")
Back to Journals » Infection and Drug Resistance » Volume 14
Acquisition of the mcr-1 Gene Lowers the Target Mutation to Impede the Evolution of a High-Level Colistin-Resistant Mutant in Escherichia coli
Authors Huang C, Shi Q, Zhang S, Wu H, Xiao Y
Received 14 June 2021
Accepted for publication 3 August 2021
Published 10 August 2021 Volume 2021:14 Pages 3041—3051
DOI https://doi.org/10.2147/IDR.S324303
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Chen Huang,1,2 Qingyi Shi,2 Shuntian Zhang,2 Hongcheng Wu,1 Yonghong Xiao2
1Department of Respiratory Medicine, Ningbo Medical Center Lihuili Hospital, Ningbo, People’s Republic of China; 2State Key Laboratory for Diagnosis and Treatment of Infectious Disease, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, People’s Republic of China
Correspondence: Hongcheng Wu
Department of Respiratory Medicine, Ningbo Medical Center Lihuili Hospital, Ningbo, 315000, People’s Republic of China
Email [email protected]
Yonghong Xiao
State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, 310003, People’s Republic of China
Tel +865 718 723 6421
Email [email protected]
Objective: The spread of the plasmid-mediated colistin resistance gene mcr-1 poses a significant public health threat. Little information is available on the development of high-level colistin-resistant mutants (HLCRMs) in MCR-1-producing Escherichia coli (MCRPEC). The present study was designed to evaluate the impact of chromosomal modifications in pmrAB, phoPQ, and mgrB combined with mcr-1 on colistin resistance in E. coli.
Methods: Five MCRPEC and three non-MCRPEC (E. coli ATCC25922 and two plasmid-curing) strains were used. The HLCRMs were selected through multi-stepwise colistin exposure. Moreover, two E. coli C600-pMCRs were constructed and used for selection of HLCRMs. Further analysis included mutation rates and DNA sequencing. Transcripts of pmrABC, phoP, mgrB, and mcr-1 were quantified by real-time quantitative PCR.
Results: All tested HLCRMs were successfully isolated from their parental strains. Non-MCRPEC strains had higher minimum inhibitory concentrations (MICs) and mutation rates than MCRPEC strains. Nineteen amino acid substitutions were identified: seven in PmrA, six in PmrB, one in PhoP, three in PhoQ, and two in MgrB. Most were detected in non-MCRPEC strains. Sorting Intolerant From Tolerant predicted that four substitutions, PmrA Gly15Arg, Gly53Arg, PmrB Pro94Gln, and PhoP Asp86Gly, affected protein function. Two HLCRM isolates did not show amino acid substitutions in contrast to their parental MCRPEC isolates. No further mutations were detected in the second- and third-step mutants. Further transcriptional analysis showed that the up-regulation of pmrCAB expression was greater in the mutant of E. coli C600 than in E. coli C600-pMCR.
Conclusion: Acquisition of the mcr-1 gene had a negative impact on the development of HLCRMs in E. coli, but was associated with low-level colistin resistance. Thus, colistin-based combination regimens may be effective against infections with MCR-1-producing isolates.
Keywords: mutation, mcr-1, chromosomal resistance mechanisms, high-level colistin resistance, Escherichia coli
Introduction
The overuse of antibiotics and the widespread development of antibiotic resistance genes have facilitated the evolution of multidrug-resistant (MDR) Gram-negative bacteria.1 Owing to its toxicity and narrow therapeutic window, colistin has been approved for treatment only for infections in certain patients, including those with cystic fibrosis.2,3 However, the increased incidence of infections with MDR pathogens has led to increased interest in the use of colistin as a last-resort option in a larger number of patients.
Colistin is a positively charged, polypeptide drug that exerts a strong bactericidal effect against a broad-spectrum of Gram-negative bacteria by integration into the negatively charged lipid A, thereby destabilizing the outer membrane lipopolysaccharide (LPS) and leading to cell death.3 However, exposure of Enterobacterales to colistin both in vivo and in vitro has been reported to induce the emergence of colistin resistance in these strains.4,5 The main mechanism of colistin resistance occurs via the addition of cationic groups (ie, phosphoethanolamine [PEtN] or 4-amino-4-deoxy-L-arabinose [L-Ara4N]) to the LPS on bacterial membranes, preventing the high-affinity binding of colistin to LPS.3 The two-component system (TCS) of pmrAB and phoPQ, and the regulator of TCS (ie, mgrB), are primarily responsible for the development of colistin resistance in Enterobacterales.3,6 Moreover, a recently identified plasmid carrying mcr-1 resulted in the addition of PEtN to lipid A.7 Studies have assessed the development of high-level colistin-resistant mutants (HLCRMs) in MCR-1-producing Escherichia coli (MCRPEC). It is not known whether the mcr-1 gene has effects similar to those of plasmid-mediated quinolone resistance genes, which promote the evolution of strains with higher quinolone resistance.8,9 The aim of this study was to determine the impact of chromosomal modifications in pmrAB, phoPQ, and mgrB, combined with mcr-1, on colistin resistance in E. coli.
Materials and Methods
Bacterial Isolates
Six E. coli isolates, five mcr-1-positive clinical strains of E. coli and E. coli ATCC25922, obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) were used in this study. Isolates were re-identified as E. coli by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS).10 The mcr-1 gene was amplified by PCR, and its DNA sequence was determined (Table S1). Multi-Locus Sequence Typing (MLST) was performed by comparing sequences of the seven housekeeping genes adk, fumC, gyrB, icd, mdh, purA and recA (https://enterobase.readthedocs.io/en/latest/mlst/mlst-legacy-info-ecoli.html) with the E. coli MLST database (https://enterobase.warwick.ac.uk/species/ecoli/allele_st_search) to determine the allelic types and STs of the tested isolates. None of the data in this study were linked to clinical information.
