")
Back to Journals » Cancer Management and Research » Volume 12
Abnormal Activations of Super-Enhancers Enhance the Carcinogenicity in Lung Adenocarcinoma
Authors Zhou J, Wang D, Tang D, Huang W
Received 8 May 2020
Accepted for publication 29 July 2020
Published 15 September 2020 Volume 2020:12 Pages 8509—8518
DOI https://doi.org/10.2147/CMAR.S258497
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Seema Singh
Jianlong Zhou,1,2,* Dingxue Wang,3,* Dongxin Tang,3 Wenhua Huang1
1Department of Oncology, Zhujiang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 2Department of Oncology, The Affiliated Xinhui Hospital, Southern Medical University, Xinhui, People’s Republic of China; 3Department of Oncology, The First Affiliated Hospital, Guizhou University of Traditional Chinese Medicine, Guiyang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dongxin Tang
The First Affiliated Hospital, Guizhou University of Traditional Chinese Medicine, No. 71 Baoshanbei Road, Guiyang 550000, People’s Republic of China
Email [email protected]
Wenhua Huang
Department of Oncology, Zhujiang Hospital, Southern Medical University, No.253 Gongye Middle Avenue, Guangzhou 510280, People’s Republic of China
Email [email protected]
Background: Lung tumors and normal lung tissues show large differences in epigenetic modification which can affect the chromosome structure and expression of genes. However, the epigenetic reprogramming in lung adenocarcinoma remains unclear.
Methods and Results: With the bioinformatics analysis, we found that some activated super-enhancers (SEs) only appear in lung adenocarcinoma cells, and 781 abnormal activated super-enhancers (AASEs) were found. Not only are the traditional oncogenes found to be activated by AASEs, such as MET and SLC2A1, but also some new genes were activated by AASEs, which probably contributes to the carcinogenic process in lung cancer. The enrichment analysis of the genes activated by AASEs shows that the glycolysis process and cell proliferation were enhanced and the apoptotic process was negatively regulated. Two AASEs were separately knockout by CRISPR/Cas9 in A549, PC-9, and H1299 cell lines and the expression of target genes decreased. The motif of CTCF, SMARCA1, SOX4, FOXM1, IRF3, IRF7, and STAT2 was enriched in AASEs, supporting that the chromosome structure changed and these transcription factors would be the master regulators on the formation of AASEs.
Conclusion: This study provided comprehensive insight into the mechanisms of SEs, as well as a potential therapeutic target for lung cancer.
Keywords: lung adenocarcinoma, epigenetic, super-enhancer, transcription factors
Introduction
Lung cancer is the most common cause of cancer-related deaths worldwide, and lung adenocarcinoma is the most frequent pathological type in lung cancer.1,2 It is important to investigate the underlying mechanisms of lung tumorigenesis and tumor progression. Lung tumor cells and normal lung cells have very different characteristics, including the histological features, expression of genes, and epigenetic modification of genome.3,4 Super-Enhancers (SEs) are regions of genome comprising multiple individual enhancers which are the key regulatory elements to control tissue-specific transcription. SEs have been identified by locating genomic regions that are highly enriched in the H3K27ac ChIP-Seq signal.5 Compared to typical enhancers, SEs are larger, and exhibit a higher transcription factor and histone modification density. An array of transcription factor proteins bind on SEs and drive transcription of genes involved in cell identity.6–8 In cancer cells, abnormal SEs enhance the expression of critical oncogenes such as CACNA1H, MYC, LMO1, and RARA.9–12 Cancer cells generate new SEs nearoncogenes that are involved in tumor pathogenesis, and the absence of these SEs would reduce by at least 50% the survival rate of cancer cells.9 Accordingly, targeting the SEs and transcription factors on it will be the approaches to cancer diagnosis and clinical therapeutics, including small-molecule inhibitors against super-enhancers binding proteins and gene therapy strategies.13
In the study of SEs in lung cancer, we found hundreds of emerging SEs in lung adenocarcinoma. Abnormal activation of these super-enhancers enhance the expression of oncogenes which would be the cause of lung adenocarcinoma pathogenesis. With the analysis and mining of bioinformation, we got all AASEs and their associated oncogenes in lung adenocarcinoma. The activated biological processes and pathways will show us the major characteristic and new therapeutic target of lung adenocarcinoma. Furthermore, we predicted the transcription factors binding on AASEs, which could be the pathogenic elements and help us understand pathogenesis in lung cancer.
