")
Back to Journals » OncoTargets and Therapy » Volume 14
Aberrant Factors of Fibrinolysis and Coagulation in Pancreatic Cancer
Authors Fang L, Xu Q, Qian J, Zhou JY
Received 9 September 2020
Accepted for publication 11 December 2020
Published 6 January 2021 Volume 2021:14 Pages 53—65
DOI https://doi.org/10.2147/OTT.S281251
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Leo Jen-Liang Su
Lianghua Fang,1,* Qing Xu,2,* Jun Qian,1 Jin-Yong Zhou3
1Department of Oncology, Jiangsu Province Hospital of Chinese Medicine, Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing 210029, People’s Republic of China; 2Women’s Hospital of Nanjing Medical University, Nanjing Maternity and Child Health Care Hospital, Nanjing 210029, People’s Republic of China; 3Central Laboratory, Jiangsu Province Hospital of Chinese Medicine, Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing 210029, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jin-Yong Zhou
Central Laboratory, Jiangsu Province Hospital of Chinese Medicine, Affiliated Hospital of Nanjing University of Chinese Medicine, No. 155, Han Zhong Road, Nanjing, People’s Republic of China
Email [email protected]
Jun Qian
Department of Oncology, Jiangsu Province Hospital of Chinese Medicine, Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing 210029, People’s Republic of China
Email [email protected]
Abstract: Aberrant factors associated with fibrinolysis and thrombosis are found in many cancer patients, which can promote metastasis and are associated with poor prognosis. The relationship between tumor-associated fibrinolysis and thrombosis is poorly understood in pancreatic cancer. This review provides a brief highlight of existing studies that the fibrinolysis and coagulation systems were activated in pancreatic cancer patients, along with aberrant high concentrations of tissue plasminogen activator (t-PA), urine plasminogen activator (u-PA), D-dimer, fibrinogen, or platelets. These factors cooperate with each other, propelling tumor cell shedding, localization, adhesion to distant metastasis. The relationship between thrombosis or fibrinolysis and cancer immune escape is also investigated. In addition, the potential prevention and therapy strategies of pancreatic cancer targeting factors in fibrinolysis and coagulation systems are also been discussed, in which we highlight two effective agents aspirin and low-molecular weight heparin (LMWH). Summarily, this review provides new directions for the research and treatment of pancreatic cancer.
Keywords: fibrinolysis, thrombosis, pancreatic cancer, immune escape
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related death in men and fifth in women in the United States from 2013 to 2017.1 Its prognosis remains very poor with a five-year net survival of less than 10%.2 Several reasons are responsible for this poor prognosis, including poor early diagnosis, a high rate of relapse after curative surgery, and strong resistance to chemotherapy and radiotherapy. Venous thrombosis has been identified as the second leading cause of death in patients with cancer, inferior only to the progression of cancer.3
Pancreatic cancer has the highest risk of venous thromboembolism (VTE).4 In systematic analysis, thromboembolic event in patients with pancreatic cancer predicted excess premature (3 months) mortality,5 and symptomatic VTE is an independent risk factor for death.6 However, anticoagulation is not associated with longer survival6 and should not be used to extend the survival of patients with cancer in the absence of other indications.7 This suggested that thrombosis is a late event in the process of cancer. Control of thrombosis cannot impede cancer progression. Interestingly, the fibrin degradation product, D-dimer, could be found in resectable pancreatic cancer without thrombosis and is associated with poor prognosis in these patients.8 D-dimer is the product of secondary fibrinolysis, which aims to disintegrate the thrombus and maintain patency of the vascular system. This suggested that the pathological state of thrombosis already exists, although the thrombus has not yet formed in patients with pancreatic cancer.
Primary hyperfibrinolysis is uncommon in the setting of solid tumors and only isolated cases of primary fibrinolysis have been reported in metastatic prostate cancer9,10 and breast cancer,11 which can be reversed by anti-tumor therapy.11 Although it was not reported in pancreatic cancer so far, factors associated with primary hyperfibrinolysis, such as tissue plasminogen activator (t-PA)12 and urine plasminogen activator (u-PA),13 were found at high concentrations in tissue homogenates and sera of patients with pancreatic cancer patients. This indicated that plasminogen was more likely to be activated because of these high concentrations of plasminogen activator, which would lead to a fibrinolysis cascade in pancreatic cancer.
Understanding the aberrant factors associated with tumor-associated thrombosis and fibrinolysis has generated novel hypotheses regarding the mechanisms involved in pancreatic ductal adenocarcinoma (PDAC) growth and dissemination. We will initially review studies identifying the factors associated with the clotting and bleeding system in PDAC and speculate how these two distinct systems might be related to one another and promote metastasis of PDAC. We will then discuss potential strategies to target the molecules associated with the clotting and bleeding system in pancreatic cancer and the development of new directions for the research and treatment of PDAC.
Correlation Between Factors of the Fibrinolysis System and Tumor Cells Free from Their Original Site
The Activation of t-PA and u-PA in Patients with Pancreatic Cancer
The fibrinolytic system is a highly regulated enzymatic process that prevents the unnecessary accumulation of intravascular fibrin and enables the removal of thrombi, which is more appropriately referred to as the plasminogen activator system. In humans, t-PA and u-PA are the two activators of this system. High u-PA expression in tumor tissues14–16 and increased plasma levels of uPA17 have long been noted in colonic cancer, lung cancer, basal cell carcinoma, endometrial cancer, and cervical cancer. The overexpression of u-PA in tumor tissue or increased u-PA levels in serum have strong independent prognostic value in terms of relapse-free and/or overall survival in patients with breast, colorectal, esophageal, gastric, hepatocellular, prostate cancer, sarcoma, head and neck squamous cell carcinoma.17–25
In pancreatic cancer, the importance of the plasminogen activator system has also been demonstrated.13,26–30 The first study on u-PA in pancreatic cancer was conducted in 1993, which showed that 78% of pancreatic cancers overexpressed u-PA and this overexpression correlated with decreased survival.31 Ten years later, these results were confirmed by another study, which showed a higher rate of u-PA expression, with 93% in archival paraffin sections. The u-PA staining was also found in pancreatic intraepithelial neoplasia (Pan IN) lesions, but not in normal tissue,29 which indicated that u-PA was an early event in the malignant transformation of pancreatic cancer. Furthermore, in situ hybridization experiments revealed the presence of u-PA mRNA, not only in the cytoplasm of tumor cells but also in the vessels of the tumor stroma,29 which suggested that u-PA should be detectable via serum analysis. Indeed, u-PA overexpression in tumor tissue correlated with the serum levels of u-PA in patients with pancreatic or biliary cancer.30 In summary, overexpression of u-PA correlates closely with the rapid progression, invasiveness, and short overall survival of pancreatic cancer.