Plasmid Eradication Assay
Plasmid eradication for mcr-1-positive E. coli was performed as previously described.11 Briefly, 5 mL aliquots of Luria–Bertani (LB) medium were inoculated with 50 µL of a suspension of wild-type E. coli. To each suspension was added 7.5 µL, 15 µL, or 30 µL 10% SDS, and the cultures were incubated with shaking at 37°C for 12 h. Subsequently, 50 µL of these bacterial suspensions was inoculated into 5 mL fresh LB medium, and the cultures were incubated at 43°C for 8 h. Both steps were repeated, and the incubation at 37°C was performed a third time. These plasmid-cured derivative strains were plated onto Mueller–Hinton agar (MHA) plates with and without 4 mg/L colistin. The elimination of the mcr-1-bearing plasmid was confirmed by pulsed-field gel electrophoresis (PFGE), S1-nuclease PFGE (S1-PFGE), and Southern blotting, as described.12,13
Antimicrobial Susceptibility Testing
Antibiotic susceptibility, except for colistin, was evaluated by Vitek2 (bioMérieux, Marcy-l’Etoile, France). The results were in accordance with Clinical and Laboratory Standards Institute (CLSI) guidelines.14 The minimum inhibitory concentrations (MICs) of colistin against the tested strains were determined using the broth microdilution according to CLSI. In addition, the MICs of colistin against the multi-stepwise solutions were determined using the agar dilution method.
In vitro Multi-Step Selection of Colistin-Resistant Mutants
The parental and plasmid-curing strains were grown in antibiotic-free Mueller–Hinton broth at 37°C for 6–8 h, and ~1010 CFU/mL of each strain was spread onto MHA in the presence or absence of colistin. The colistin concentrations used for mutant induction ranged from 1×MIC to the concentration at which growth of the parental strain or a sub-parental mutant strain isolated from the prior induction step was fully inhibited. After 48–72 h incubation at 37°C, colonies growing on the plates were randomly selected, and their MICs of colistin were determined using both the broth microdilution and agar dilution methods. Isolates with the highest MIC were subjected to next-step induction. These induction/selection cycles were terminated when mutants with significantly high MIC were selected, or when their growth on plates with 1×MIC colistin concentration was completely inhibited.
Screening for Mutations in the pmrAB, phoPQ, mgrB, and mcr-1 Genes
The TCS of pmrAB and phoPQ, the negative regulator of the phoPQ system (mgrB) and mcr-1 in parental strains, and their respective mutants, were PCR amplified using primers (listed in Table S1) and 2X A9 LongHiFi PCR MasterMix (Aidlab Biotechnologies Co., Ltd.). Following DNA sequencing, the presumed amino acid sequences of the mutants were compared with those of parental strains using the web platforms of the NCBI (National Center for Biotechnological Information) and ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/). Sorting Intolerant From Tolerant (SIFT) scores were calculated (http://sift.jcvi.org) to evaluate whether amino acid alterations in PmrAB and PhoPQ affected protein function. Moreover, the TCS domains of PmrA/PmrB and PhoP/PhoQ were subjected to SMART analysis (http://smart.embl.de/).
Complementation Experiments and Transcriptional Analysis
To determine the effect of mcr-1-bearing plasmids on the evolution of HLCRMs, conjugation experiments were performed as previously described.15 Briefly, a culture of mcr-1-producing isolates was mixed 1:9 with a culture of the recipient strain E. coli C600 in LB broth, followed by overnight incubation on LB agar plates. The resulting transconjugants were selected on MHA plates containing 150 µg/mL sodium azide and 2 µg/mL colistin. The colonies were identified as E. coli via MALDI-TOF MS, and the DNA of these colonies were sequenced to determine the presence of the mcr-1 gene. Plasmid sizes and numbers were determined using S1-nuclease PFGE. The colonies containing only mcr-1-bearing plasmids (E63-C600 and E66-C600) and E. coli C600 were used to select for colistin-resistant mutants (MuC600, MuE63-C600, and MuE66-C600). Total RNA was extracted from cells grown to mid-log phase in drug-free MHB using the TaKaRa RNAiso Plus (TaKaRa, Japan), according to the manufacturer’s instructions. The RNA was reverse transcribed to cDNA using PrimeScriptTM RT Reagent kits (TaKaRa). Transcripts of the pmrABC, phoP, mgrB, and mcr-1 genes were quantified by RT-PCR using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) on an ABI7300 Sequence Detection System (Applied Biosystems), using the primers listed in Table S1. Transcription abundance was calculated by the 2−ΔΔCT method16 using gapA as the internal control, and the respective wild-type pmrABC, phoP, mgrB, and mcr-1 genes as references.
Results
Isolate Characterization
Four MCRPECs were subjected to the plasmid eradication test, which successfully eliminated the mcr-1 gene from strains EC18398 and EC26207 (Figure S1). The MICs and STs of the parental and plasmid-cured strains are shown in Table 1. Loss of the mcr-1 gene had little effect on the susceptibility of plasmid-curing strains to other antimicrobial agents, but reduced colistin MIC 4–16 fold, resulting in MICs of 0.5 and 2 mg/L for strains EC18398E and EC26207E, respectively. In addition, the strains EC1002 and EC2474, co-harboring the mcr-1, blaNDM-1, and blaCTX-M genes, were resistant to colistin, carbapenems, and cephalosporins.
