NLRP1 to suppress inflammasome assembly, thereby preventing caspase-1 maturation and IL-1β release.162 Additionally, various other proteins from numerous viruses can inhibit inflammasomes and prevent inflammasome formation by interacting with inflammasome components.117,163 The MV V protein can inhibit the IL-1β secretion induced by MV infection.117 A study showed that LPS- and ATP-treated THP-1 cells expressing the MV V protein only secreted a very small quantity of IL-1β, indicating that NLRP3 inflammasome activation was blocked by the MV V protein. In contrast, when the THP-1 cells expressing the MV V protein were treated with poly(dA:dT), the IL-1β secretion was much higher than in the prior experiment, which indicates that the MV V protein does not disturb the IL-1β secretion pathway, only the NLRP3 inflammasome pathway. The V protein binds to the C-terminal domain of NLRP3 to prevent NLRP3-mediated IL-1β secretion.117 The V protein co-immunoprecipitated with the NLRP3 inflammasome when HEK293T cells were co-transfected with plasmids encoding the V protein, further indicating that the V protein interacts with NLRP3.117 Thus, the V protein inhibits NLRP3 inflammasome activation by binding with the C-terminal domain of NLRP3, thereby preventing the secretion of IL-1β.
The SeV V protein can inhibit the release of IL-1β by preventing NLRP3 inflammasome assembly.164 THP-1 cells infected with SeV without the V gene secrete a large amount of IL-1β.164 NLRP3-deficient cells infected with this mutant SeV exhibited reduced IL-1β secretion, indicating the essential role of NLRP3 inflammasomes in the abovementioned IL-1β secretion. The SeV V protein interacts with NLRP3, which inhibits NLRP3 self-oligomerization, and subsequently inhibits ASC recruitment, thereby inhibiting NLRP3-dependent ASC oligomerization. This successively prevents full activation of the NLRP3 inflammasome and thereby prevents IL-1β release. Thus, the SeV V protein inhibits NLRP3 inflammasome assembly and IL-1β release.164
Picornaviridae Family (+)ssRNA Virus
The EV-71 2A protein, along with the EV-71 3C protein, inhibits NLRP3 inflammasome activation.147 EV-71 2A and 3C proteases were transfected into 293T cells to assess the resultant NLRP3 levels and cleavage sites in NLRP3. After the 2A and 3C proteases cleaved NLRP3, 65-kDa and 90-kDa fragments, respectively, were produced. Co-immunoprecipitation assays in co-transfected 293T cells confirmed the interactions of 2A and 3C with NLRP3. It was reported that the Q225–G226 pair is the cleavage site for 3C, while the G493–L494 pair is the cleavage site for 2A. Additionally, 3C and 2A expression dose-dependently decreased the IL-1β secretion by HEK293T cells, but the IL-1β secretion was increased after transfection of 293T cells with pro-IL-1β, ASC, pro-caspase-1, and NLRP3. These results indicated that the EV-71 2A and 3C proteases cleave NLRP3, thus inhibiting NLRP3 inflammasomes.147
Conclusions
Recent research demonstrated that, during certain pathogen infections, NLRP3 is able to detect specific ligands, activate caspase-1, and induce the release of various pro-inflammatory cytokines with major roles against viral infection.117
In the past few years, intense efforts have been put into the investigation of the relationship between virus and NLRP3 inflammasome. Viral RNA, viroporin, and infectious viral particles activate the NLRP3 inflammasome.154 Recent progress has been made to reveal the critical roles for RNA viruses in activation and inhibition of NLRP3 inflammasome. Most RNA viral infection activates or inhibits NLRP3 inflammasome by regulating ion channels and ROS model. The studies showed that K+ efflux plays a critical role in NLRP3 activation, however, Ca2+ channel and ROS model remain controversial. Notably, Nek7 as an undisputed essential positive regulator which is required for the stimuli or ion channel-mediated NLRP3 inflammasome activation.165,166 Clearly, further work is needed to understand the mechanisms by which viruses regulate Nek-NLRP3 inflammasome. Another study has shown that DNA virus can also induce NLRP3 activation.167 During HSV-1 infection, the stimulation of interferon genes (STING) recruits NLRP3 and allows NLRP3 to locate an endoplasmic reticulum, thereby promoting the development of NLRP3inflammasome. Again, STING integrates with NLRP3 and eliminates interactions between K48 and K63 NLRP3 polyubiquitination, encouraging the activation of inflammasome. Signals from cGAS-STING-NLRP3 are important for host security HSV-1-free infections.167
NLRP3 inflammasomes have been explored in research on colonic epithelium homeostasis168 and they are implicated in the development and invasion of various human cancers such as gastric,169 breast,170 lung,171 and skin172 cancer. NLRP3 is a primary tissue damage sensor and a sterile inflammatory activator.173 NLRP3 Inflammasome activation enables homeostasis restoration after traumatic tissue damage by promoting damage clearance, tissue recovery and regeneration.173,174 Some research indicates that activation of NLRP3 facilitates epithelial integrity and restoration during infection through IL-1β and IL-18 production.175,176 There is almost no doubt that the dysregulated NLRP3 inflammasome is involved in the pathogenesis of several infectious, allergic and inflammatory disorders. Therefore, it is crucial to consider the molecular processes by which NLRP3 inflammasome activation is triggered and terminated. These pathways demonstrate that the body generates an adequate inflammatory response that prevents unnecessary collateral harm, which may otherwise contribute to autoimmune and autoinflammatory pathology.