t-PA is one of the better studied compounds of the fibrinolysis pathway, which has been detected in multiple tumors, such as ovarian cancer,32 lung cancer,33 breast cancer,34 glioma,35 hepatocellular cancer,36 laryngeal tumors,37 malignant melanoma,38 prostate cancer,39 gastric cancer,40 and colonic cancer.41 However, t-PA antigen levels in cancer tissue varied in different tumor types. t-PA levels were significantly lower in non-small cell lung cancer, gastric cancer, and colonic cancer than in normal tissue; However, t-PA levels were significantly higher in patients with breast carcinoma compared with that in the control group. The significance of t-PA for prognosis is also controversial, some studies showed that overall survival was significantly worse for patients with tumors showing tPA in the lowest quartile of activity in primary breast cancer;42,43 however, others showed that no significance was found in gastric cancer,40 hepatocellular carcinoma (HCC).36
Although the value of t-PA varied in many cancers, multiple studies showed that t-PA was overexpressed in pancreatic cancer tissue.12,44,45 The mean t-PA concentration in tissue homogenates gradually increased in cases with a normal pancreas, chronic pancreatitis, and pancreatic cancer.12 Additionally, t-PA plays an important role in the growth, invasion and angiogenesis of pancreatic tumor cells.44 Increasing tissue homogenate t-PA concentrations were associated with blood vessel infiltration, and t-PA concentrations were higher in sera than in tissue homogenates in patients with pancreatic cancer.12 In addition, vulnerability to t-PA increased in patients with pancreatic cancer,46 which might explain the higher incidence of hemorrhage after surgery in pancreatic cancer (9.9%; 42/423),47 but was only 1.3 (4/316) in patients with lung cancer after surgery.48
Tumor Cells are Freed from Their Original Site Through the Fribrinosis System
The plasminogen-plasmin system, also known as the fibrinolysis system, has been reported to be associated with various processes of tumor development, proliferation, invasion, and tumor angiogenesis.14,49,50 The overexpression of t-PA and u-PA in tumor cells locally activated plasminogen to plasmin, which in turn activates latent metalloproteinases, latent growth factors, and the degradation of extracellular matrix (ECM) proteins, such as laminin and fibronectin.51,52 It is believed that the resulting proteolysis of ECM frees the cell from its adhesion site allowing cell migration, which is manifested as invasion at local sites, and as metastasis at distant sites via circulating tumor cells (CTC) migrating to the blood system. Therefore, it was hypothesized that the u-PA system could be responsible for CTC generation and D-dimer production. Previous studies showed that D-dimer was an essential accompaniment of CTCs in gastric cancer53 and metastatic breast cancer.54,55 Plasma u-PA was highly associated with CTCs in early breast cancer and u-PA overexpressed in CTC_EP (epithelial phenotype), but not in CTC_EMT (epithelial to mesenchyme transition phenotype).55 These results indicated that the CTC_EP cells that overexpressed u-PA become detached from the lesion and invade directly to the blood system.
In pancreatic cancer, high concentrations of D-dimer,8,56 uPA,30 and tPA12 were found in plasma and were associated with reduced survival in pancreatic cancer and predict non-resectability. Although the relationships among D-dimer, u-PA, t-PA, and CTCs remain unknown, studies have shown that t-PA and u-PA could promote metastasis by activating plasminogen in pancreatic cancer.57 t-PA binds to Annexin II, which is overexpressed in pancreatic cancer and accelerates cell surface conversion of plasminogen to plasmin.58,59 Disruption of the t-PA/annexin II interaction using a specific hexapeptide significantly decreased the invasive capacity of pancreatic cancer cells (SK-PC-1) in vitro.45 Meanwhile, u-PA binds to its receptor (u-PAR) to activate plasminogen.60 Importantly, both u-PAR and u-PA are highly expressed in human pancreatic cancer tissues, and the expression of u-PAR always correlated with the expression of its ligand u-PA. Furthermore, suppression of the u-PAR–u-PA system prevented invasion,61 and upregulation of the u-PAR–u-PA system promoted EMT and metastasis in pancreatic cancer.13
Therefore, we speculated that activation of the fibrinolysis system comprises the first step for local invasion and distant metastasis in pancreatic cancer (Figure 1). From this hypothesis, we suggested that CTCs would be detected in patients with pancreatic cancer. Most patients of pancreatic cancer have no symptoms at the early stage and lack an effective early detection method; therefore, detecting CTCs might be an effective method to detect pancreatic cancer earlier, especially in cases with a high risk of developing pancreatic cancer. Furthermore, cancer tissues are difficult to access because of their deep and hidden site; thus, detection of CTCs might help to clarify the diagnosis and guide therapy although there still remain a number of challenges to the routine implementation of CTCs in the clinical management of pancreatic cancer.62
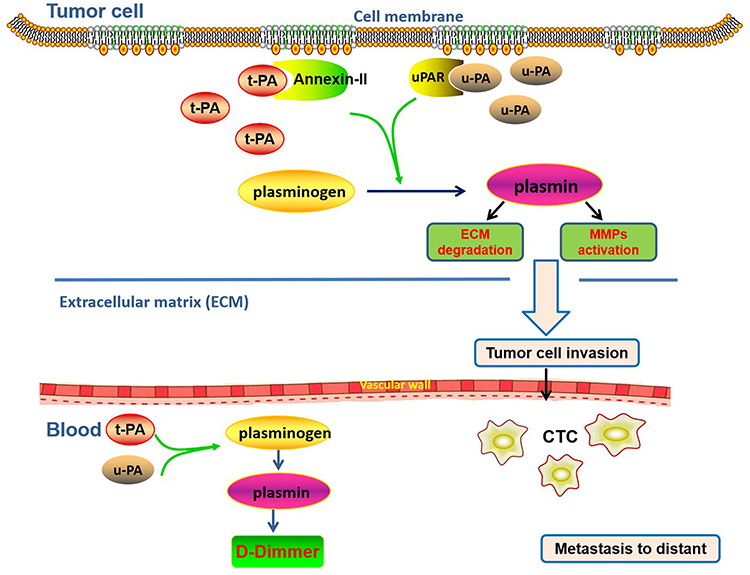 |
Figure 1 The diagram of the plasminogen activator system associated with local invasion and distant metastasis in pancreatic cancer. In this system, t-PA and u-PA are overexpressed in tumor cells; (t-PA)-(annexin II) binding and (u-PA)-(u-PAR) binding can both activate plasminogen to plasmin, which in turn activates MMPs and ECM degradation; the resulting proteolysis of ECM frees the cells from their original site allowing cell invasion, which is manifested as metastasis at distant sites via CTC migrating to the blood system; high concentration of D-dimer in plasma is an essential accompaniment of CTCs. |
The Coagulation System and the Anchoring/Adherence of Tumor Cells
Abnormality of Platelets and Fibrinogen in Pancreatic Cancer
Coagulation is a dynamic system, representing a balance between clotting and bleeding that is always maintained in normal physiology and is generally altered under disease conditions. About 60–100% of patients with malignant neoplasia were found to have hemostatic alterations, as detected by laboratory tests, including those without thrombotic manifestations. These changes comprise different levels of blood coagulation abnormalities, such as shortened activated partial thromboplastin time, thrombocytosis, elevated levels of circulating blood coagulation proteins [fibrinogen, factor V, VIII, IX, and X], and increased concentrations of fibrin or fibrinogen, or both, degradation products.63