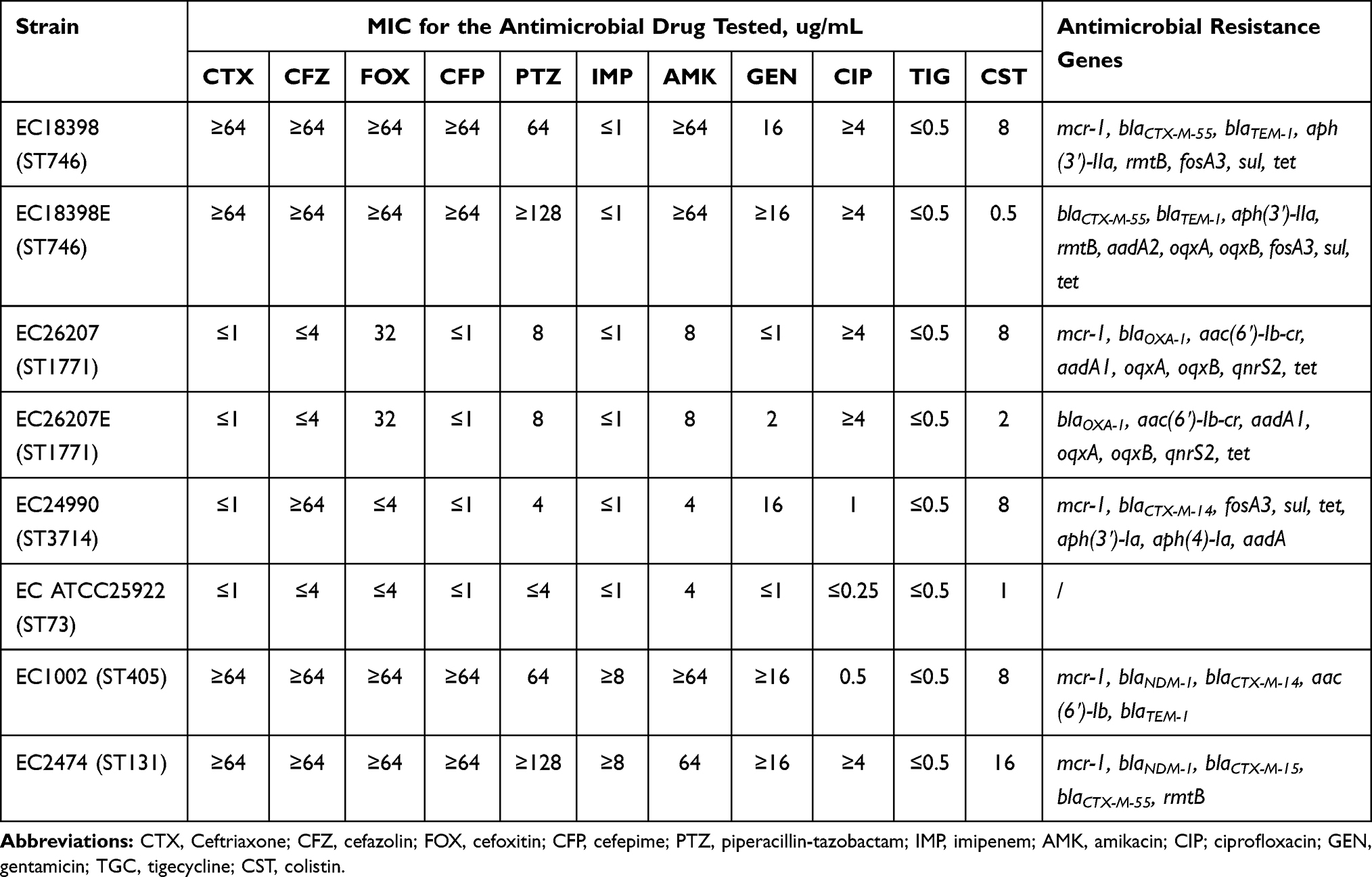 |
Table 1 Antimicrobial Susceptibility of the E. coli Strains Used in This Study |
In vitro Selection of Resistant Mutants
Following a series of in vitro colistin selection steps, all the tested strains, including MCRPEC and non-MCRPEC strains, successfully evolved to HLCRMs, with MICs of 32 and 64 mg/L, respectively, as determined by the broth microdilution method, and 64 and 64 mg/L, respectively, as determined by the agar dilution method (Table 2 and Figure 1). Colistin inhibited first-step mutants at concentrations of 8 to 32 mg/L, resulting in a 16- to 32-fold increase in susceptibility for these non-MCRPEC mutants compared with their parental strains. By contrast, the in vitro first-step induction had little effect on the MCRPECs, which had MICs equal to or 2-fold higher than their parental strains. For the second cycle, the colistin MIC of all mutants was 32 mg/L, as determined by the broth microdilution method. Second-step mutants were subjected to further repeated inductions, while Mu2EC26207, Mu2EC24990, and Mu3EC18398 failed to grow on the plates containing 1×MIC (64 mg/L). Interestingly, all three non-MCRPECs successfully grew on plates containing 1×MIC (64 mg/L) after in vitro multi-stepwise induction and selection. The MICs of these non-MCRPEC mutants were 64- to 128-fold higher than those of their parental strains (Figure 2 and Table 2). In addition to determining MIC for colistin, the MICs of various antibiotics with diverse modes of action were also evaluated. Compared with their parental strains, the mutants had equivalent MICs for carbapenems, cephalosporins, levofloxacin, and tigecycline.