Materials and Methods
Cell Culture
The human A549, PC-9, and H1299 cell lines were obtained from ATCC (American Type Culture Collection). A549 were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS, HyColne, UT, USA) at 37°C in a humidified atmosphere containing 5% CO2. PC-9 and H1299 were cultured in Roswell Park Memorial Institute (RPMI) 1640 (Gibco) containing 10% FBS at 37°C in a humidified atmosphere containing 5% CO2.
Genome Editing
For the knockout of AASEs in A549, two expression cassettes encoding the sgRNA sequences (sgRNA1-MET-AASE CTCCGTTAGAGGCGTGTCAG, sgRNA2-MET-AASE TACTCTTAGTATCCTGACTA; sgRNA1-EGFR-AASE TCATCTTCGATGACTCTGCT, sgRNA1-EGFR-AASE CTCACATGTGTCTATTCGGG) flanking the deletion region were cloned into a plasmid that expresses a codon-optimized version of Cas9. Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) was used to transfect the plasmid into the A549 cells. The transfected A549 cells were selected with puromycin for 2 days. The genotyping primers are as follows: MET-AASE: 5ʹ-AAAGCAGTGGGCATATGGGA-3ʹ (forward), 5ʹ-GTGCTCTAATGCAGGTTGGG-3ʹ (reverse1), 5ʹ-ACCTACTTAGCCTACTCA-3ʹ (reverse2); EGFR-AASE: 5ʹ-TGAGTAGGCTAAGTAGGT-3ʹ (forward), 5ʹ-AAGGCACAGCCCGTGAAAT-3ʹ (reverse1), 5ʹ-CACATTCATGCCACAGAA-3ʹ (reverse2).
Quantitative RT-PCR
Total RNA was purified from A549 with RNeasy Mini Kit (QIAGEN), and total RNA (1 µg) was reversely transcribed into cDNA and analyzed by qPCR. The primers for β-Actin are 5ʹ-GCCAACACAGTGCTGTCT-3ʹ (forward) and 5ʹ-AGGAGCAATGATCTTGATCTT-3ʹ (reverse). The primers for MET are 5ʹ-TGCACAGTTGGTCCTGCCATGA-3ʹ (forward) and 5ʹ-CAGCCATAGGACCGTATTTCGG-3ʹ (reverse). The primers for EGFR are 5ʹ-AACACCCTGGTCTGGAAGTACG-3ʹ (forward) and 5ʹ-TCGTTGGACAGCCTTCAAGACC-3ʹ (reverse). The levels of MET and EGFR mRNA were normalized to those of β-Actin. Changes in mRNA expression were calculated according to the 2−ΔΔCT method (CT, cycle threshold).
Analysis of Gene Expression Profile
The gene expression profiles of lung cancers and matched adjacent non-malignant lung were downloaded from the GEO database (accession: GSE75037 and GSE32863) (https://www.ncbi.nlm.nih.gov/geo/). GSE75037 and GSE32863 were based on Illumina WG6-V3 expression arrays, and GSE75037 included 83 lung adenocarcinomas and 83 matched adjacent non-malignant lung tissues, GSE32863 included 58 lung adenocarcinomas and 58 matched adjacent non-malignant lung tissues. Differentially expressed genes between tumors and non-malignant lung tissues were identified using two-group comparisons, and P<0.05 was set as the cutoff criterion.