On the whole, PRRs and inflammasome expression in the tissues play a monitoring and triggering role for the pathogens. Except for the inflammation activation, in general, PPRs and NLRP3 promote the tissue recovery.177,178 Therefore, we have reason to speculate that virus infection might destroy the NLRP3-mediated tissue repairing. Upon viruses infection, viruses use their own genomes, proteins or by-products to regulate inflammation, different conditions produced in different infection stages, in-depth further study based on different infection stages and status plays an important role in the control of inflammation.
Many RNA viral proteins have been shown to mediate NLRP3 activation or inhibition (Table 1), and viral induction of NLRP3 inhibition might facilitate virus rescue of the antiviral immune defenses of the host. More research on the association between viral infections and NLRP3 activation will likely promote the development of novel therapeutics (including inhibitors) for managing RNA viral infections and associated diseases. Our review provides the summarization of research on the consequences of infections involving RNA viruses.
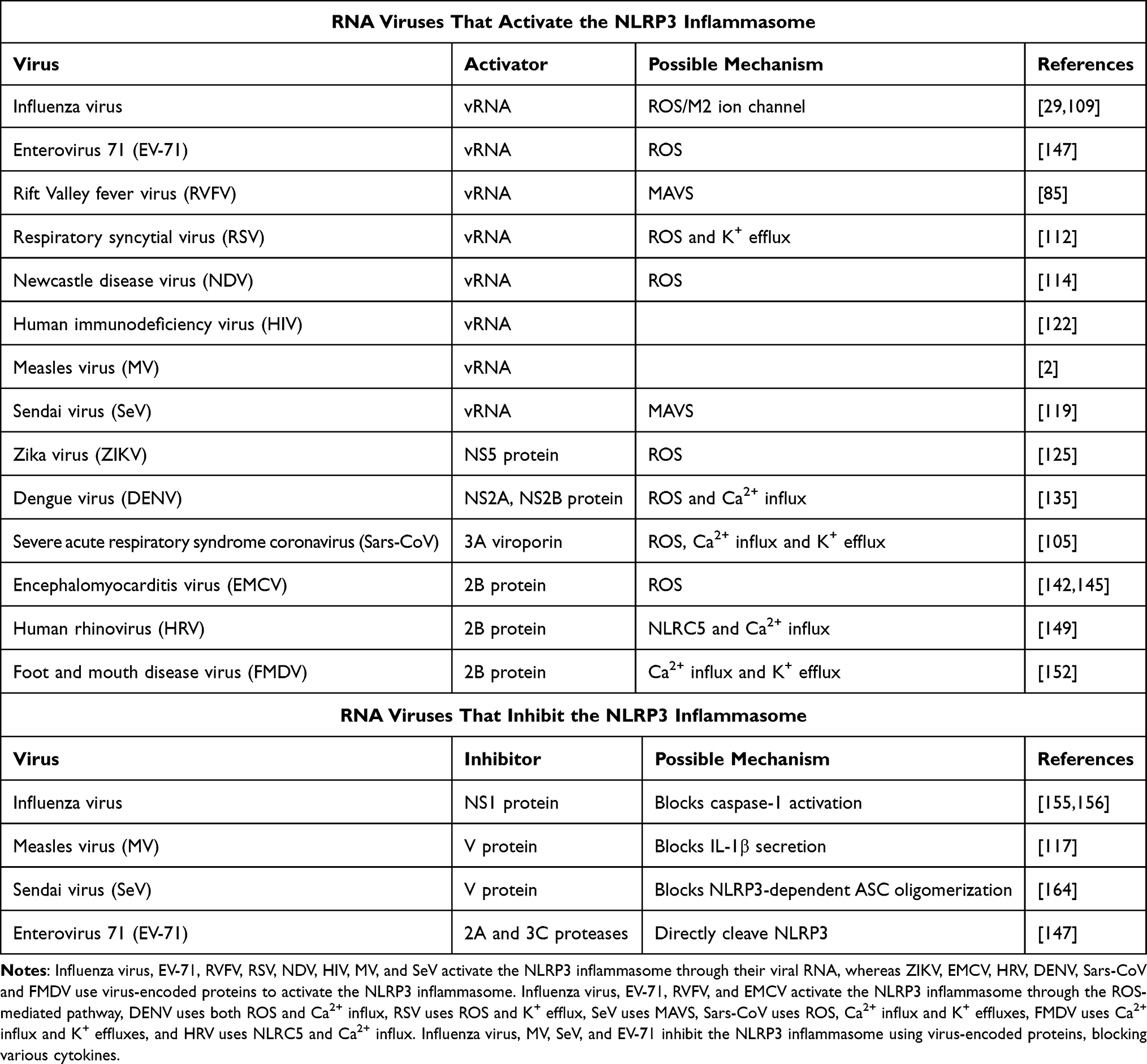 |
Table 1 Summary of the Components of RNA Viruses That Affect the NLRP3 Inflammasome |
Acknowledgments
We sincerely thank Dr. Zhidong Zhang for the revision of the manuscript.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by a grant from the National Natural Sciences Foundation of China (no. 31602037).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Medzhitov R, Janeway CA
2. Medzhitov R, Janeway CA
3. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30(1):16–34. doi:10.3109/08830185.2010.529976
4. Leemans JC, Cassel SL, Sutterwala FS. Sensing damage by the NLRP3 inflammasome. Immunol Rev. 2011;243(1):152–162. doi:10.1111/j.1600-065X.2011.01043.x
5. Tominello TR, Oliveira ERA, Hussain SS, et al. Emerging roles of autophagy and inflammasome in ehrlichiosis. Front Immunol. 2019;10:1011. doi:10.3389/fimmu.2019.01011
6. Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21(4):317–337. doi:10.1093/intimm/dxp017
7. Huang Q, Pope RM. Toll-like receptor signaling: a potential link among rheumatoid arthritis, systemic lupus, and atherosclerosis. J Leukoc Biol. 2010;88(2):253–262. doi:10.1189/jlb.0310126
8. Oliveira-Nascimento L, Massari P, Wetzler LM. The role of TLR2 in infection and immunity. Front Immunol. 2012;3:79. doi:10.3389/fimmu.2012.00079
9. Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. 2013;45:e66. doi:10.1038/emm.2013.97
10. Gehring T, Seeholzer T, Krappmann D. BCL10 - bridging CARDs to immune activation. Front Immunol. 2018;9:1539. doi:10.3389/fimmu.2018.01539