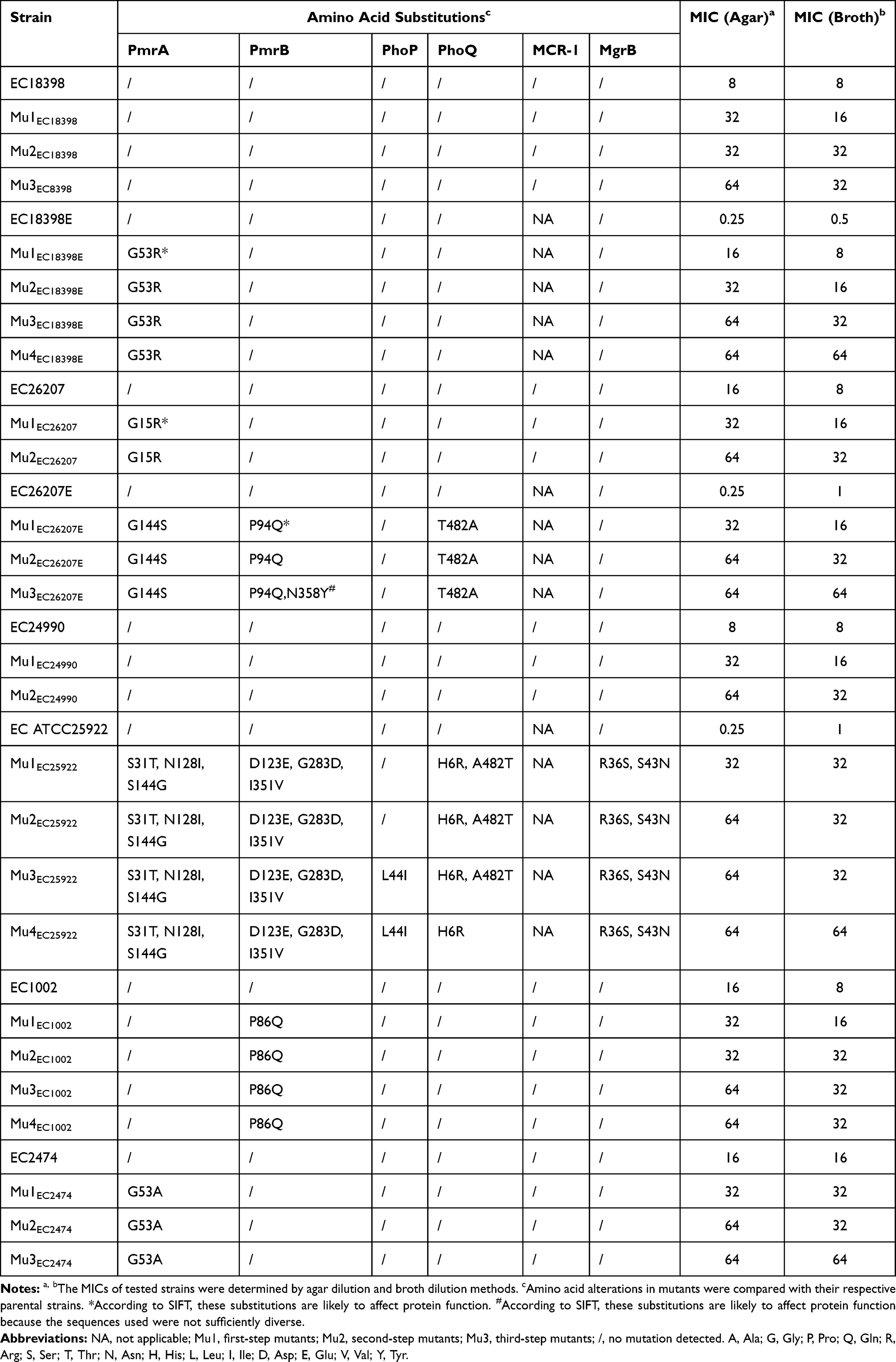 |
Table 2 Phenotypic and Genotypic Profiles of the in vitro Selected Mutants of mcr-1-Positive and mcr-1-Negative E. coli |
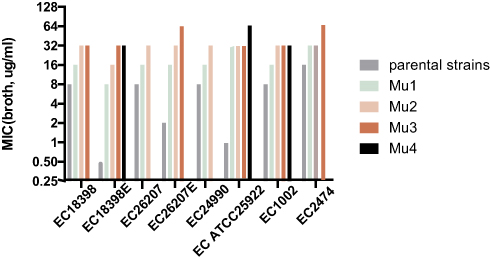 |
Figure 1 Changes in the colistin susceptibility of selected mutants. Five mcr-1-positive and three mcr-1-negative E. coli strains were exposed to colistin in a multi-stepwise manner. MIC was measured by the broth dilution method. Mutants with the highest MIC were used for next-step induction and selection processes. Mu1, Mu2, Mu3, and Mu4 indicate the first, second, third, and fourth cycles of induction, respectively. |
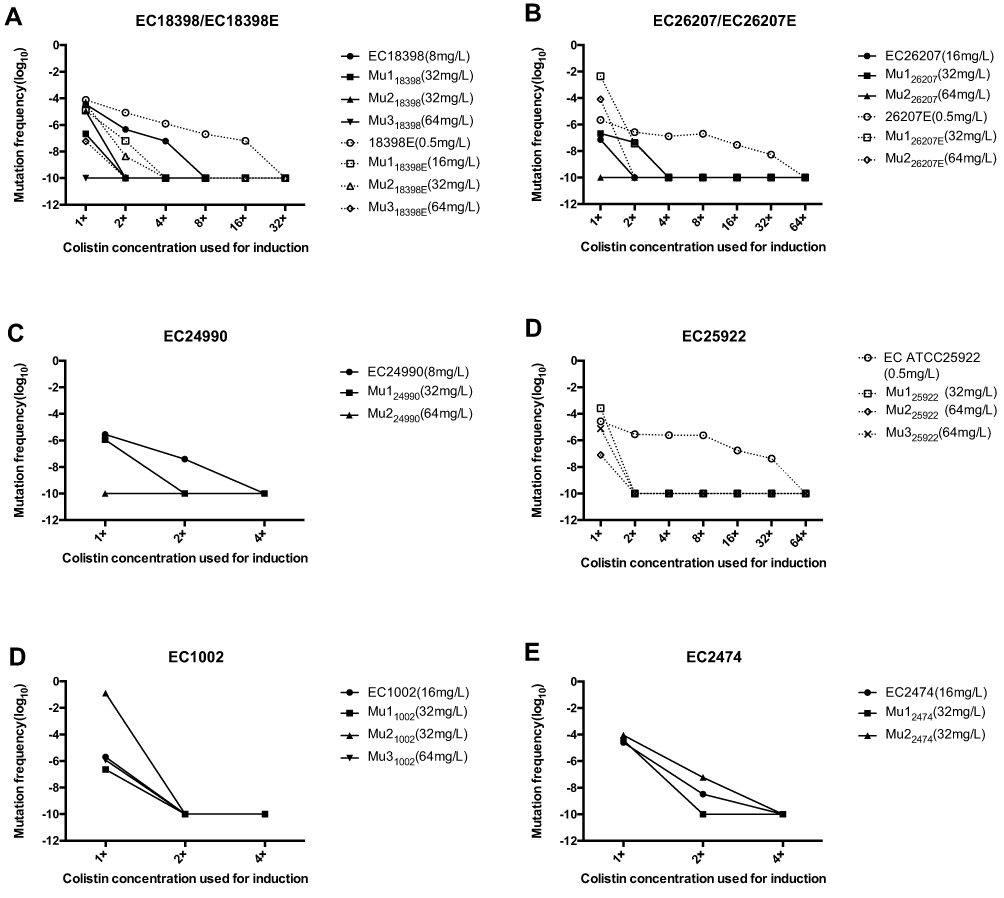 |
Figure 2 Mutation frequencies of mcr-1-positive and negative strains when cultured with colistin at its MICs for the parent strains and sub-parental mutants. The colistin MICs of the tested strains were determined by the agar dilution method. Solid line, mcr-1-positive strains; dotted line, mcr-1-negative strains. |
In the tested E. coli strains, the mutation rates decreased significantly with increasing colistin concentrations on the selection plates (Figure 2). These results revealed that the frequency of mutation of non-MCRPEC strains to colistin resistance ranged from 10−6 to 10−2, whereas the frequency of mutation of MCRPEC strains to colistin resistance was 10–8 to 10.−4 The non-MCRPEC strains could grow on plates containing colistin concentrations of 16 or 32 mg/L, and showed higher mutation rates than their parental strains. For example, EC26207E had a mutation rate of 10−2 to 10−6 at 1×MIC, which was much higher than that of EC26207 (10–8 to 10−6). Additionally, the frequency of non-MCRPEC mutants on plates containing 32 or 64 mg/L was higher than that of MCRPEC mutants.