The Analysis of Super Enhancer and ChIP-Seq Data
The super-enhancer regions of the lung cancer cell line (A549) and normal lung tissue (lung_30y). AASEs were found by calculating the super-enhancer regions which only occur in A549 compared to normal lung tissue though BEDTools.30 The super-enhancer corresponded ChIP-seq data of H3K27ac were obtained from the Encyclopedia of DNA Elements (ENCODE).31 The ChIP-Seq data were displayed using the UCSC Genome Browser (http://genome.ucsc.edu/). All analyses were performed using human (build hg19, GRCh37) RefSeq annotations downloaded from the UCSC genome browser.
Functional and Pathway Enrichment Analysis
Panther (http://www.pantherdb.org/) was used to class the genes based on Gene Ontology (GO) molecular function. DAVID (https://david.ncifcrf.gov/home.jsp, version 6.8) was used for the KEGG pathway and GO analysis for the activated genes. EASE Score, a modified Fisher Exact P-value, is used to measure the gene-enrichment in annotation terms. P-value<0.05 to be considered strongly enriched in the annotation categories.
Construction of Protein–Protein Interaction Network
To further explore the relationships between genes activated by AASEs, the protein–protein interaction network was mapped by (STRING) database (http://string-db.org/) with default parameter and visualized though Cytoscape software (https://cytoscape.org/).
Analysis of Motif Enrichment
The AME tool in MEME suite is used to identify motifs that are relatively enriched in the sequences of AASEs with default parameter.32 The motif database is HOCOMOCO Human (v11 CORE).33
Survival Analysis
Kaplan-Meier Plotter is a web tool that predicts the prognostic values of genes in lung cancer patients (http://kmplot.com/analysis/). The patients with lung cancer were divided into two groups according to the particular gene expression level (high vs low expression). Based on these categories, overall survival (OS) analysis of the two patient groups was compared by the tool. The HR with 95% CIs and log-rank P-value were calculated and shown.
Results
Abnormal Activation of Super-Enhancers in Lung Adenocarcinoma
Lung tissue and lung tumor share the same genome, their histone modifications such as H3K27ac H3K4me1 are distinct.4 In the research of epigenetic processes in lung cancer, we found some regions of chromosome were overactivated in lung cancer. These regions were largely marked with H3K27ac which is an active enhancer marker in genome and clusters of these enhancers formed new super-enhancers in lung cancers (Figure 1A). The super-enhancers data of lung normal tissue and lung cancer cell lines were obtained from the SEdb database.14 After analysis, we find 781 emerging super-enhancer regions in lung cancer line (A549) (Table S1). We can find the abnormally activated super-enhancers (AASEs) emerged in lung cancer and the nearby genes, such as ENO1, MET, EGFR, SLC2A1, and TKT, were over-expression in lung cancer tissues (Figure 1B). These genes are widely recognized for their importance in cancer.
 |
Figure 1 Abnormal activation of super-enhancers in lung cancer. (A) A hypothetical model of how SEs are abnormally activated in lung cancer and induce the overexpression of genes distantly (enhancer) or closely (promoter). (B) Examples of the abnormal activation of super enhancers and the involved genes are overexpressed in lung cancer tissues. The expression of genes came from GSE32863 (58 lung adenocarcinoma and 58 adjacent non-tumor lung tissues). |
AASEs Cause Oncogenesis and Promote Tumor Development
The closest active genes of SEs were obtained from the SEdb database.14 Combining the gene expression data of GSE75037 and GSE32863 which were tested from lung adenocarcinoma tumors and their matched histologically normal adjacent lung tissue samples, we screened 261 genes which were activated by AASEs and over-expressed in lung cancer (Figure 2A and B, Table S2). The major molecular functions of these genes are binding and catalytic activity (Figure 2C). From the results of the protein–protein interaction network, we could find the the HSPA4, HSPB1, EGFR, MAPK6, and MET were in the center of the network (Figure 2D). From the results of the GO biological process and KEGG pathway enrichment analysis, we found these genes were significantly enriched in the glycolysis process, metabolic pathway, negative regulation of apoptotic process, cell proliferation, TGF-β signaling pathway, and VEGF signaling pathway (Figure 3A and B). The over-activated of these genes formed the major characteristic in lung cancer cells.
