")
Back to Journals » International Journal of Women's Health » Volume 15
About a Rare Association Between Vulvar Dowling Degos Disease and HS
Authors Dupont M, Parent M, Vanhooteghem O
Received 22 November 2022
Accepted for publication 20 February 2023
Published 9 March 2023 Volume 2023:15 Pages 355—359
DOI https://doi.org/10.2147/IJWH.S398604
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Elie Al-Chaer
Manon Dupont,1 Muriel Parent,2 Olivier Vanhooteghem1
1Dermatology Department, CHU UCL Namur, Site Sainte Elisabeth, Namur, 5000, Belgium; 2Department of Pathology, Institute of Pathology and Genetics (IPG), Gosselies, 6041, Belgium
Correspondence: Olivier Vanhooteghem, Email [email protected]
Abstract: Dowling Degos disease (DDD) is a rare genodermatosis that manifests itself as acquired, reticulated hyperpigmentation of the folds. We report the case of a 45-year-old woman who presented since the age of 30 with hyperpigmented macules of reticulated appearance of vulvar, perianal and bilateral axillary location associated with hidradenitis suppurativa (HS) at Hurley stage 2 of later onset. DDD is classically described in the flexural folds and, to our knowledge; less than a dozen cases of vulvar location are published in the literature. We postulate that DDD is responsible for the development of HS in susceptible patients. Indeed, this association seems to be explained by a common pathophysiological mechanism, targeting the Notch signalling pathway, involved in the proliferation and differentiation of epidermal cells that can induce the development of HS. DDD should be considered as a comorbid factor of HS.
Keywords: hidradenitis suppurativa, Dowling Degos, vulvar, hyperpigmentation, comorbidities
Introduction
Dowling Degos disease (DDD), also known as reticular dermatosis of the folds, is a rare genodermatosis that manifests itself by the progressive appearance in adulthood of small isolated or confluent millimetric brownish macules with a reticular appearance and progressive hyperpigmentation over time. The lesions are asymptomatic, rarely pruritic and classically described in the flexural folds of the axillae, neck, inguinal and sub mammary folds. We report the case of a 45-year-old female patient who presented since the age of 30 with bilateral hyperpigmented macules of reticulated appearance in vulvar, perianal and axillary locations associated with Hurley stage 2 hidradenitis suppurativa (HS) that appeared later. Less than ten cases of genital DDD have been described in the literature. This atypical location is probably underestimated because it is asymptomatic. To our knowledge, the association of vulvar DDD with HS has only been reported in 3 cases; we report the fourth case.
Case Reports
A 45-year-old female patient presented with pigmented lesions that had appeared around the age of 30, during a pregnancy, in genital, perianal and bilateral axillary locations. In her history, she reports an HS that started at the age of 35, manifesting itself as multiple and recurrent painful nodules of the axillary, inguinal and sub mammary folds. The HS was previously treated by lymecycline. The patient had no family history of HS or DDD. She was not taking any medication and did not smoke. She was not overweight. Her body mass index was 24. On clinical examination, numerous isolated or confluent pigmented macules with a reticulated appearance are present. The pigmentation is bilateral and symmetrical, located in the axillary, vulvar and perianal folds and does not affect the mucous membranes (Figures 1 and 2).