Amino Acid Substitutions in Colistin Resistance Genes
Comparative genomic analysis of parental and mutants strains showed that non-synonymous mutations in the major TCS associated with colistin resistance were more frequent in non-MCRPEC than in MCRPEC strains. None of the TCS mutations were found in any mutants of EC18398 and EC24990, with only single amino acid changes found in PmrA at position 15 (Gly15Arg) in EC26207, and in PmrB at position 86 (Pro86Gln) in EC1002. By contrast, the mutants of non-MCRPEC strains acquired more non-synonymous mutations in the target regions, including in PmrAB, PhoPQ, and MgrB in EC25922; PmrA in EC18398E; and PmrAB and PhoQ in EC26207E (Table 2). No amino acid substitutions were observed in MCR-1, and neither frameshift mutations nor deletions were identified in any of these strains. The alterations in the TCS regions of EC25922 were predicted to have little impact on protein function, as determined by SIFT score. Interestingly, the amino acid substitution in the mutant of EC25922 was also detected in other tested parental strains, including both MCRPEC and non-MCRPEC strains, suggesting that non-synonymous mutations may occur frequently in PmrA at positions 31, 128, and 144; in PmrB at positions 123 and 351; in PhoQ at positions 6 and 482; and in MgrB at position 36. Interestingly, PmrA at position 144 (Ser144Gly) and PhoQ at position 482 (Ala482Thr) could convert to each other when exposed to colistin plates (Table 2 and S2). The non-synonymous mutations in the plasmid-curing isolates were easily detected when compared with their parental strains. Amino acid alterations were observed in PmrA Gly144Ser, PmrB Pro94Gln, Asn358Tyr, and PhoQ Thr482Ala in the EC26207E mutant, and in PmrA Gly53Arg in the EC18398E mutant. The EC26207 and 18398 mutants had 1 or 0 amino acid variations, respectively. However, the second- and third-step mutants showed no further mutational changes in PmrAB, PhoPQ, and MgrB, except for those in EC26207E and EC25922.
SMART analysis revealed the major domains of the PmrA/PmrB and PhoP/PhoQ TCS, and the positions of all the mutations in colistin-resistant mutants (Figure 3). Our results showed that non-synonymous mutations were mainly found in the HAMP and ATPase domains of PmrB and PhoQ, and in the receiver domain of PmrA and PhoQ. SIFT analysis predicted that the PmrA Gly15Arg, Gly53Arg, PmrB Pro94Gln, and PhoP Asp86Gly mutations would affect protein function.
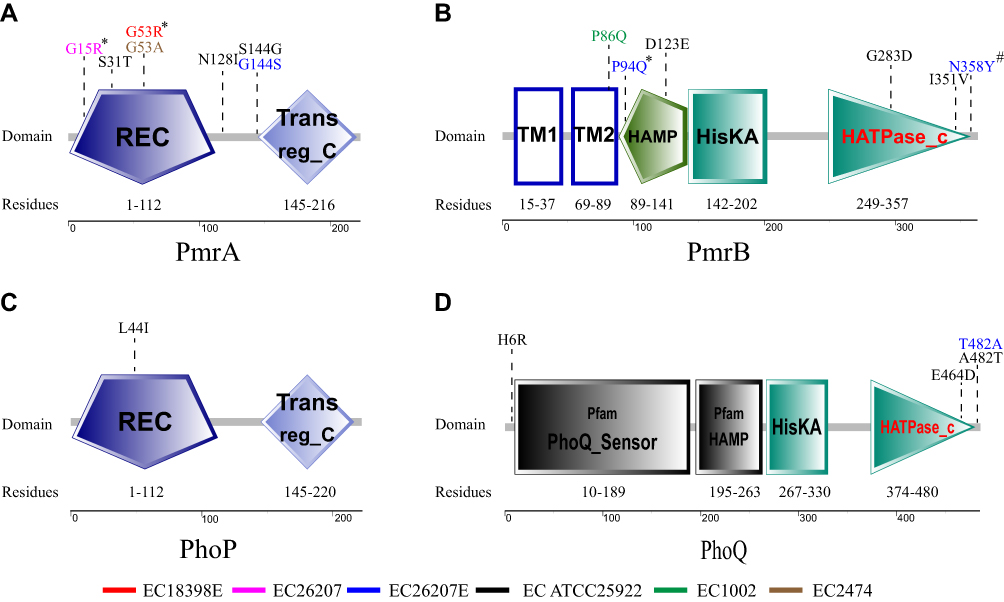 |
Figure 3 Domains of the PmrA/PmrB and PhoP/PhoQ two-component system and the positions of all mutations in colistin-resistant mutants. *These substitutions are predicted to affect protein function by SIFT. #These substitutions are predicted to affect protein function by SIFT because the sequences used were not sufficiently diverse. Red, EC18398E; Fuchsin, EC26207; Blue, EC26207E; Black, EC25922; Green, EC1002; Brown, EC2474. Domains of PmrA/PmrB and PhoP/PhoQ are indicated as REC, CheY-homologous receiver domain; Trans_reg_c, transcriptional regulatory C-terminal domain; TM1, first transmembrane domain; TM2, second transmembrane domain; HAMP, histidine kinases, adenylyl cyclases, methyl-binding proteins, and phosphatases domain; HisKA, histidine kinase domain; HATPase_c, histidine kinase-like ATPase C-terminal domain. |
pmrABC, phoP, mgrB, and mcr-1 Expression
To better understand the impact of mcr-1-bearing plasmids on the evolution of HLCRMs, E63-C600, E66-C600, and E. coli C600 were used for the selection of HLCRMs. Transcription of the pmrCAB, phoP, mgrB, and mcr-1 genes in HLCRMs was evaluated by qRT–PCR. The levels of expression of pmrCAB and phoP were higher in MuC600 than in E. coli C600, with the level of expression of pmrA being 200-fold higher in MuC600 than in E. coli C600 (Figure 4). Moreover, the magnitude of pmrCAB up-regulation was higher than that of phoP and mgrB, indicating that pmrCAB may play more important roles in the evolution of HLCRMs than phoPQ and mgrB. However, the levels of expression of the pmrCAB, phoP, mgrB, and mcr-1 genes in MuE63-C600 and MuE66-C600 were not significantly higher than those in their parental strains.