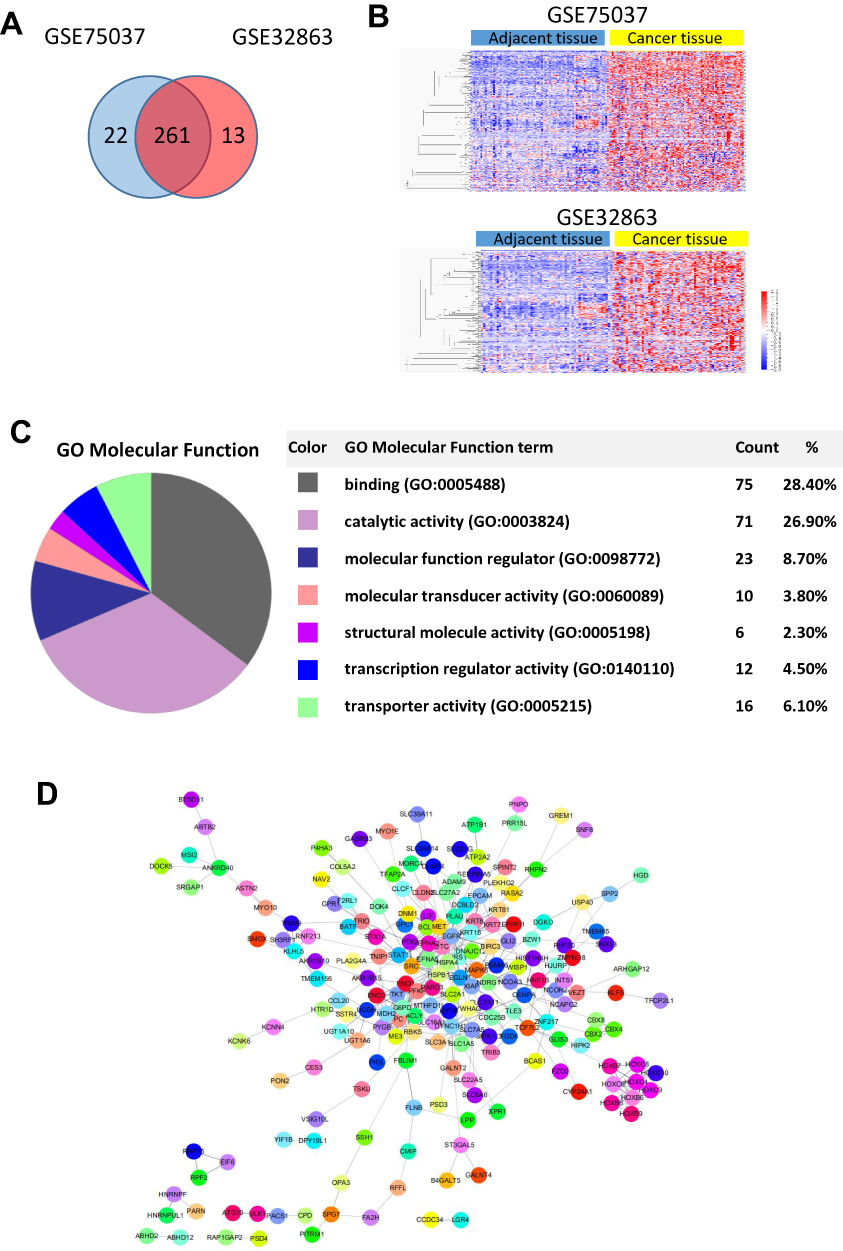 |
Figure 2 Genes activated by abnormal SEs in lung cancer. (A) Screening the genes activated by SE which is over-expressed in both two lung cancer databases (GSE75037 and GSE32863). (B) Heatmaps show the expression of screened genes in (A). (C) Molecular function of over-expressed genes activated by AASEs. (D) The protein–protein interaction network of the genes activated by AASEs. |
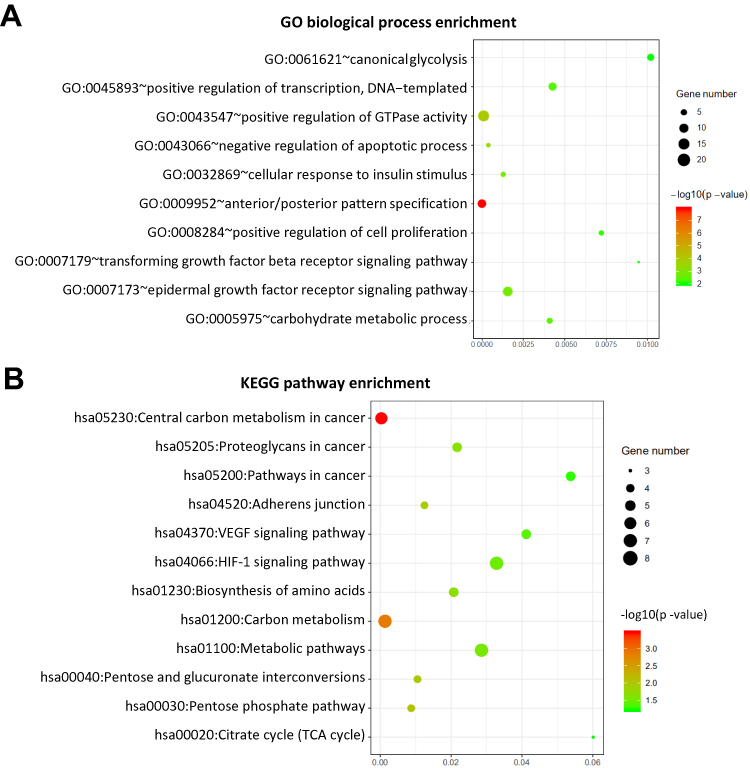 |
Figure 3 AASEs enhance the carcinogenicity through up-regulation of the involved gene. (A) Enriched Gene Ontology (GO) biological process of over-expressed genes activated by AASEs. (B) Enriched KEGG pathway of over-expressed genes activated by abnormal AASEs. |
Identify the Activation of AASEs in Lung Adenocarcinoma
To determine whether AASEs could activate the transcription of nearby genes, we knockout the AASEs of MET and EGFR in A549, PC-9, and H1299 cell lines though CRISPR/Cas9-based genome engineering (Figure 4A). The pool of cells after transfection and screening were collected to identity the activation of AASEs near MET and EGFR. The results showed the deletion of AASEs decreased the expression of the MET and EGFR mRNA in A549, PC-9, and H1299 cell lines (Figure 4B).
 |