11. Staal J, Driege Y, Haegman M, et al. Ancient origin of the CARD-coiled coil/Bcl10/MALT1-like paracaspase signaling complex indicates unknown critical functions. Front Immunol. 2018;9:1136. doi:10.3389/fimmu.2018.01136
12. Zhao XQ, Zhu LL, Chang Q, et al. C-type lectin receptor dectin-3 mediates trehalose 6,6ʹ-dimycolate (TDM)-induced mincle expression through CARD9/Bcl10/MALT1-dependent nuclear factor (NF)-kappaB activation. J Biol Chem. 2014;289(43):30052–30062. doi:10.1074/jbc.M114.588574
13. Onomoto K, Onoguchi K, Yoneyama M. Regulation of RIG-I-like receptor-mediated signaling: interaction between host and viral factors. Cell Mol Immunol. 2021:1–7.
14. Li Y, Li Y, Cao X, Jin X, Jin T. Pattern recognition receptors in zebrafish provide functional and evolutionary insight into innate immune signaling pathways. Cell Mol Immunol. 2017;14(1):80–89. doi:10.1038/cmi.2016.50
15. Zhu W, Zu X, Liu S, Zhang H. The absent in melanoma 2 (AIM2) inflammasome in microbial infection. Clin Chim Acta. 2019;495:100–108. doi:10.1016/j.cca.2019.04.052
16. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–73, Table of Contents. doi:10.1128/CMR.00046-08
17. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi:10.1016/j.cell.2010.01.040
18. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5. doi:10.1189/jlb.0306164
19. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249(1):158–175. doi:10.1111/j.1600-065X.2012.01146.x
20. Tsai SY, Segovia JA, Chang TH, et al. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog. 2014;10(1):e1003848. doi:10.1371/journal.ppat.1003848
21. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–1022. doi:10.1016/j.cell.2014.04.007
22. Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015;265(1):6–21. doi:10.1111/imr.12296
23. Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213(6):617–629. doi:10.1083/jcb.201602089
24. Bankers-Fulbright JL, Kalli KR, McKean DJ. Interleukin-1 signal transduction. Life Sci. 1996;59(2):61–83. doi:10.1016/0024-3205(96)00135-X
25. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi:10.1146/annurev.immunol.21.120601.141126
26. Shrivastava G, Leon-Juarez M, Garcia-Cordero J, Meza-Sanchez DE, Cedillo-Barron L. Inflammasomes and its importance in viral infections. Immunol Res. 2016;64(5–6):1101–1117. doi:10.1007/s12026-016-8873-z
27. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–791. doi:10.4049/jimmunol.0901363
28. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi:10.1146/annurev.immunol.021908.132715
29. Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. 2010;11(5):404–410. doi:10.1038/ni.1861
30. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303(5663):1529–1531. doi:10.1126/science.1093616