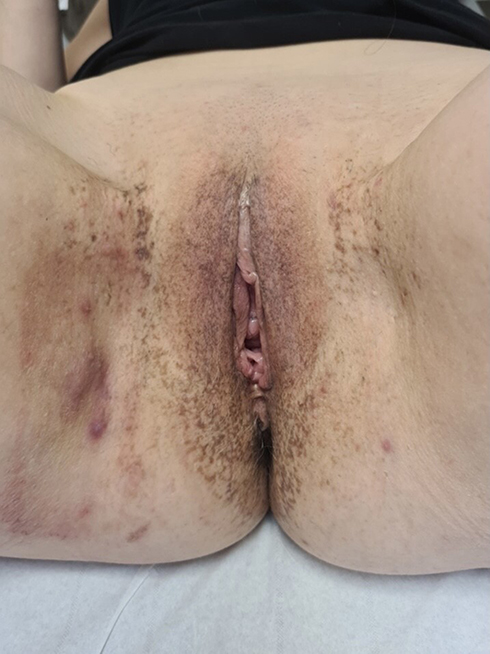 |
Figure 1 Numerous isolated or confluent perivulvar and perianal pigmented macules of reticulated appearance with the presence of inguinal HS nodules and fistulas. |
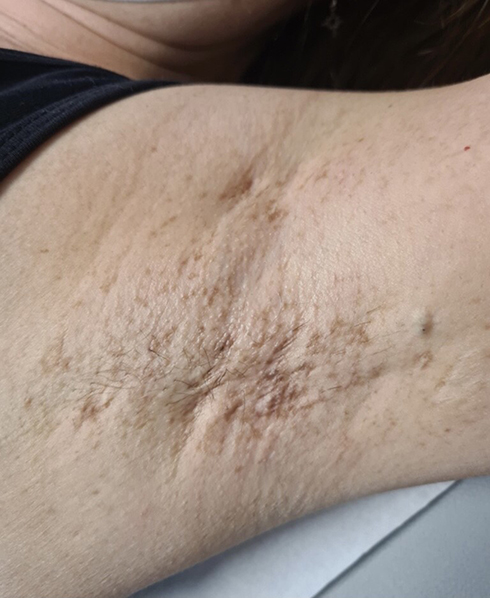 |
Figure 2 Discrete isolated brownish macules of the axillae with HS cysts and fistulae. |
The nails are free of pigmentation. The macules are asymptomatic but increase with time. In addition to the classic HS lesions in the form of nodules, sometimes simple, sometimes fistulised, characterising the disease at stage 2 of Hurley’s scale, we observe pseudo-comedonal lesions and atrophic scars on the back (Figure 3). She has no perioral or mucosal lesions. A skin biopsy of the reticular pigmentation shows discrete orthokeratotic hyperkeratosis with epidermal bud elongation and basal hyperpigmentation without associated melanocytic hyperplasia (Figure 4). The histological and clinical diagnosis of DDD with an atypical genital and perianal location was made in association with Hurley stage 2 HS.
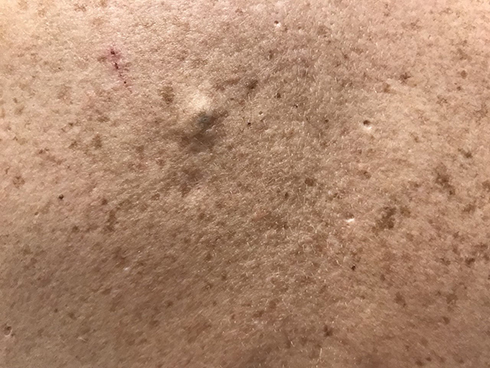 |
Figure 3 Pseudo-comedonal lesions and atrophic scars on the back. |
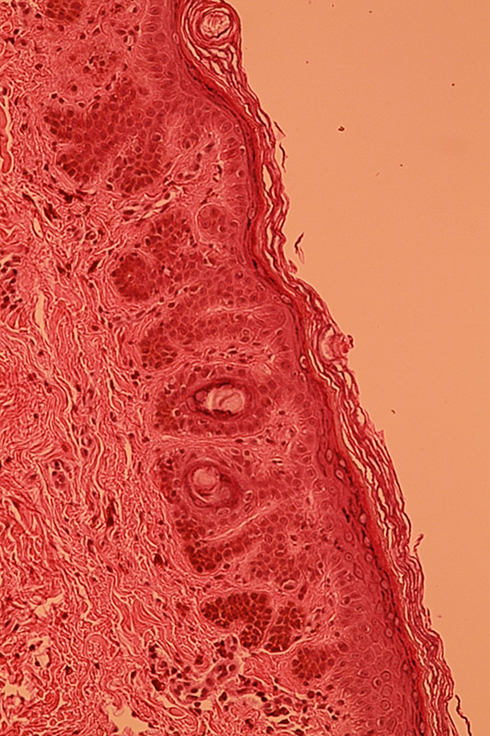 |
Figure 4 Elongation of epidermal buds, hyperpigmentation of basal keratinocytes without associated melanocytic hyperplasia (HES x 20). |
Discussion
DDD is an acquired or inherited reticular genodermatosis of the folds, autosomal dominant or sporadic, characterised by reticular hyperpigmentation of the axillae, neck, inguinal and sub mammary folds. It is more common among women than men.1 Pseudo-comedonal lesions of the back and neck as well as perioral atrophic scars and palmar pits are sometimes found.