Figure 4 Identify the activation of AASEs though CRISPR/Cas9-based genome engineering. (A) Knockout the AASEs of MET and EGFR in A549 cell line (MET-E-KO and EGFR-E-KO). Scissors sketch shows the regions that knockout through CRISPR/Cas9 (left). PCR-based genotyping of MET-E-KO and EGFR-E-KO (right). (B) The RNA expressions of MET and EGFR were decreased when their AASEs were deleted (*P<0.05). |
Master Transcription Factors Activate AASEs
Master transcription factors could bind on the SEs and determine the cell type-specific.6 To find which master transcription factors bound on AASEs, we analyzed the motifs of transcription factors which were enriched in the sequences of AASEs (Figure 5A). The most enriched transcription factor is CTCF, which is thought to be in regulating the 3D structure of chromatin and forming enhancer-promoter loops (Figure 5B).15,16 The other enriched transcription factors are SMARCA1, SOX4, FOXM1, IRF3, IRF7, and STAT2, and these factors function in regulating the transcription of genes. Consistent with the mRNA expression data of lung tumor tissues and adjacent non-tumor tissues, SMARCA1, SOX4, FOXM1, IRF3, IRF7, and STAT2 are overexpressed in lung cancer and correlated with the poor prognosis of lung cancer patients, but not CTCF (Figure 5C and D).