31. Lund JM, Alexopoulou L, Sato A, et al. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101(15):5598–5603.
32. Franchi L, Eigenbrod T, Munoz-Planillo R, et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J Immunol. 2014;193(8):4214–4222. doi:10.4049/jimmunol.1400582
33. Qiao Y, Wang P, Qi J, Zhang L, Gao C. TLR-induced NF-kappaB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 2012;586(7):1022–1026. doi:10.1016/j.febslet.2012.02.045
34. Guarda G, Zenger M, Yazdi AS, et al. Differential expression of NLRP3 among hematopoietic cells. J Immunol. 2011;186(4):2529–2534. doi:10.4049/jimmunol.1002720
35. O’Connor W
36. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11(5):373–384. doi:10.1038/ni.1863
37. Becker CE, O’Neill LAJ. Inflammasomes in inflammatory disorders: the role of TLRs and their interactions with NLRs. Semin Immunopathol. 2007;29(3):239–248. doi:10.1007/s00281-007-0081-4
38. Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7(1):31–40. doi:10.1038/nri1997
39. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi:10.1016/S1097-2765(02)00599-3
40. Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell. 2006;126(4):659–662. doi:10.1016/j.cell.2006.08.002
41. Yu HB, Finlay BB. The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe. 2008;4(3):198–208. doi:10.1016/j.chom.2008.08.007
42. Poeck H, Bscheider M, Gross O, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11(1):63–69. doi:10.1038/ni.1824
43. Cui S, Eisenacher K, Kirchhofer A, et al. The C-terminal regulatory domain is the RNA 5ʹ-triphosphate sensor of RIG-I. Mol Cell. 2008;29(2):169–179. doi:10.1016/j.molcel.2007.10.032
44. Saito T, Gale M
45. Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227(1):54–65. doi:10.1111/j.1600-065X.2008.00727.x
46. Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286(5):C1100–C1108. doi:10.1152/ajpcell.00494.2003
47. Kanneganti TD, Body-Malapel M, Amer A, et al. Critical role for cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281(48):36560–36568. doi:10.1074/jbc.M607594200
48. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. doi:10.1016/j.immuni.2013.05.016
49. Franchi L, Kanneganti TD, Dubyak GR, Nunez G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem. 2007;282(26):18810–18818. doi:10.1074/jbc.M610762200
50. Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14(9):1583–1589. doi:10.1038/sj.cdd.4402195
51. Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem. 1994;269(21):15195–15203. doi:10.1016/S0021-9258(17)36591-2
52. da Costa LS, Outlioua A, Anginot A, Akarid K, Arnoult D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis. 2019;10(5):346. doi:10.1038/s41419-019-1579-0
53. Lee GS, Subramanian N, Kim AI, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492(7427):123–127. doi:10.1038/nature11588
54. Murakami T, Ockinger J, Yu J, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012;109(28):11282–11287. doi:10.1073/pnas.1117765109
55. Rossol M, Pierer M, Raulien N, et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun. 2012;3:1329. doi:10.1038/ncomms2339
56. Brough D, Le Feuvre RA, Wheeler RD, et al. Ca2+ stores and Ca2+ entry differentially contribute to the release of IL-1 beta and IL-1 alpha from murine macrophages. J Immunol. 2003;170(6):3029–3036. doi:10.4049/jimmunol.170.6.3029
57. Chu J, Thomas LM, Watkins SC, Franchi L, Nunez G, Salter RD. Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J Leukoc Biol. 2009;86(5):1227–1238. doi:10.1189/jlb.0309164
58. Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol. 2007;17(13):1140–1145. doi:10.1016/j.cub.2007.05.074
59. Katsnelson MA, Rucker LG, Russo HM, Dubyak GR. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol. 2015;194(8):3937–3952. doi:10.4049/jimmunol.1402658
60. Verhoef PA, Kertesy SB, Lundberg K, Kahlenberg JM, Dubyak GR. Inhibitory effects of chloride on the activation of caspase-1, IL-1beta secretion, and cytolysis by the P2X7 receptor. J Immunol. 2005;175(11):7623–7634. doi:10.4049/jimmunol.175.11.7623
61. Channappanavar R, Fehr AR, Vijay R, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19(2):181–193. doi:10.1016/j.chom.2016.01.007
62. Daniels MJ, Rivers-Auty J, Schilling T, et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against alzheimer’s disease in rodent models. Nat Commun. 2016;7:12504. doi:10.1038/ncomms12504
63. Tang T, Lang X, Xu C, et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun. 2017;8(1):202. doi:10.1038/s41467-017-00227-x
64. Schorn C, Frey B, Lauber K, et al. Sodium overload and water influx activate the NALP3 inflammasome. J Biol Chem. 2011;286(1):35–41. doi:10.1074/jbc.M110.139048