To our knowledge, less than ten cases of genital involvement are described in the literature.2 In our patient’s case, axillary and perianal lesions were also observed. However, DDD can manifest itself as isolated genital hyperpigmentation.2 This location, mainly reported among women, is also reported among men on the penis or scrotum.3 DDD should be included in the differential diagnosis of vulvar hyperpigmentation with acanthosis nigricans and lentiginosis syndromes (Laugier’s disease, Leopard’s disease, Peutz Jeghers disease or Carney’s complex).4
DDD may be associated with HS.5 Among the reported cases of DDD with vulvar involvement, to our knowledge only three cases evoke the concomitant presence of epidermoid cysts suggesting association with HS.2,6,7 The coexistence of these two diseases seems to be explained by a common pathophysiological mechanism, targeting the functioning of the Notch signalling pathway, involved in epidermal cell proliferation and differentiation. Defects in Notch signalling result in inhibition of the hair growth cycle with conversion of hair follicles to epidermal cysts and inhibition of sebaceous gland differentiation, which are responsible for HS’s lesions. Notch signalling pathway seems to be also implicated in the regulation of melanocyte homeostasis which may underlie the hyperpigmentation in DDD.8 The genes mutated in Dowling Degos disease code for keratin 5 (KRT 5), protein GDP-fucose O-fucosyltransferase 1 (POFUT1), protein O-glucosyltransferase 1 (POGLUT1) and presenilin enhancer protein 2 (PSENEN) which is a subunit of the gamma secretase complex.
The role of this complex is to catalyse the cleavage of the intracellular part of the Notch receptor and thus to induce the transcription of the target genes of the Notch pathway. Inhibition of the gamma secretase complex is thus responsible for abnormalities in the Notch signalling pathway leading to abnormal pigmentation as well as epidermal hyperkeratosis and follicular occlusion which may explain the association between DDD and HS in genetically predetermined patients.9–11 HS can arise as an isolated condition, in a syndromic form or in association with DDD. There is a high proportion of pathogenic genetic variants, responsible for HS. Heterozygous variants in PSENEN on 19q13 seems to be most commonly reported in the association between DDD and HS.12,13 The lack of genetic analysis of this case is a limitation of our work.
Since DDD is asymptomatic, no treatment is necessary. Nevertheless, topical treatments based on retinoids, hydroquinone or cortisone can be tried but their efficacy remains limited. The ErbiumYAG laser seems promising.14 However, HS does require treatment and it seems like the patients with underlying HS-associated genetic variant or syndromic forms may be more resistant to the recommended therapeutics options.15 In the case of our patient, she had a good response to lymecycline treatment.
Conclusion
DDD is a rare genodermatosis, classically described in the axillae, neck, inguinal and sub mammary folds in the same way as HS. Its vulvar location is much more rarely reported but probably underestimated. DDD should be suspected in the presence of vulvar hyperpigmentation. As for HS, its appearance a few years after the appearance of DDD could be explained by the pathophysiological mechanism of the Notch signalling pathway anomaly inducing epidermal hyperkeratosis and follicular occlusion. This may explain the overlapping locations of the two diseases, which are also more common in women. We postulate that DDD is responsible for the development of HS in predisposed patients and would be a comorbid factor of HS as part of the follicular triad.16 Gynecologists should think about DDD in case of vulvar hyperpigmentation and refer to a dermatologist to explore.