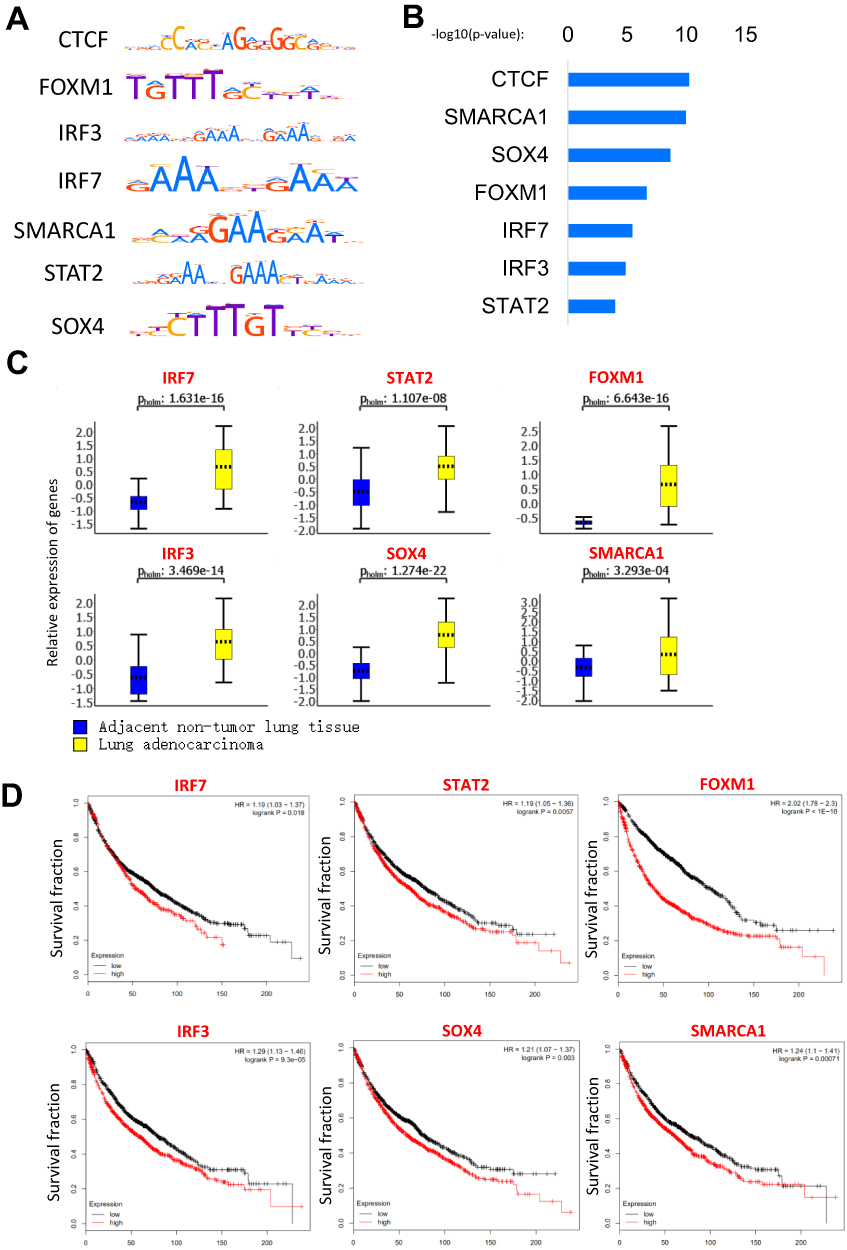 |
Figure 5 The master transcription factors contribute to the AASEs in lung cancer. (A) Motif of enriched transcription factors in AASEs. (B) The enrichment P-value of transcription factors in AASEs. (C) The overexpression of master transcription factors in lung cancer. (D) Log-rank (Mantel Cox) survival test of lung cancer patients based on the levels of IRF7, STAT2, FOXM1, IRF3, SOX4, and SMARCA1 (low expression n=963, high expression n=963). |
Discussion
Our results identified the AASEs that contribute to the pathogenesis in lung cancers. AASEs promote the initiation and progression of lung cancer by enhancing the expression of genes such as ENO1, MET, EGFR, SLC2A1, and TKT (Figure 1B). New SEs appeared around these genes in lung cancer cells, but not in normal lung tissues. The genes we show in Figure represent the well-known oncogenes: ENO1 is up-regulated in several tumors including breast, lung, prostate, and pancreas, and plays an important role in the Warburg effect in cancer cells.17 Another Warburg effect gene SLC2A1, also known as GLUT1, plays a key role in glucose uptake in many cell types including cancer cells. High expression of SLC2A1 will promote growth and proliferation in cancer cells.18 MET is a proto-oncogene and plays a role in cellular survival, embryogenesis, and cellular migration and invasion.19 Overexpression of MET is also associated with multiple human cancers.20 EGFR (Epidermal growth factor receptor) has been reported to be implicated in the pathogenesis of many human malignancies and promote the metastasis of cancer.21 TKT is a thiamine-dependent enzyme involved in glycolysis and the pentose phosphate pathway which is essential in cancer energy metabolism.22 Furthermore, with the visualization of AASEs regions and ChIP-Seq signal data of H3K27ac, we could find a new activated mechanism of the oncogenes and more genes involved in cancer pathogenesis. We choose two AASEs and knockout them in three lung adenocarcinoma cell lines, the expression of target genes MET and EGFR fell by half. MET and EGFR were well-known oncogenes, and their AASEs regions will be the therapeutic target in lung cancer.