It was reported that a long duration of thrombocytosis was a major risk factor for thrombosis.64 The prevalence of thrombocytosis was 11.4% in gastric cancer,65 32% in lung cancer,66 and 8% in rectal cancer,67 and predict worse prognosis in non-small cell lung cancer (NSCLC)68 and gastric cancer.69 Also, high plasma fibrinogen levels (≥400 mg/dL) were another important risk factor for thrombosis, which were found in 45.8% (55/120) of small-cell lung cancer,70 in 34% (34/100) of ovarian cancer,71 and in 56.7% (68/120) of renal cell carcinoma (levels ≥343 mg/dL).72 High plasma fibrinogen was also considered as a poor prognosis factor and predicted shorter survival.
In advanced pancreatic cancer, VTE represents a frequent complication, with an estimated incidence between 12.1% and 42%. Symptomatic VTE was an independent risk factor for death.6 Many factors associated with thrombosis had existed in patients with pancreatic cancer without VTE. Thrombocytosis was reported in 15.2% (19/125) of pancreatic cancer.73 It was higher in patients with pancreatic cancer with T3 and T4 tumor than in patients with T1–2 tumors.74 Furthermore, a higher concentration of platelets was associated with a worse prognosis and a high risk of developing distant organ metastasis in pancreatic cancer.73,75 The other factor associated with thrombus, high plasma fibrinogen level, was found in 32.8% (39/119) of postoperative patients with resectable pancreatic cancer and in 24.8% (31/125) of patients with stage I–IV disease.73 Similarly, plasma fibrinogen levels ≥400 mg/dL were independent predictors of poor progression-free survival (PFS) and OS in locally advanced pancreatic adenocarcinoma.73,76 Notably, both platelet and fibrinogen levels were simultaneously and significantly elevated in the plasma of patients with pancreatic cancer. There was also a significant correlation between higher fibrinogen/platelet levels and distant organ metastasis (p < 0.05, respectively).73 Therefore, we suggested that high levels of fibrinogen and platelets in patients with pancreatic cancer would help the CTCs anchor and adhere to the vascular wall, promoting the formation of blood clots with cancer cells, and thus resulting in distant metastasis.
Tumor Cells Anchor/Adhere Though Platelets and Fibrinogen
Following their entrance into the circulation, CTCs must adhere to the microvasculature of a target organ prior to growth.77 Obviously, adhesion or anchoring to the microvasculature is the key step in hematogenous metastasis. Platelets express a number of cell-surface receptors for adhesion and aggregation, including the glycoprotein (GP) Ib-IX-V complex, which serves as a receptor for von Willebrand factor (vWF), and GP IIb-IIIa integrin, which binds to fibrinogen and fibronectin.78,79 They might also contribute to the physical interaction between CTCs and vascular endothelial cells by supporting the stable adhesion to endothelium and/or transmigration of tumor cells out of the vasculature.
The molecular coordination between platelets and tumor cells adhered to the vascular wall, which supported metastasis from the bloodstream, is well described.80 The platelet membrane glycoprotein (GP) αIIbß3 complex,81 GP αIb,82 and GP VI83 facilitate platelet adhesion and aggregation to tumor cells, thus promoting metastatic seeding. P-selectin, a platelet surface protein, is known to bind to CD24 on tumor cells, causing their adherence to endothelial cells, and mediate adhesion of leukocytes, platelets and cancer cells in inflammation, thrombosis, cancer growth and metastasis.84,85 P-selectin-mediated adhesion of cancer cells to immobilized platelets under dynamic flow conditions86 and immobilized platelets support cancer cell tethering, rolling, and firm adhesion under dynamic flow conditions via P-selectin and GPαIIbß3.87
However, in pancreatic cancer, there are still many opportunities for research into the role of platelets. Platelets adhere to vWF through the platelet receptor GPIb/IX/V complex bound to endothelial cells. This interaction is responsible for the tethering, rolling, and activation of platelets that eventually become firmly adhered, leading to thrombus formation.88 Recently, a novel molecule, vimentin, was found to be expressed on the platelet surface and serve as an adhesive receptor for vWF.89 In pancreatic cancer, there was a 3-fold increase in vimentin expression compared with that in other tumors, and a more specific antigenic isoform of vimentin was found at 5–10 fold higher levels in pancreatic cancer.90 Therefore, we hypothesized that the pancreatic cancer cells in circulating blood were also combined with the vWF molecule by vimentin. This suggested that platelets enriched around circulating pancreatic cancer cells because vWF molecule captured CTCs to adhere to the capillary wall, as well as captured the platelets. Pancreatic cancer cells would tightly adhere together by the platelets that were around them, making it easier to form a thrombus. Meanwhile, platelets form a protective network, which impedes natural killer cell-mediated elimination of tumor cells.91
Fibrinogen acts as a “molecular bridge” between specific receptors on tumor cells and the vascular endothelium or adherent platelets and leukocytes to promote cancer cell metastasis. Although fibrinogen deficiency was found to have no effect on the time required for the formation of palpable tumors and tumor growth, it markedly reduced the incidence of spontaneous macroscopic metastases in the lung and regional lymph nodes.92 Fibrinogen appears to facilitate metastasis by enhancing the sustained adherence and survival of individual tumor cell emboli in the vasculature of target organs.93
It has been demonstrated that fibrinogen binding to intercellular adhesion molecule-1 (ICAM-1) on endothelial cells could mediate the attachment of leukocytes and platelets.94 Tumor-derived ICAM-1 serves as an important docking point for tumor infiltration of immune cells that functionally promote pancreatic cancer cell metastasis.95 Interestingly, expression of ICAM-1 was increased by five-fold in surgical specimens from patients with pancreatic cancer compared with healthy controls, and this increased level of ICAM-1 correlates with increased nodal metastasis, advanced tumor stage, and shorter survival time.96 However, the normal human pancreas expresses low levels of ICAM-1.97 This indicated that ICAM-1 might be a promising target for therapy and could play an important role in pancreatic cancer progression and metastasis. Therefore, we postulated that high fibrinogen levers in patients with pancreatic cancer might promote CTC adhesion and immobilization to the endothelium through ICAM-1.