Data Sharing Statement
On request.
Consent for Publication
Patients gave written informed consent to publish their data in this observational study. Institutional approval was not required to publish this observational study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was obtained for this study.
Disclosure
All authors certified that they have no conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject of this manuscript.
References
1. Stephan C, Kurban M, Abbas O. Dowling-Degos disease: a review. Int J Dermatol. 2021;60(8):944–950. doi:10.1111/ijd.15385
2. Kang HS, Hur J, Lee JW, et al. A case of Dowling-Degos disease on the vulva. Ann Dermatol. 2011;23(2):205–208. doi:10.5021/ad.2011.23.2.205
3. Taskapan O, Gokce E, Akin O, Erkilinc AC, Demirkesen C. Dowling-Degos disease with diffuse penile pigmentation. J Eur Acad Dermatol Venereol. 2014;28(10):1405–1406. doi:10.1111/jdv.12367
4. Horner ME, Parkinson KE, Kaye V, Lynch PJ. Dowling-Degos disease involving the vulva and back: case report and review of the literature. Dermatol Online J. 2011;17(7):1. doi:10.5070/D38XQ4S916
5. Agut-Busquet E, González-Villanueva I, Romani de Gabriel J, et al. Dowling-Degos Disease and hidradenitis suppurativa. Epidemiological and clinical study of 15 patients and review of the literature. Acta Derm Venereol. 2019;99(10):917–918. doi:10.2340/00015555-3225
6. Wong LS, Yang YC. Dowling-Degos Disease localized on vulva mimicking condyloma acuminata. Indian J Dermatol. 2018;63(6):521–522. doi:10.4103/ijd.IJD_10_18
7. Arjona-Aguilera C, Linares-Barrios M, Albarrán-Planelles C, Jiménez-Gallo D. Dowling-Degos disease associated with hidradenitis suppurativa: a case report. Actas Dermosifiliogr. 2015;106(4):337–338. doi:10.1016/j.ad.2014.09.010
8. Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178(2):502–508. doi:10.1111/bjd.16000
9. Nokdhes YN, Rutnumnoi T, Patthamalai P, Leeyaphan C. Follicular Dowling-Degos Disease with hidradenitis suppurativa: a case report and review of the literature. Case Rep Dermatol. 2021;13(3):530–536. doi:10.1159/000520541
10. McGrath JA. Concurrent hidradenitis suppurativa and Dowling-Degos disease taken down a “Notch”. Br J Dermatol. 2018;178(2):328. doi:10.1111/bjd.16068
11. Gratton R, Tricarico PM, Moltrasio C, et al. Pleiotropic role of notch signaling in human skin diseases. Int J Mol Sci. 2020;21(12):4214. doi:10.3390/ijms21124214
12. Peter DCV, Smith FJD, Wilson NJ, Dandi S. PSENEN mutation in coexistent hidradenitis suppurativa and Dowling-Degos disease. Indian Dermatol Online J. 2020;12(1):147–149. doi:10.4103/idoj.IDOJ_218_20
13. Pace NP, Mintoff D, Borg I. The genomic architecture of hidradenitis suppurativa-A systematic review. Front Genet. 2022;13:861241. doi:10.3389/fgene.2022.861241
14. Yun JH, Kim JH, Choi JS, Roh JY, Lee JR. Treatment of Dowling-Degos disease with fractional Er: yAGlaser. J Cosmet Laser Ther. 2013;15(6):336–339. doi:10.3109/14764172.2013.764437
15. Mintoff D, Pace NP, Ceci M. Management of patients with hidradenitis suppurativa having underlying genetic variation: a systematic review and a call for precision medicine. Clin Exp Dermatol. 2022;47(2):444–445. doi:10.1111/ced.14938
16. Loo WJ, Rytina E, Todd PM. Hidradenitis suppurativa, Dowling-Degos and multiple epidermal cysts: a new follicular occlusion triad. Clin Exp Dermatol. 2004;29(6):622–624. doi:10.1111/j.1365-2230.2004.01631.x
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.