To investigate the function and pathway affected by AASEs, enrichment analysis of the genes activated by AASEs was undertaken. The results showed the energy-related processes and pathways appeared frequently. Increased glucose uptake and aerobic glycolysis are the major characteristic in cancer cells.23 The energy metabolism associated genes such as ENO1, SLC2A1, ENO3, G6PD, and TKT were activated by AASEs and over-expressed in lung cancer cells. Meanwhile, cell proliferation was positively regulated and the apoptotic process was negatively regulated in lung cancer cells.
In the results of enriched transcription factors on AASEs, we found CTCF to have the minimum P-value. CTCF (CCCTC-Binding Factor) is a multifunctional protein in genome regulation and gene expression.24,25 CTCF can bind and remodel the three-dimensional structure of the genome which promotes the formation of long-range interaction of chromosome.26 A number of studies have demonstrated the co-localization of CTCF and cohesion on chromosomes, suggesting they can steady the chromatin loops between enhancers and promoters and also promote the binding of transcription factors at enhancers.27–29 Therefore, the enrichment of CTCF on AASEs means large changes of chromosome structure happened on AASEs and help the formation of the connections between SEs and promoter of genes. The transcription factors such as IRF7, SOX4, STAT2, and FOXM1 will promote the activation of AASEs and regulate the transcription of downstream genes through long-range interaction. High expression of these transcription factors means poor survival rates in lung cancer patients. They can be biomarkers and therapeutic targets in lung cancer. However, further studies are required to find the mechanism that these transcription factors are activating AASEs by in lung cancer.
Acknowledgments
The following grants and foundations supported this work: Guangdong Provincial Health Department Fund of China (20122190, 20131134); and Basic and Applied Basic Research Key Project of Jiangmen (2019030102500013071).