In summary, a high concentration of platelets and fibrinogen might work together to help the adhesion of CTCs to the vascular wall in pancreatic cancer, which is shown in a diagram in Figure 2.
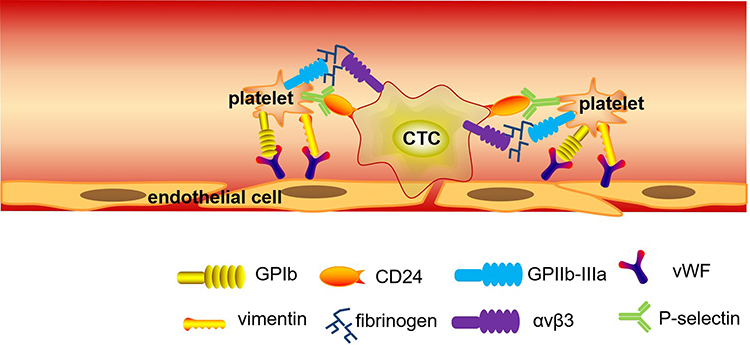 |
Figure 2 The diagram of the aberrant coagulation system associated with the anchoring and adherence in pancreatic cancer. In this system, platelets express a number of cell-surface receptors for adhesion and aggregation, including the GP Ib-IX-V complex and vimentin for vWF binding, the GP IIb-IIIa integrin for binding to fibrinogen and fibronectin, and the P-selectin for binding to CD24 on CTCs, supporting their adherence to endothelial cells, thus causing thrombosis and metastasis in pancreatic cancer; Fibrinogen acts as a “molecular bridge” between specific receptors on CTCs and the vascular endothelium or adherent platelets and leukocytes to promote cell metastasis. |
Platelets and Immune Escape in Pancreatic Cancer
Tumor cell adhesion via platelet receptors leads to platelet activation and release of secondary mediators to create a positive feedback activation mechanism. Once activated, platelets change shape and degranulate to release high local concentrations of soluble factors, including transforming growth factor-β (TGF-β), platelet-activating factor (PAF), histamine, 5-HT, and serotonin, which act as signals for target cells, including cells of the innate and adaptive immune systems.98 Lam et al recently reviewed the interaction of platelets with other immune cells.99
A previous study demonstrated that TGF-β from platelets diminished natural killer (NK) cell granule mobilization, downregulated NKG2D receptor on the NK cells, and interferon-γ (IFN-γ) secretion.100 Moreover, tumor cell coating by platelets might cause the transfer of MHC class I onto the tumor cell surface, resulting in impaired cytotoxicity and IFN-γ production by NK cells.101 In other words, downregulation of NKG2D in NK cells and the “pseudonormal” phenotype of platelet-coated tumor cells impairs NK cell tumor immune surveillance by disturbing “induced self” and “missing self” tumor cell recognition and elimination.102 Apart from NK cells, TGF-β from platelets could completely block T cell proliferation, blastogenesis, and IFN-γ production. T cell immunity against cancer was subverted by platelets via the glycoprotein A repetitions predominant (GARP)-TGF-β axis. Furthermore, anti-platelet agents can enhance the anti-tumor effect of immunotherapy with adoptive T cell transfer.103
Lam recently reviewed the interaction of platelets with neutrophils.99 A previous study showed that activated platelets present high mobility group box 1(HMGB1) to neutrophils, promoting the extrusion of neutrophil extracellular traps (NETs),104 which was able to trap CTCs, thus promoting early adhesion of tumor cells to distant organ sites in a lung cancer model.105 Although platelet-mediated tumor immune escape has been demonstrated in many tumor models, little research has been done in pancreatic cancer. Recently, Boone et al showed that NETs could be detected in both human and murine pancreatic tumors,106 suggesting that infiltrating neutrophils release NETs into the tumor microenvironment. Genetic ablation of the receptor for advanced glycation end products (RAGE) resulted in a decreased propensity for the NET formation and decreased serum DNA (one of the compositions of NETs). RAGE is a class III MHC protein receptor and mediates platelet–neutrophil interactions, which is increasingly expressed on the surface of activated platelets. HMGB1 binds to activated platelets via RAGE in platelet-rich human coronary artery thrombi.107 Interestingly, circulating HMGB1 is elevated in pancreatic cancer, which has been shown to stimulate NET formation;108,109 therefore, we hypothesized that NETs might be found in the blood system, where they trap cancer cells to protect them from NK cells, thus promoting metastasis in patients with pancreatic cancer.
In addition, fibrinogen helps platelets to adhere to tumor cells, and platelets in turn promote more fibrinogen to aggregate around tumor cells by forming thrombin. Their mutual facilitation protects tumor cells from NK cytotoxicity.91,110 However, the exact mechanisms of how platelet activation and fibrin clotting mediates immune escape still need to be defined in pancreatic cancer.