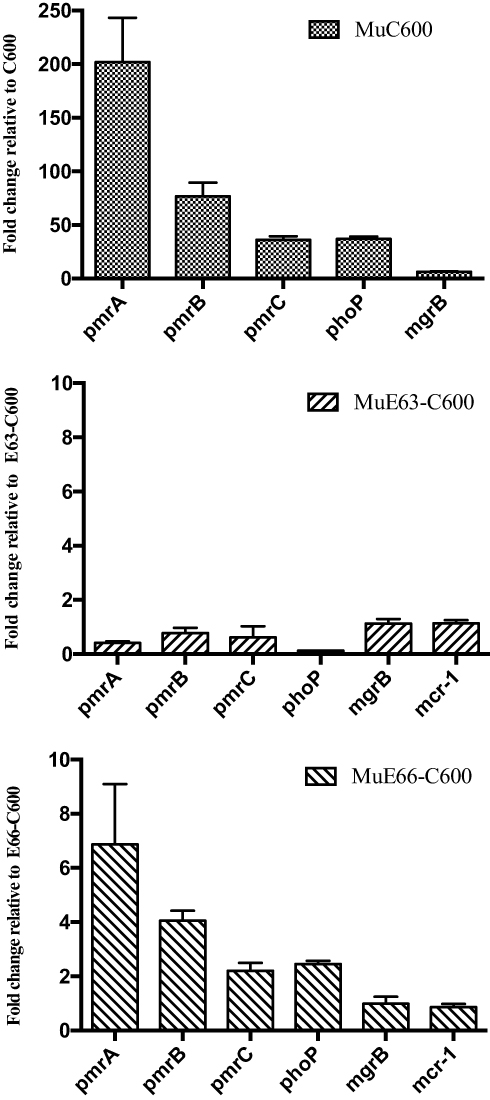 |
Figure 4 Transcriptional activities of pmrABC, phoP, mgrB, and mcr-1 in wild-type isolates and their derivative colistin-resistant mutants (MuC600, MuE63-C600, and MuE66-C600) grown in drug-free MHB. The fold change in transcription was calculated as 2−ΔΔCT. Means and standard deviations were determined for three independent replicates. |
Discussion
The clinical use of colistin is being re-evaluated because of the increasing prevalence of infections caused by MDR organisms.17 Plasmid-mediated colistin resistance via the mcr-1 gene was found to provide a horizontal transfer mechanism for rapid dissemination.7 The prevalence of colistin resistance has become of great concern because of the location of the mcr-1 gene on highly mobile genetic elements and its coexistence with other resistance determinants. However, the phenotype of HLCRMs in mcr-1-harboring E. coli is not fully understood. Moreover, the impact of chromosomal modifications in TCS combined with mcr-1 on colistin resistance has not been determined.18–20
The present study found that HLCRMs could be successfully isolated from MCRPEC and non-MCRPEC strains by multi-stepwise induction under conditions of colistin exposure. Unexpectedly, the absence of the mcr-1 gene from E. coli resulted in higher mutation rates and facilitated the selection of HLCRMs, in contrast to the role of plasmid-mediated quinolone resistance genes in Enterobacteriae. Quinolone resistance may be due to the presence of a plasmid-carried quinolone resistance determinant Qnr, which has been shown to bind to and protect both DNA gyrase and topoisomerase IV from inhibition by ciprofloxacin. In addition, because of their additive nature, the concentration required for mutant prevention is increased.8,21,22 Conversely, mcr-1, which encodes a pEtN transferase, confers colistin resistance via the addition of pEtN to LPS, similar to the chromosomal colistin resistance mechanism that constitutively activates PhoPQ and PmrAB.19,23 Thus MCR-1-associated LPS modifications may impair the role of TCS in the evolution of HLCRMs. These findings demonstrated that non-synonymous mutations by TCS were more easily observed in non-MCRPECs than in MCRPECs. Furthermore, pmrABC and phoP expression levels were higher in non-MCRPECs. Taken together, these findings indicated that the presence of mcr-1 limited the up-regulation of TCS genes related to colistin resistance. Usually, the MIC of colistin against MCRPECs is 2 to 8 mg/L, whereas the MIC of colistin mediated by chromosomal resistance mechanisms, such as mutations in pmrAB or phoPQ, is 16 to 256 mg/L.3,23 Because chromosomal resistance mechanisms, rather than mcr-1, may have an important impact on the evolution of HLCRMs, HLCRMs in the present study were more easily generated by non-MCRPECs. The presence of the mcr-1 gene may, however, facilitate the selection of HLCRMs. These findings suggest that the dilution of overnight cultures was too low (105 CFU/mL) to prevent E. coli TOP10 from generating HLCRMs.24
Mutations related to colistin resistance in PmrAB and PhoPQ TCS play crucial roles in the development of MCRPEC and non-MCRPEC into HLCRMs, as mutations in these systems can cause their constitutive overexpression, resulting in the activation of arnBCADTEF and pmrCAB and the modification of lipid A.23 Various genetic alterations have been associated with an increased MIC of colistin, including Ser39Ile and Arg81Ser in PmrA; Glu375lys in PhoQ; several mutations in PmrB, including Leu10Gly, Glu, 41::Tn5 (insertion of Tn5 at nucleotide 41), Cys84Tyr, a 12 bp deletion from nucleotide 258 to nucleotide 269 (GlnAlaValArgArg), Ile91Thr92 ins Ile (an insertion of isoleucine at position 92), Asp149Tyr, Thr156Lys, Ala159Val, and Val161Gly.3,4,25 Although none of these non-synonymous mutations were detected in the present study, SIFT determined that the Gly15Arg and Gly53Arg mutations in PmrA, the Pro94Gln mutation in PmrB, and the Asp86Gly mutation in PhoP affect protein function. Except for PmrA Gly15Arg, which was found in MCRPEC strains, these mutations were found in non-MCRPEC strains. The Gly15Arg and Gly53Arg mutations in PmrA, and the Pro94Gln mutation in PmrB, were found to be involved in colistin resistance in Salmonella enterica.6 Position 53 in the PmrA has also been described as being responsible for acquired colistin resistance in Klebsiella pneumoniae and Enterobacter aerogenes.3 Gly53 of PmrA is located in its phosphate receiver domain, close to the active site at Asp51.26 An amino acid substitution at Gly53, whether to Arg or Ala, prevented the Asp active site from being dephosphorylated by the phosphatase activity of PmrB. Pro94 of PmrB is located in its HAMP domain, which is crucial for signal transduction from the periplasmic input to the kinase domain.27 A mutation in the HAMP domain might therefore lead to constitutive activation of PmrA. In addition, several non-synonymous mutations were identified in PmrAB and PhoPQ, especially in non-MCRPEC strains, but SIFT showed that these mutations had little impact on protein function.