Author Contributions
This manuscript was written by Jianlong Zhou, Dingxue Wang, Dongxin Tang, and Wenhua Huang, who made significant contributions in conception and design, data acquisition, data analysis and interpretation, writing articles, or critical modification of important intellectual content. All authors gave final approval of the version to be published, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi:10.3322/caac.21254
3. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299–311. doi:10.1016/S0140-6736(16)30958-8
4. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–641. doi:10.1038/nrg.2016.93
5. Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107(50):21931–21936. doi:10.1073/pnas.1016071107
6. Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi:10.1016/j.cell.2013.03.035
7. Huang J, Liu X, Li D, et al. Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev Cell. 2016;36(1):9–23. doi:10.1016/j.devcel.2015.12.014
8. Yu L, Ji KY, Zhang J, et al. Core pluripotency factors promote glycolysis of human embryonic stem cells by activating GLUT1 enhancer. Protein Cell. 2019. doi:10.1007/s13238-019-0637-9
9. Mack SC, Pajtler KW, Chavez L, et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature. 2018;553(7686):101–105. doi:10.1038/nature25169
10. Oldridge DA, Wood AC, Weichert-Leahey N, et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature. 2015;528(7582):418–421. doi:10.1038/nature15540
11. McKeown MR, Corces MR, Eaton ML, et al. Superenhancer analysis defines novel epigenomic subtypes of non-APL AML, including an RARalpha dependency targetable by SY-1425, a potent and selective RARalpha agonist. Cancer Discov. 2017;7(10):1136–1153. doi:10.1158/2159-8290.CD-17-0399
12. Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. doi:10.1016/j.cell.2013.09.053
13. Shin HY. Targeting super-enhancers for disease treatment and diagnosis. Mol Cells. 2018;41(6):506–514. doi:10.14348/molcells.2018.2297
14. Jiang Y, Qian F, Bai X, et al. SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res. 2019;47(D1):D235–D243. doi:10.1093/nar/gky1025
15. Ren G, Jin W, Cui K, et al. CTCF-mediated enhancer-promoter interaction is a critical regulator of cell-to-cell variation of gene expression. Mol Cell. 2017;67(6):1049–1058. doi:10.1016/j.molcel.2017.08.026
16. Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137(7):1194–1211. doi:10.1016/j.cell.2009.06.001
17. Capello M, Ferri-Borgogno S, Riganti C, et al. Targeting the warburg effect in cancer cells through ENO1 knockdown rescues oxidative phosphorylation and induces growth arrest. Oncotarget. 2016;7(5):5598–5612. doi:10.18632/oncotarget.6798
18. Carvalho KC, Cunha IW, Rocha RM, et al. GLUT1 expression in malignant tumors and its use as an immunodiagnostic marker. Clinics (Sao Paulo). 2011;66(6):965–972. doi:10.1590/S1807-59322011000600008
19. Salgia R. MET in lung cancer: biomarker selection based on scientific rationale. Mol Cancer Ther. 2017;16(4):555–565. doi:10.1158/1535-7163.MCT-16-0472
20. Mo HN, Liu P. Targeting MET in cancer therapy. Chronic Dis Transl Med. 2017;3(3):148–153. doi:10.1016/j.cdtm.2017.06.002
21. Bethune G, Bethune D, Ridgway N, Xu Z. Epidermal growth factor receptor (EGFR) in lung cancer: an overview and update. J Thorac Dis. 2010;2(1):48–51.
22. Xu IM, Lai RK, Lin SH, et al. Transketolase counteracts oxidative stress to drive cancer development. Proc Natl Acad Sci U S A. 2016;113(6):E725–E734. doi:10.1073/pnas.1508779113
23. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152. doi:10.1186/1476-4598-12-152
24. Ling JQ, Li T, Hu JF, et al. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science. 2006;312(5771):269–272. doi:10.1126/science.1123191
25. Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–380. doi:10.1038/nature11082
26. Handoko L, Xu H, Li G, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet. 2011;43(7):630–638. doi:10.1038/ng.857
27. Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467(7314):430–435. doi:10.1038/nature09380
28. Faure AJ, Schmidt D, Watt S, et al. Cohesin regulates tissue-specific expression by stabilizing highly occupied cis-regulatory modules. Genome Res. 2012;22(11):2163–2175. doi:10.1101/gr.136507.111
29. Gosalia N, Neems D, Kerschner JL, Kosak ST, Harris A. Architectural proteins CTCF and cohesin have distinct roles in modulating the higher order structure and expression of the CFTR locus. Nucleic Acids Res. 2014;42(15):9612–9622. doi:10.1093/nar/gku648
30. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi:10.1093/bioinformatics/btq033
31. ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi:10.1038/nature11247
32. Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–W208. doi:10.1093/nar/gkp335
33. Kulakovskiy IV, Medvedeva YA, Schaefer U, et al. HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 2013;41(Databaseissue):D195–D202. doi:10.1093/nar/gks1089
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.