Prevention and Therapy of Cancer Targeting the Factors in Thrombosis or Fibrinolysis
As it is well known, classical anti-platelet/anti-thrombotic agents include aspirin and low-molecular-weight heparin (LMWH). Currently, LMWH was suggested as the standard of care. The American Society of Clinical Oncology (ASCO) recommendations propose unfractionated heparin, fondaparinux or LMWH as a first-line treatment, unless contraindicated due to high bleeding risk or active bleeding.111 However, little clinical information is available regarding other anti-platelet agents in cancer, but some of them may be beneficial for cancer patients. In other words, these drugs are in the preclinical stage or different phases of clinical trials, and part of them may be available in the market in the near future. Studies pointed out that targeting specific molecules on platelet membranes, such as protease-activated receptor 4 (PAR-4),112 glycoprotein (GP) GPIIb-GPIIIa complex,81,113–115 the GPIb-V-IX complex,116 GPVI,83 integrins α6ß1 and αIIbß3,117 and p-selectin,118,119 showed a significant experimental anti-metastatic effect. Interestingly, these molecules all mediated the characteristic of platelet adhesion, which further confirmed the importance of platelet adhesion in tumor cell metastasis. In Heyu Ni group’s review, P-selectin inhibitors, GPVI and GPIbα antagonists are under development.120 Other emerging anti-platelet agents include those targeting platelet-activating receptors and inhibitory receptors, anti-coagulants and plant-based food products (eg anthocyanins) may also have anti-tumor effects. Interestingly, Heyu Ni et al comprehensively characterized these new agents that may affect tumor metastasis and tumorigenesis in Table 1, which will certainly advance our knowledge for tumor treatment.120 It is also possible that platelet inhibitors could have other antitumor mechanisms, such as blocking angiogenesis and immune-suppressive prostaglandins,121 which would contribute to their antitumor activity. Another review paper by Nikolaos Arkadopoulos’s group highlights the family of Novel Oral Anticoagulants (NOACs), including dabigatran etexilate, rivaroxaban, apixaban and edoxaban, each one with their own special pharmacokinetics and pharmacodynamics, which were detailedly summarized in Table 3.111 However, the use of NOACs for VTE prophylaxis is certainly debatable. The potential drug interactions with chemotherapeutic components, GI abnormalities, and hepatic and renal insufficiency remain significant determinants of NOACs administration,122–124 which indicate NOACs’ limited bioavailability and the main reason not carried out in clinical. Here, we will highlight aspirin and LMWH, whose molecular mechanisms, efficiency and benefits for patients have been well discussed.
Aspirin
Aspirin, an antithrombotic drug, has become the focus of intense research on cancer prevention and outcome. Some of these studies have shown a favorable effect, while others failed to find any significant decrease in cancer incidence. These studies have been reviewed.125 Furthermore, regular use of aspirin reduces the long-term risk of several cancers and the risk of distant metastasis.126
In pancreatic cancer, evidence from 15 clinical studies or analyses that aspirin has chemo-preventive effects is somewhat controversial: Five trials endorsed the protective effect of aspirin, one indicated a harmful effect, and the others revealed no significant correlation.127 Among the studies that showed a protective effect, all of them indicated that long-term aspirin use, or use for 5 to 10 years or more in the past, was associated with reduced risk of cancer incidence.128 Furthermore, this trend of decreasing risk with duration of use was evident.129 In addition, high-dose aspirin, rather than low-dose aspirin, might be associated with a decreased risk for pancreatic cancer, especially for Americans.130 Further efforts should be made to explore the chemo-preventive effects as well as the dose, frequency, and duration of aspirin use for better clinical practice. A previous meta-analysis of 20 studies conducted a decade ago estimated that individuals with diabetes have a two-fold greater relative risk (RR) of pancreatic cancer compared with individuals without diabetes.131 Therefore, the combination of metformin and aspirin could provide additive and possibly synergistic effects for the prevention and treatment of pancreatic cancer.132,133 Prospective clinical trials are urgently required, whose results might form a milestone for the prevention of pancreatic cancer in patients with diabetes.
The therapeutic effects of aspirin in pancreatic cancer involve multiple molecular targets, including: suppressing sustained cell survival and proliferation (eg, via COX, IKBKB, and PRKA), attenuating genome instability and mutations, regulating the cancer immune and inflammatory profile, anti-tumor angiogenesis, and impeding the accommodation of cancer cells to the microenvironment. An animal study showed that aspirin strongly inhibited the formation of metastasis via inhibition of αvβ1/β3 integrins and p-selectin in an orthotopic model of pancreatic cancer.134 Inhibition of platelets by aspirin could affect their ability to induce cell proliferation through the modulation of the c-MYC oncoprotein in pancreatic cancer.135 Furthermore, activated platelet-derived TGF-β1 resulted in a reduction of cisplatin sensitivity in pancreatic cancer cells through stimulating PI3K/AKT and MEK/ERK signaling.136 This suggested that inhibition of platelet activation could reverse the resistance to cisplatin in pancreatic cancer.
Low-Molecular Weight Heparin (LMWH)
In 1992, Green et al found that LMWH could reduce mortality in patients with cancer.137 Thereafter, a number of data showed the potential anti-metastatic activity of heparin and LMWH via in vitro and in vivo studies.138–143 These in vitro studies showed that heparin or LMWH treatment attenuated tumor metastasis in mice by inhibiting P-selectin-mediated interactions of platelets with carcinoma cell-surface mucin ligands, which reinforced the importance of adherence between the cancer cells and platelets.
In patients with advanced pancreatic cancer, the in vitro study showed that dalteparin could decrease serum-mediated induction of cancer cell invasion.144 Sulfated non-anticoagulant heparins (S-NACHs) distinctly increased tumor necrosis, enhanced gemcitabine response in mouse pancreatic cancer models,145 and inhibited pancreatic cancer cell adhesion and metastasis in human umbilical cord vessel segments and in a mouse model.146 Clinical data showed that nadroparin147 and dalteparin144 could prevent thromboembolic events in patients with advanced pancreatic cancer receiving chemotherapy;144 however, the survival data were not analyzed.
von Delius et al reported that treatment with LMWH did not produce a survival difference in 213 patients with advanced pancreatic adenocarcinoma. However, in the sub-group with metastatic pancreatic adenocarcinoma, treatment with LMWH was a survival advantage for patients. The median survival was 6.6 months and 3.8 months for the LMWH group and the non-LMWH group, respectively.148 Another study, which involved 69 patients with advanced pancreatic cancer with a follow-up time of less than 25 months, showed that the addition of LMWH to chemotherapy significantly improved the response to the chemotherapy (response rate: 58.8% vs 12.1%, p = 0.0001) and prolonged survival (13.0 vs 5.5 months, p = 0.0001) patients with advanced pancreatic cancer.149 However, another prospective clinical study, which was conducted in 312 patients with pancreatic cancer, with a follow-up time up to 60 months, did not show a significant difference in progression-free survival (4.99 vs 5.42 months, p = 0.64) or overall survival (8.51 vs 8.02 months, p = 0.44) in these patients receiving chemotherapy together with LMWH or without LMWH. However, the overall cumulative incidence rate of symptomatic VTEs was lower in the patients with APC that received chemotherapy with LMWH than in those not receiving LMWH (6.4% vs 15.1%). The numbers of major bleeding events were similar between the two groups (7/160 vs 5/152, P = 1.0).150 Then, inconsistent survival data in these studies might be caused by differences between the number of enrolled patients and the follow-up duration.