65. Summersgill H, England H, Lopez-Castejon G, et al. Zinc depletion regulates the processing and secretion of IL-1beta. Cell Death Dis. 2014;5:e1040. doi:10.1038/cddis.2013.547
66. Chen J, Chen ZJ. PtdIns4P on dispersed trans-golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564(7734):71–76. doi:10.1038/s41586-018-0761-3
67. Brydges SD, Mueller JL, McGeough MD, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30(6):875–887. doi:10.1016/j.immuni.2009.05.005
68. Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30(6):860–874. doi:10.1016/j.immuni.2009.04.012
69. Heo WD, Inoue T, Park WS, et al. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314(5804):1458–1461. doi:10.1126/science.1134389
70. Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443(7112):651–657. doi:10.1038/nature05185
71. Gross CJ, Mishra R, Schneider KS, et al. K(+) efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity. 2016;45(4):761–773. doi:10.1016/j.immuni.2016.08.010
72. Kanneganti TD, Ozoren N, Body-Malapel M, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440(7081):233–236. doi:10.1038/nature04517
73. Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9(8):857–865. doi:10.1038/ni.1636
74. Dostert C, Guarda G, Romero JF, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One. 2009;4(8):e6510. doi:10.1371/journal.pone.0006510
75. Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9(8):847–856. doi:10.1038/ni.1631
76. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple cathepsins promote pro-IL-1beta synthesis and NLRP3-mediated IL-1beta activation. J Immunol. 2015;195(4):1685–1697. doi:10.4049/jimmunol.1500509
77. Cassel SL, Eisenbarth SC, Iyer SS, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105(26):9035–9040. doi:10.1073/pnas.0803933105
78. Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16(11):1323–1367. doi:10.1089/ars.2011.4123
79. Kelley N, Jeltema D, Duan Y, The HY. NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13). doi:10.3390/ijms20133328
80. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282(5):2871–2879. doi:10.1074/jbc.M608083200
81. Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187(2):613–617. doi:10.4049/jimmunol.1100613
82. Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi:10.1038/ni.1980
83. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–361. doi:10.1016/j.cell.2013.02.054
84. Allam R, Lawlor KE, Yu EC, et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014;15(9):982–990. doi:10.15252/embr.201438463
85. Ermler ME, Traylor Z, Patel K, et al. Rift valley fever virus infection induces activation of the NLRP3 inflammasome. Virology. 2014;449:174–180. doi:10.1016/j.virol.2013.11.015
86. Park S, Juliana C, Hong S, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol. 2013;191(8):4358–4366. doi:10.4049/jimmunol.1301170
87. Dubois H, Sorgeloos F, Sarvestani ST, et al. Nlrp3 inflammasome activation and gasdermin D-driven pyroptosis are immunopathogenic upon gastrointestinal norovirus infection. PLoS Pathog. 2019;15(4):e1007709. doi:10.1371/journal.ppat.1007709
88. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci U S A. 2013;110(44):17963–17968. doi:10.1073/pnas.1312571110
89. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. 2012;76(1):16–32. doi:10.1128/MMBR.05015-11
90. Clark IA, Vissel B. The meteorology of cytokine storms, and the clinical usefulness of this knowledge. Semin Immunopathol. 2017;39(5):505–516. doi:10.1007/s00281-017-0628-y
91. Bone RC, Sibbald WJ, Sprung CL. The ACCP-SCCM consensus conference on sepsis and organ failure. Chest. 1992;101(6):1481–1483. doi:10.1378/chest.101.6.1481
92. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39(5):529–539. doi:10.1007/s00281-017-0629-x
93. Guo XJ, Thomas PG. New fronts emerge in the influenza cytokine storm. Semin Immunopathol. 2017;39(5):541–550. doi:10.1007/s00281-017-0636-y
94. Basler CF. Molecular pathogenesis of viral hemorrhagic fever. Semin Immunopathol. 2017;39(5):551–561. doi:10.1007/s00281-017-0637-x
95. Martina BE, Koraka P, Osterhaus AD. Dengue virus pathogenesis: an integrated view. Clin Microbiol Rev. 2009;22(4):564–581. doi:10.1128/CMR.00035-09
96. Rajapakse S. Dengue shock. J Emerg Trauma Shock. 2011;4(1):120–127. doi:10.4103/0974-2700.76835
97. Puerta-Guardo H, Glasner DR, Harris E. Dengue virus NS1 disrupts the endothelial glycocalyx, leading to hyperpermeability. PLoS Pathog. 2016;12(7):e1005738. doi:10.1371/journal.ppat.1005738
98. Machhi J, Herskovitz J, Senan AM, et al. The natural history, pathobiology, and clinical manifestations of SARS-CoV-2 infections. J Neuroimmune Pharmacol. 2020;15(3):359–386.
99. Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1953–1966. doi:10.1056/NEJMoa030781
100. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in china: summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA. 2020;323(13):1239–1242. doi:10.1001/jama.2020.2648
101. Chi Y, Ge Y, Wu B, et al. Serum cytokine and chemokine profile in relation to the severity of coronavirus disease 2019 in China. J Infect Dis. 2020;222(5):746–754. doi:10.1093/infdis/jiaa363
102. Han Y, Zhang H, Mu S, et al. Lactate dehydrogenase, an independent risk factor of severe COVID-19 patients: a retrospective and observational study. Aging. 2020;12(12):11245–11258. doi:10.18632/aging.103372
103. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi:10.1016/S0140-6736(20)30183-5
104. Rodrigues TS, de Sa KSG, Ishimoto AY, et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. 2021;218(3). doi:10.1084/jem.20201707
105. Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019;10:50. doi:10.3389/fmicb.2019.00050
106. Nieto-Torres JL, Verdia-Baguena C, Jimenez-Guardeno JM, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–339. doi:10.1016/j.virol.2015.08.010
107. Kumar B, Asha K, Khanna M, Ronsard L, Meseko CA, Sanicas M. The emerging influenza virus threat: status and new prospects for its therapy and control. Arch Virol. 2018;163(4):831–844. doi:10.1007/s00705-018-3708-y
108. Owen DM, Gale M
109. Pang IK, Iwasaki A. Inflammasomes as mediators of immunity against influenza virus. Trends Immunol. 2011;32(1):34–41. doi:10.1016/j.it.2010.11.004
110. Ahsan NA, Sampey GC, Lepene B, et al. Presence of viral RNA and proteins in exosomes from cellular clones resistant to Rift Valley fever virus infection. Front Microbiol. 2016;7:139. doi:10.3389/fmicb.2016.00139
111. Mandal A, Singh A, Sahi PK, Rishi B. Bronchiolitis: comparative study between Respiratory Synctial Virus (RSV) and non RSV aetiology. J Clin Diagn Res. 2017;11(2):SL01–SL2.
112. Segovia J, Sabbah A, Mgbemena V, et al. TLR2/MyD88/NF-kappaB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS One. 2012;7(1):e29695. doi:10.1371/journal.pone.0029695
113. Sabbah A, Chang TH, Harnack R, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10(10):1073–1080. doi:10.1038/ni.1782
114. Wang B, Zhu J, Li D, et al. Newcastle disease virus infection induces activation of the NLRP3 inflammasome. Virology. 2016;496:90–96. doi:10.1016/j.virol.2016.05.023
115. Naim HY. Measles virus. Hum Vaccin Immunother. 2015;11(1):21–26. doi:10.4161/hv.34298
116. Devaux P, von Messling V, Songsungthong W, Springfeld C, Cattaneo R. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology. 2007;360(1):72–83. doi:10.1016/j.virol.2006.09.049
117. Komune N, Ichinohe T, Ito M, Yanagi Y. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1beta secretion. J Virol. 2011;85(24):13019–13026. doi:10.1128/JVI.05942-11
118. Russell CJ, Hurwitz JL. Sendai virus as a backbone for vaccines against RSV and other human paramyxoviruses. Expert Rev Vaccines. 2016;15(2):189–200. doi:10.1586/14760584.2016.1114418
119. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146(3):448–461. doi:10.1016/j.cell.2011.06.041
120. Ganar K, Das M, Sinha S, Kumar S. Newcastle disease virus: current status and our understanding. Virus Res. 2014;184:71–81. doi:10.1016/j.virusres.2014.02.016
121. Fanales-Belasio E, Raimondo M, Suligoi B, Butto S. HIV virology and pathogenetic mechanisms of infection: a brief overview. Ann Ist Super Sanita. 2010;46(1):5–14. doi:10.4415/ANN_10_01_02
122. Pontillo A, Silva LT, Oshiro TM, Finazzo C, Crovella S, Duarte AJ. HIV-1 induces NALP3-inflammasome expression and interleukin-1beta secretion in dendritic cells from healthy individuals but not from HIV-positive patients. AIDS. 2012;26(1):11–18. doi:10.1097/QAD.0b013e32834d697f
123. Musso D, Gubler DJ. Zika virus. Clin Microbiol Rev. 2016;29(3):487–524. doi:10.1128/CMR.00072-15
124. Pierson TC, Graham BS. Zika virus: immunity and vaccine development. Cell. 2016;167(3):625–631. doi:10.1016/j.cell.2016.09.020
125. Wang W, Li G, De W, et al. Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin-1beta secretion. Nat Commun. 2018;9(1):106. doi:10.1038/s41467-017-02645-3