These results are in agreement with studies showing that not all the mutations in pmrAB and phoPQ result in colistin resistance.4,28 The MICs of mutants were progressively elevated by in vitro multi-stepwise induction and selection, whereas the second- and third-steps did not yield further mutations in pmrAB, phoPQ, and mgrB. This analysis may have been unable to identify mutations in other regulatory pathways that led to colistin resistance.
Previous genetic analysis revealed that Etk, a tyrosine-kinase, can phosphorylate Ugd, the starting material for L-Ara4N synthesis and can activate the PmrAB system, resulting in colistin resistance and the deletion of mgrR (influenced by the PhoPQ system).29–31 Therefore, different mechanisms mediating or contributing to colistin resistance may be responsible for the development of greater resistance to colistin, especially for MCRPEC strains, inasmuch as non-synonymous mutations in pmrAB, phoPQ, and mgrB were not detected in EC24990 or EC18398. Thus, increasing the clinical use of colistin may result in the spread of colistin-resistant organisms. The present findings suggested that acquisition of the mcr-1 gene partly lowered the target mutation to impede the evolution of HLCRMs. The difficulty of a chromosomal mutation related to further colistin resistance in MCRPEC strains may provide further support for the use of colistin-based combination strategies to treat infections caused by MCR-1-producing isolates. Exposure of MCR-1- and NDM-5-producing E. coli to polymyxin B monotherapy did not result in the acquisition of a chromosomal polymyxin resistance mutation, with polymyxin B MIC remaining stable at 4 mg/L in the hollow-fiber infection model.18 The triple combination of polymyxin B, aztreonam, and amikacin resulted in undetectable bacterial counts and suppression of colistin resistance.
The present study had several limitations. First, the number of tested strains in this study was limited. Moreover, our findings showed that the presence of the mcr-1 gene may limit the evolution of MCRPEC strains into HLCRMs. Further investigations are required to determine the effects on colistin resistance of a combination of chromosomal modifications in TCS and the mcr-1 gene. Additionally, the colistin MICs of mutants in this study were further improved by in vitro multi-stepwise induction and selection, with non-synonymous mutations and other resistance mechanisms not detected. Further research is required to determine the internal molecular mechanisms of colistin resistance.
Conclusion
The acquisition by E. coli of the mcr-1 gene usually results in a low-level colistin resistance (2–8 mg/L), while having a negative impact on the development of HLCRMs. This may support the use of colistin-based combination regimens to combat infections with MCR-1-producing isolates.
Data Sharing Statement
The datasets used and analyzed during the current study are available from the corresponding author, Yonghong Xiao, upon reasonable request.
Acknowledgments
All named authors meet the criteria of the International Committee of Medical Journal Editors (ICMJE) for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
All authors contributed to the data analysis, and the drafting and revising of the article; agreed on the journal to which the article will be submitted; gave final approval for the version to be published; and agreed to be accountable for all aspects of this work.