One meta-analysis showed that the use of prophylactic LMWH did not have a discernible effect on overall survival in patients with solid malignancy without VTE.151 The result was confirmed by the most recent meta-analysis,152 which was the second update by Cochrane in 2016. It showed that primary thromboprophylaxis with LMWH significantly reduced the incidence of symptomatic VTE in patients with ambulatory cancer treated with chemotherapy. Therefore, LMWH as an agent to prevent metastasis and improve the survival duration requires further investigation.
Concluding Remarks
Increased attention has focused on the effect of activation of the fibrinolysis and coagulation systems in cancer. Our review showed that the fibrinolysis and coagulation systems were activated in patients with pancreatic cancer, manifested as high concentrations of t-PA, u-PA, D-dimer, fibrinogen, or platelets. These factors cooperate with each other and encourage cancer immune escape to achieve local invasion and distant metastasis. Prevention and treatment of cancer metastasis by the agents of aspirin and LMWH targeting molecules of these two systems have been well discussed. However, other novel anti-platelet/anti-thrombotic agents are still in the preclinical stage or different phases of clinical trials, but some of them may be beneficial for cancer patients and available in the market in the near future.
Funding
This work was supported by the Natural Science Foundation of China (No.81603566) and Natural Science Foundation of Jiangsu Province (No. BK20141034 and No. BK20201499), by peak academic talents plan in Jiangsu Province Hospital of Chinese Medicine (No. k2018yrc25) and by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Henley SJ, Ward EM, Scott S, et al. Annual report to the nation on the status of cancer, part I: national cancer statistics. Cancer. 2020;126(10):2225–2249. doi:10.1002/cncr.32802
2. Drouillard A, Manfredi S, Lepage C, Bouvier A-M. [Epidemiology of pancreatic cancer]. Bull Cancer. 2018;105(1):63–69. French. doi:10.1016/j.bulcan.2017.11.004
3. Donnellan E, Khorana AA. Cancer and venous thromboembolic disease: a review. Oncologist. 2017;22(2):199–207. doi:10.1634/theoncologist.2016-0214
4. Hisada Y, Mackman N. Cancer-associated pathways and biomarkers of venous thrombosis. Blood. 2017;130(13):1499–1506. doi:10.1182/blood-2017-03-743211
5. Sgouros J, Maraveyas A. Excess premature (3-month) mortality in advanced pancreatic cancer could be related to fatal vascular thromboembolic events. A hypothesis based on a systematic review of Phase III chemotherapy studies in advanced pancreatic cancer. Acta Oncol (Madr). 2008. doi:10.1080/02841860701687267
6. Kruger S, Haas M, Burkl C, et al. Incidence, outcome and risk stratification tools for venous thromboembolism in advanced pancreatic cancer - a retrospective cohort study. Thromb Res. 2017;157:9–15. doi:10.1016/j.thromres.2017.06.021
7. Lyman GH, Bohlke K, Khorana AA, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update 2014. J Clin Oncol. 2015;33(6):654–656. doi:10.1200/JCO.2014.59.7351
8. Cao J, Fu Z, Gao L, et al. Evaluation of serum D-dimer, fibrinogen, and CA19-9 for postoperative monitoring and survival prediction in resectable pancreatic carcinoma. World J Surg Oncol. 2017;1–8. doi:10.1186/s12957-017-1104-9
9. Kulic A, Cvetkovic Z, Libek V. Primary hyperfibrinolysis as the presenting sign of prostate cancer: a case report. Vojnosanit Pregl. 2016;73(9):877–880. doi:10.2298/VSP150525076K
10. Jafri MA, Cohen JV, Much MA, Petrylak DP, Podoltsev NA, Patient With A. Pancytopenia, intractable epistaxis, and metastatic prostate cancer: how correct diagnosis of primary hyperfibrinolysis helps to stop the bleeding. Clin Genitourin Cancer. 2016;14(5):e545–e548. doi:10.1016/j.clgc.2016.05.002
11. Naina HVK, Patnaik MM, Ali UA, Chen D, Ashrani AA. Systemic fibrinolysis caused by tissue plasminogen activator-producing metastatic breast cancer. J Clin Oncol. 2010;28(11):e167–e168. doi:10.1200/JCO.2009.25.2502
12. Baluka D, Urbanek T, Lekstan A, et al. The role of the tissue plasminogen activator as a prognostic and differentiation factor in patients with pancreatic cancer and chronic pancreatitis. J Physiol Pharmacol. 2016;67(1):93–101.
13. Huang C, Xie D, Cui J, Li Q, Gao Y, Xie K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin Cancer Res. 2014;20(6):1477–1488. doi:10.1158/1078-0432.CCR-13-2311
14. McMahon BJ, Kwaan HC. Components of the plasminogen-plasmin system as biologic markers for cancer. Adv Exp Med Biol. 2015;867:145–156.