126. Klema VJ, Padmanabhan R, Choi KH. Flaviviral replication complex: coordination between RNA synthesis and 5ʹ-RNA capping. Viruses. 2015;7(8):4640–4656. doi:10.3390/v7082837
127. Grant A, Ponia SS, Tripathi S, et al. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe. 2016;19(6):882–890. doi:10.1016/j.chom.2016.05.009
128. Faustin B, Lartigue L, Bruey JM, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25(5):713–724. doi:10.1016/j.molcel.2007.01.032
129. Wang H, Mao L, Meng G. The NLRP3 inflammasome activation in human or mouse cells, sensitivity causes puzzle. Protein Cell. 2013;4(8):565–568. doi:10.1007/s13238-013-3905-0
130. Screaton G, Mongkolsapaya J, Yacoub S, Roberts C. New insights into the immunopathology and control of dengue virus infection. Nat Rev Immunol. 2015;15(12):745–759. doi:10.1038/nri3916
131. Uno N, Ross TM. Dengue virus and the host innate immune response. Emerg Microbes Infect. 2018;7(1):167. doi:10.1038/s41426-018-0168-0
132. Hottz ED, Lopes JF, Freitas C, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. 2013;122(20):3405–3414. doi:10.1182/blood-2013-05-504449
133. Wu MF, Chen ST, Yang AH, et al. CLEC5A is critical for dengue virus-induced inflammasome activation in human macrophages. Blood. 2013;121(1):95–106. doi:10.1182/blood-2012-05-430090
134. Pan P, Zhang Q, Liu W, et al. Dengue virus M protein promotes NLRP3 inflammasome activation to induce vascular leakage in Mice. J Virol. 2019;93(21). doi:10.1128/JVI.00996-19
135. Shrivastava G, Visoso-Carvajal G, Garcia-Cordero J, et al. Dengue virus serotype 2 and its non-structural proteins 2A and 2B activate NLRP3 inflammasome. Front Immunol. 2020;11:352. doi:10.3389/fimmu.2020.00352
136. Morozov VA, Lagaye S. Hepatitis C virus: morphogenesis, infection and therapy. World J Hepatol. 2018;10(2):186–212. doi:10.4254/wjh.v10.i2.186
137. Negash AA, Ramos HJ, Crochet N, et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9(4):e1003330. doi:10.1371/journal.ppat.1003330
138. Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. 2006;1:23–61. doi:10.1146/annurev.pathol.1.110304.100230
139. Tang H, Grise H. Cellular and molecular biology of HCV infection and hepatitis. Clin Sci. 2009;117(2):49–65.
140. Chen W, Xu Y, Li H, et al. HCV genomic RNA activates the NLRP3 inflammasome in human myeloid cells. PLoS One. 2014;9(1):e84953. doi:10.1371/journal.pone.0084953
141. Carocci M, Bakkali-Kassimi L. The encephalomyocarditis virus. Virulence. 2012;3(4):351–367. doi:10.4161/viru.20573
142. Ito M, Yanagi Y, Ichinohe T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 2012;8(8):e1002857.
143. Rajan JV, Rodriguez D, Miao EA, Aderem A. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol. 2011;85(9):4167–4172. doi:10.1128/JVI.01687-10
144. de Jong AS, de Mattia F, Van Dommelen MM, et al. Functional analysis of picornavirus 2B proteins: effects on calcium homeostasis and intracellular protein trafficking. J Virol. 2008;82(7):3782–3790. doi:10.1128/JVI.02076-07
145. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
146. Bessaud M, Razafindratsimandresy R, Nougairede A, et al. Molecular comparison and evolutionary analyses of VP1 nucleotide sequences of new African human enterovirus 71 isolates reveal a wide genetic diversity. PLoS One. 2014;9(3):e90624. doi:10.1371/journal.pone.0090624
147. Wang H, Lei X, Xiao X, et al. Reciprocal regulation between enterovirus 71 and the NLRP3 inflammasome. Cell Rep. 2015;12(1):42–48. doi:10.1016/j.celrep.2015.05.047
148. Jacobs SE, Lamson DM, St George K, Walsh TJ. Human rhinoviruses. Clin Microbiol Rev. 2013;26(1):135–162.
149. Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am J Respir Cell Mol Biol. 2013;49(6):923–934. doi:10.1165/rcmb.2013-0032OC
150. Shrivastava S, Mukherjee A, Ray R, Ray RB. Hepatitis C virus induces interleukin-1beta (IL-1beta)/IL-18 in circulatory and resident liver macrophages. J Virol. 2013;87(22):12284–12290. doi:10.1128/JVI.01962-13
151. Carrillo C, Tulman ER, Delhon G, et al. Comparative genomics of foot-and-mouth disease virus. J Virol. 2005;79(10):6487–6504. doi:10.1128/JVI.79.10.6487-6504.2005
152. Zhi X, Zhang Y, Sun S, et al. NLRP3 inflammasome activation by foot-and-mouth disease virus infection mainly induced by viral RNA and non-structural protein 2B. RNA Biol. 2020;17(3):335–349.