Funding
This work was supported by the Key research and development program of Zhejiang province (no. 2021C03068) and the Natural Science Foundation of Ningbo (no. 2019A610232).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Baker S, Duy PT, Nga TVT, et al. Fitness benefits in fluoroquinolone-resistant Salmonella Typhi in the absence of antimicrobial pressure. Elife. 2013;2. doi:10.7554/eLife.01229
2. Tangden T, Giske CG. Global dissemination of extensively drug-resistant carbapenemase-producing Enterobacteriaceae: clinical perspectives on detection, treatment and infection control. J Intern Med. 2015;277(5):501–512. doi:10.1111/joim.12342
3. Poirel L, Jayol A, Polymyxins: NP, Activity A, Testing S. Resistance mechanisms encoded by plasmids or chromosomes. Clin Microbiol Rev. 2017;30:557–596. doi:10.1128/CMR.00064-16
4. Phan MD, Nhu NTK, Achard MES, et al. Modifications in the pmrB gene are the primary mechanism for the development of chromosomally encoded resistance to polymyxins in uropathogenic Escherichia coli. J Antimicrobial Chemother. 2017;72:2729–2736. doi:10.1093/jac/dkx204
5. Cannatelli A, Di Pilato V, Giani T, et al. In vivo evolution to colistin resistance by PmrB sensor kinase mutation in KPC-producing Klebsiella pneumoniae is associated with low-dosage colistin treatment. Antimicrob Agents Chemother. 2014;58(8):4399–4403. doi:10.1128/AAC.02555-14
6. Olaitan AO, Morand S, Rolain J-M. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front Microbiol. 2014;5:643. doi:10.3389/fmicb.2014.00643
7. Liu YY, Wang Y, Walsh TR, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16(2):161–168. doi:10.1016/S1473-3099(15)00424-7
8. Briales A, Rodríguez-Martínez JM, Velasco C, et al. In vitro effect of qnrA1, qnrB1, and qnrS1 genes on fluoroquinolone activity against isogenic Escherichia coli isolates with mutations in gyrA and parC. Antimicrob Agents Chemother. 2011;55(3):1266–1269. doi:10.1128/AAC.00927-10
9. Robicsek A, Strahilevitz J, Jacoby GA, et al. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med. 2006;12(1):83–88. doi:10.1038/nm1347
10. Wattal C, Oberoi JK, Goel N, Raveendran R, Khanna S. Matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) for rapid identification of micro-organisms in the routine clinical microbiology laboratory. Eur J Clin Microbiol. 2017;36(5):807–812. doi:10.1007/s10096-016-2864-9
11. Sun Y, Liu Q, Chen S, et al. Characterization and plasmid elimination of NDM-1-producing Acinetobacter calcoaceticus from China. PLoS One. 2014;9(9):e106555. doi:10.1371/journal.pone.0106555
12. Shen C, Feng S, Chen H, et al. Transmission of mcr-1-producing multidrug-resistant enterobacteriaceae in public transportation in Guangzhou, China. Clin Infect Dis. 2018;67(suppl_2):S217–S224. doi:10.1093/cid/ciy661
13. Shen P, Wei Z, Jiang Y, et al. Novel genetic environment of the carbapenem-hydrolyzing β-Lactamase KPC-2 among enterobacteriaceae in China. Antimicrob Agents Chemother. 2009;53:4333–4338. doi:10.1128/AAC.00260-09
14. Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing.
15. Zheng B, Huang C, Xu H, et al. Occurrence and genomic characterization of ESBL-producing, MCR-1-harboring Escherichia coli in farming soil. Front Microbiol. 2017;8:2510. doi:10.3389/fmicb.2017.02510
16. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
17. Falagas ME, Rafailidis PI, Matthaiou DK. Resistance to polymyxins: mechanisms, frequency and treatment options. Drug Resist Updat. 2010;13(4–5):132–138. doi:10.1016/j.drup.2010.05.002
18. Bulman ZP, Chen L, Walsh TJ, et al. Polymyxin combinations combat Escherichia coli harboring mcr-1 and blaNDM-5: preparation for a postantibiotic era. MBio. 2017;8. doi:10.1128/mBio.00540-17
19. Smith NM, Bulman ZP, Sieron AO, et al. Pharmacodynamics of dose-escalated ‘front-loading’ polymyxin B regimens against polymyxin-resistant mcr-1-harbouring Escherichia coli. J Antimicrob Chemother. 2017;72(8):2297–2303. doi:10.1093/jac/dkx121
20. Zhou Y-F, Tao M-T, Feng Y, et al. Increased activity of colistin in combination with amikacin against Escherichia coli co-producing NDM-5 and MCR-1. J Antimicrob Chemother. 2017;72(6):1723–1730. doi:10.1093/jac/dkx038
21. Rodriguez-Martinez JM, Velasco C, García I, et al. Mutant prevention concentrations of fluoroquinolones for Enterobacteriaceae expressing the plasmid-carried quinolone resistance determinant qnrA1. Antimicrob Agents Chemother. 2007;51(6):2236–2239. doi:10.1128/AAC.01444-06
22. Jacoby GA. Mechanisms of resistance to quinolones. Clin Infect Dis. 2005;41 Suppl 2:S120–126. doi:10.1086/428052
23. Jeannot K, Bolard A, Plesiat P. Resistance to polymyxins in gram-negative organisms. Int J Antimicrob Agents. 2017;49(5):526–535. doi:10.1016/j.ijantimicag.2016.11.029
24. Yang Q, Li M, Spiller OB, et al. Balancing mcr-1 expression and bacterial survival is a delicate equilibrium between essential cellular defence mechanisms. Nat Commun. 2017;8(1):2054. doi:10.1038/s41467-017-02149-0
25. Cannatelli A, Giani T, Aiezza N, et al. An allelic variant of the PmrB sensor kinase responsible for colistin resistance in an Escherichia coli strain of clinical origin. Sci Rep. 2017;7(1):5071. doi:10.1038/s41598-017-05167-6
26. Sun S, Negrea A, Rhen M, Andersson DI. Genetic analysis of colistin resistance in Salmonella enterica Serovar Typhimurium. Antimicrob Agents Chemother. 2009;53(6):2298–2305. doi:10.1128/AAC.01016-08
27. Aravind L, Ponting CP. The cytoplasmic helical linker domain of receptor histidine kinase and methyl-accepting proteins is common to many prokaryotic signalling proteins. FEMS Microbiol Lett. 1999;176(1):111–116. doi:10.1111/j.1574-6968.1999.tb13650.x
28. Agerso Y, Torpdahl M, Zachariasen C, et al. Tentative colistin epidemiological cut-off value for Salmonella spp. Foodborne Pathog Dis. 2012;9(4):367–369. doi:10.1089/fpd.2011.1015
29. Lacour S, Doublet P, Obadia B, Cozzone AJ, Grangeasse C. A novel role for protein-tyrosine kinase Etk from Escherichia coli K-12 related to polymyxin resistance. Res Microbiol. 2006;157(7):637–641. doi:10.1016/j.resmic.2006.01.003
30. Lacour S, Bechet E, Cozzone AJ, Mijakovic I, Grangeasse C. Tyrosine phosphorylation of the UDP-glucose dehydrogenase of Escherichia coli is at the crossroads of colanic acid synthesis and polymyxin resistance. PLoS One. 2008;3:e3053. doi:10.1371/journal.pone.0003053
31. Moon K, Gottesman S. A PhoQ/P-regulated small RNA regulates sensitivity of Escherichia coli to antimicrobial peptides. Mol Microbiol. 2009;74(6):1314–1330. doi:10.1111/j.1365-2958.2009.06944.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.