15. Chen Q, Fei J, Wu L, et al. Detection of cathepsin B, cathepsin L, cystatin C, urokinase plasminogen activator and urokinase plasminogen activator receptor in the sera of lung cancer patients. Oncol Lett. 2011;2(4):693–699. doi:10.3892/ol.2011.302
16. Rubina KA, Sysoeva VY, Zagorujko EI, et al. Increased expression of uPA, uPAR, and PAI-1 in psoriatic skin and in basal cell carcinomas. Arch Dermatol Res. 2017;309(6):433–442. doi:10.1007/s00403-017-1738-z
17. Yang SF, Hsieh YS, Lin CL, et al. Increased plasma levels of urokinase plasminogen activator and matrix metalloproteinase-9 in nonsmall cell lung cancer patients. Clin Chim Acta. 2005;354(1–2):91–99. doi:10.1016/j.cccn.2004.11.011
18. Urban P, Vuaroqueaux V, Labuhn M, et al. Increased expression of urokinase-type plasminogen activator mRNA determines adverse prognosis in ErbB2-positive primary breast cancer. J Clin Oncol. 2006;24(26):4245–4253. doi:10.1200/JCO.2005.05.1912
19. Halamkova J, Kiss I, Pavlovsky Z, et al. Clinical significance of the plasminogen activator system in relation to grade of tumor and treatment response in colorectal carcinoma patients. Neoplasma. 2011;58(5):377–385. doi:10.4149/neo_2011_05_377
20. Liu Q, Li W, Yang S, Liu Z. High expression of uPA related to p38MAPK in esophageal cancer indicates poor prognosis. Onco Targets Ther. 2018;11:8427–8434. doi:10.2147/OTT.S181701
21. Ma J, Qi G, Xu J, et al. Overexpression of forkhead box M1 and urokinase-type plasminogen activator in gastric cancer is associated with cancer progression and poor prognosis. Oncol Lett. 2017;14(6):7288–7296. doi:10.3892/ol.2017.7136
22. Wang X, Wang N, Li H, et al. Up-regulation of PAI-1 and down-regulation of uPA are involved in suppression of invasiveness and motility of hepatocellular carcinoma cells by a natural compound berberine. Int J Mol Sci. 2016;17(4):577. doi:10.3390/ijms17040577
23. Kumano M, Miyake H, Muramaki M, Furukawa J, Takenaka A, Fujisawa M. Expression of urokinase-type plasminogen activator system in prostate cancer: correlation with clinicopathological outcomes in patients undergoing radical prostatectomy. Urol Oncol. 2009;27(2):180–186. doi:10.1016/j.urolonc.2008.01.012
24. Taubert H, Wurl P, Greither T, et al. Co-detection of members of the urokinase plasminogen activator system in tumour tissue and serum correlates with a poor prognosis for soft-tissue sarcoma patients. Br J Cancer. 2010;102(4):731–737. doi:10.1038/sj.bjc.6605520
25. Pavon MA, Arroyo-Solera I, Cespedes MV, Casanova I, Leon X, Mangues R. uPA/uPAR and SERPINE1 in head and neck cancer: role in tumor resistance, metastasis, prognosis and therapy. Oncotarget. 2016;7(35):57351–57366. doi:10.18632/oncotarget.10344
26. Nielsen A, Scarlett CJ, Samra JS, et al. Significant overexpression of urokinase-type plasminogen activator in pancreatic adenocarcinoma using real-time quantitative reverse transcription polymerase chain reaction. J Gastroenterol Hepatol. 2005;20:256–263. doi:10.1111/j.1400-1746.2004.03531.x
27. Shin SJ, Kim KO, Kim MK, et al. Expression of E-cadherin and uPA and their association with the prognosis of pancreatic cancer. Japan J Clin Oncol. 2005;35(6):342–348. doi:10.1093/jjco/hyi094
28. Wang W, Abbruzzese JL, Evans DB, Chiao PJ. Overexpression of urokinase-type plasminogen activator in pancreatic adenocarcinoma is regulated by constitutively activated RelA. Oncogene. 1999;18(32):4554–4563. doi:10.1038/sj.onc.1202833
29. Harvey SR, Hurd TC, Markus G, et al. Evaluation of urinary plasminogen activator, its receptor, matrix metalloproteinase-9, and von Willebrand factor in pancreatic cancer. Clin Cancer Res. 2003;9(13):4935–4943.
30. Gibbs JF, Schlieman M, Singh P, et al. A pilot study of urokinase-type plasminogen activator (uPA) overexpression in the brush cytology of patients with malignant pancreatic or biliary strictures. HPB Surg. 2009;2009:1–5. doi:10.1155/2009/805971
31. Takeuchi Y, Nakao A, Harada A, Nonami T, Fukatsu T, Takagi H. Expression of plasminogen activators and their inhibitors in human pancreatic carcinoma: immunohistochemical study. Am J Gastroenterol. 1993;88(11):1928–1933.
32. Boss EA, Massuger LFAG, Thomas CMG, et al. Clinical value of components of the plasminogen activation system in ovarian cyst fluid. Anticancer Res. 2002;22(1A):275–282.
33. Shim BS, Kang BH, Hong YK, et al. The kringle domain of tissue-type plasminogen activator inhibits in vivo tumor growth. Biochem Biophys Res Commun. 2005;327(4):1155–1162. doi:10.1016/j.bbrc.2004.12.126
34. Grebenchtchikov N, Maguire TM, Riisbro R, et al. Measurement of plasminogen activator system components in plasma and tumor tissue extracts obtained from patients with breast cancer: an EORTC Receptor and Biomarker Group collaboration. Oncol Rep. 2005;14(1):235–239.
35. Sciacca FL, Ciusani E, Silvani A, et al. Genetic and plasma markers of venous thromboembolism in patients with high grade glioma. Clin Cancer Res. 2004;10(4):1312–1317. doi:10.1158/1078-0432.ccr-03-0198
36. De Petro G, Tavian D, Copeta A, Portolani N, Giulini SM, Barlati S. Expression of urokinase-type plasminogen activator (u-PA), u-PA receptor, and tissue-type PA messenger RNAs in human hepatocellular carcinoma. Cancer Res. 1998;58(10):2234–2239.