153. Ao D, Guo HC, Sun SQ, et al. Viroporin activity of the foot-and-mouth disease virus non-structural 2B protein. PLoS One. 2015;10(5):e0125828. doi:10.1371/journal.pone.0125828
154. Chen I-Y, Ichinohe T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015;23(1):55–63. doi:10.1016/j.tim.2014.09.007
155. Stasakova J, Ferko B, Kittel C, et al. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1β and 18. J Gen Virol. 2005;86(1):185–195. doi:10.1099/vir.0.80422-0
156. Yoshizumi T, Ichinohe T, Sasaki O, et al. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat Commun. 2014;5:4713. doi:10.1038/ncomms5713
157. Cheung PH, Ye ZW, Lee TT, Chen H, Chan CP, Jin DY. PB1-F2 protein of highly pathogenic influenza A (H7N9) virus selectively suppresses RNA-induced NLRP3 inflammasome activation through inhibition of MAVS-NLRP3 interaction. J Leukoc Biol. 2020;108(5):1655–1663. doi:10.1002/JLB.4AB0420-694R
158. McAuley JL, Tate MD, MacKenzie-Kludas CJ, et al. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013;9(5):e1003392. doi:10.1371/journal.ppat.1003392
159. Dorfleutner A, Talbott SJ, Bryan NB, et al. A shope fibroma virus PYRIN-only protein modulates the host immune response. Virus Genes. 2007;35(3):685–694. doi:10.1007/s11262-007-0141-9
160. Johnston JB, Barrett JW, Nazarian SH, et al. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity. 2005;23(6):587–598. doi:10.1016/j.immuni.2005.10.003
161. Cheong WC, Kang HR, Yoon H, Kang SJ, Ting JP, Song MJ. Influenza A virus NS1 protein inhibits the NLRP3 inflammasome. PLoS One. 2015;10(5):e0126456. doi:10.1371/journal.pone.0126456
162. Gregory SM, Davis BK, West JA, et al. Discovery of a viral NLR homolog that inhibits the inflammasome. Science. 2011;331(6015):330–334. doi:10.1126/science.1199478
163. Gerlic M, Faustin B, Postigo A, et al. Vaccinia virus F1L protein promotes virulence by inhibiting inflammasome activation. Proc Natl Acad Sci U S A. 2013;110(19):7808–7813. doi:10.1073/pnas.1215995110
164. Komatsu T, Tanaka Y, Kitagawa Y, et al. Sendai virus V protein inhibits the secretion of interleukin-1beta by preventing NLRP3 inflammasome assembly. J Virol. 2018;92:19. doi:10.1128/JVI.00842-18
165. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354–357. doi:10.1038/nature16959
166. Shi H, Wang Y, Li X, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17(3):250–258. doi:10.1038/ni.3333
167. Wang W, Hu D, Wu C, et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020;16(3):e1008335. doi:10.1371/journal.ppat.1008335
168. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118(2):229–241. doi:10.1016/j.cell.2004.07.002
169. Graham DY. Helicobacter pylori update: gastric cancer, reliable therapy, and possible benefits. Gastroenterology. 2015;148(4):719–31 e3. doi:10.1053/j.gastro.2015.01.040
170. Snoussi K, Strosberg AD, Bouaouina N, Ben Ahmed S, Chouchane L. Genetic variation in pro-inflammatory cytokines (interleukin-1beta, interleukin-1alpha and interleukin-6) associated with the aggressive forms, survival, and relapse prediction of breast carcinoma. Eur Cytokine Netw. 2005;16(4):253–260.
171. Cao Z, Fang Y, Lu Y, et al. Exposure to nickel oxide nanoparticles induces pulmonary inflammation through NLRP3 inflammasome activation in rats. Int J Nanomedicine. 2016;11:3331–3346. doi:10.2147/IJN.S106912
172. Ahmad I, Muneer KM, Tamimi IA, Chang ME, Ata MO, Yusuf N. Thymoquinone suppresses metastasis of melanoma cells by inhibition of NLRP3 inflammasome. Toxicol Appl Pharmacol. 2013;270(1):70–76. doi:10.1016/j.taap.2013.03.027
173. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. 2016;166(2):288–298. doi:10.1016/j.cell.2016.05.051
174. Karin M, Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature. 2016;529(7586):307–315. doi:10.1038/nature17039
175. Dupaul-Chicoine J, Yeretssian G, Doiron K, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. 2010;32(3):367–378. doi:10.1016/j.immuni.2010.02.012
176. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32(3):379–391. doi:10.1016/j.immuni.2010.03.003
177. Harberts E, Gaspari AA. TLR signaling and DNA repair: are they associated? J Invest Dermatol. 2013;133(2):296–302. doi:10.1038/jid.2012.288
178. Pulskens WP, Butter LM, Teske GJ, et al. Nlrp3 prevents early renal interstitial edema and vascular permeability in unilateral ureteral obstruction. PLoS One. 2014;9(1):e85775. doi:10.1371/journal.pone.0085775
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.