37. Wojtukiewicz MZ, Sierko E, Zacharski LR, Rózanska-Kudelska M, Zimnoch L. Occurrence of components of fibrinolytic pathways in situ in laryngeal cancer. Semin Thromb Hemost. 2003;29(3):317–320. doi:10.1055/s-2003-40970
38. Collen D, Lijnen HR. The tissue-type plasminogen activator story. Arterioscler Thromb Vasc Biol. 2009;29(8):1151–1155. doi:10.1161/ATVBAHA.108.179655
39. Marignol L, Robson T, McCarthy HO, et al. The tissue plasminogen activator gene promoter: a novel tool for radiogenic gene therapy of the prostate? J Gene Med. 2008;10(9):1032–1038. doi:10.1002/jgm.1221
40. Sanz L, Vizoso F, Vérez P, et al. Prognostic significance of tissue-type plasminogen activator (tPA) content in gastric cancer and surrounding mucosa. Int J Biol Markers. 2002;17(3):169–176. doi:10.1177/172460080201700305
41. Neudecker J, Neudecker BA, Raue W, Stern R, Schwenk W. Hyaluronan levels during laparoscopic versus open colonic resections. Surg Endosc. 2008;22(3):660–663. doi:10.1007/s00464-007-9455-x
42. Chappuis PO, Dieterich B, Sciretta V, et al. Functional evaluation of plasmin formation in primary breast cancer. J Clin Oncol. 2001;19(10):2731–2738. doi:10.1200/JCO.2001.19.10.2731
43. Corte MD, Vérez P, Rodríguez JC, et al. Tissue-type plasminogen activator (tPA) in breast cancer: relationship with clinicopathological parameters and prognostic significance. Breast Cancer Res Treat. 2005;90(1):33–40. doi:10.1007/s10549-004-2624-x
44. Diaz VM, Planaguma J, Thomson TM, Reventos J, Paciucci R. Tissue plasminogen activator is required for the growth, invasion, and angiogenesis of pancreatic tumor cells. Gastroenterology. 2002;122(3):806–819. doi:10.1053/gast.2002.31885
45. Diaz VM, Hurtado M, Thomson TM, Reventos J, Paciucci R. Specific interaction of tissue-type plasminogen activator (t-PA) with annexin II on the membrane of pancreatic cancer cells activates plasminogen and promotes invasion in vitro. Gut. 2004;53(7):993–1000. doi:10.1136/gut.2003.026831
46. Nielsen VG, Matika RW, Ley MLB, et al. Tissue-type plasminogen activator-induced fibrinolysis is enhanced in patients with breast, lung, pancreas and colon cancer. Blood Coagul Fibrinolysis. 2014;25(3):248–253. doi:10.1097/MBC.0000000000000040
47. Gao F, Li J, Quan S, et al. Risk factors and treatment for hemorrhage after pancreaticoduodenectomy: a case series of 423 patients. Biomed Res Int. 2016;2016:2815693. doi:10.1155/2016/2815693
48. Uramoto H, Shimokawa H, Tanaka F. Postoperative bleeding after surgery in patients with lung cancer. Anticancer Res. 2014;34(2):981–984.
49. Deryugina EI, Quigley JP. Cell surface remodeling by plasmin: a new function for an old enzyme. J Biomed Biotechnol. 2012;2012:564259. doi:10.1155/2012/564259
50. Kwaan HC, Mazar AP, McMahon BJ. The apparent uPA/PAI-1 paradox in cancer: more than meets the eye. Semin Thromb Hemost. 2013;39(4):382–391. doi:10.1055/s-0033-1338127
51. Kwaan HC, McMahon B. The role of plasminogen-plasmin system in cancer. Cancer Treat Res. 2009;148:43–66. doi:10.1007/978-0-387-79962-9_4
52. Houard X, Monnot C, Dive V, Corvol P, Pagano M. Vascular smooth muscle cells efficiently activate a new proteinase cascade involving plasminogen and fibronectin. J Cell Biochem. 2003;88(6):1188–1201. doi:10.1002/jcb.10460
53. Diao D, Cheng Y, Song Y, Zhang H, Zhou Z, Dang C. D-dimer is an essential accompaniment of circulating tumor cells in gastric cancer. BMC Cancer. 2017;17(1):56. doi:10.1186/s12885-016-3043-1
54. Mego M, Zuo Z, Gao H, et al. Circulating tumour cells are linked to plasma D-dimer levels in patients with metastatic breast cancer. Thromb Haemost. 2015;113(3):593–598. doi:10.1160/TH14-07-0597
55. Mego M, Karaba M, Minarik G, et al. Relationship between circulating tumor cells, blood coagulation, and urokinase-plasminogen-activator system in early breast cancer patients. Breast J. 2015;21(2):155–160. doi:10.1111/tbj.12388
56. Stender MT, Larsen AC, Sall M, Thorlacius-Ussing O. D-dimer predicts prognosis and non-resectability in patients with pancreatic cancer: a prospective cohort study. Blood Coagul Fibrinolysis. 2016;27(5):597–601. doi:10.1097/MBC.0000000000000559
57. Vishwanatha JK, Chiang Y, Kumble KD, Hollingsworth MA, Pour PM. Enhanced expression of annexin II in human pancreatic carcinoma cells and primary pancreatic cancers. Carcinogenesis. 1993;14(12):2575–2579. doi:10.1093/carcin/14.12.2575
58. Cesarman GM, Guevara CA, Hajjar KA. An endothelial cell receptor for plasminogen/tissue plasminogen activator (t-PA). II. Annexin II-mediated enhancement of t-PA-dependent plasminogen activation. J Biol Chem. 1994;269(33):21198–21203.
59. Hajjar KA, Jacovina AT, Chacko J. An endothelial cell receptor for plasminogen/tissue plasminogen activator. I. Identity with annexin II. J Biol Chem. 1994;269(33):21191–21197.
60. Sidenius N, Blasi F. The urokinase plasminogen activator system in cancer: recent advances and implication for prognosis and therapy. Cancer Metastasis Rev. 2003;22(2–3):205–222. doi:10.1023/a:1023099415940
61. Gorantla B, Asuthkar S, Rao JS, Patel J, Gondi CS. Suppression of the uPAR-uPA system retards angiogenesis, invasion, and in vivo tumor development in pancreatic cancer cells. Mol Cancer Res. 2011;9(4):377–389. doi:10.1158/1541-7786.MCR-10-0452
62. DiPardo BJ, Winograd P, Court CM, Tomlinson JS. Pancreatic cancer circulating tumor cells: applications for personalized oncology. Expert Rev Mol Diagn. 2018;18(9):809–820. doi:10.1080/14737159.2018.1511429
63. Zwicker JI, Furie BC, Furie B. Cancer-associated thrombosis. Crit Rev Oncol Hematol. 2007;62(2):126–136. doi:10.1016/j.critrevonc.2007.01.001
64. Vannucchi AM, Barbui T. Thrombocytosis and thrombosis. Hematology Am Soc Hematol Educ Program. 2007;2007:363–370. doi:10.1182/asheducation-2007.1.363
65. Hwang SG, Kim KM, Cheong JH, et al. Impact of pretreatment thrombocytosis on blood-borne metastasis and prognosis of gastric cancer. Eur J Surg Oncol. 2012;38(7):562–567. doi:10.1016/j.ejso.2012.04.009
66. Pedersen LM, Milman N. Prognostic significance of thrombocytosis in patients with primary lung cancer. Eur Respir J. 1996;9(9):1826–1830. doi:10.1183/09031936.96.09091826
67. Cravioto-Villanueva A, Luna-Perez P, Gutierrez-de la Barrera M, et al. Thrombocytosis as a predictor of distant recurrence in patients with rectal cancer. Arch Med Res. 2012;43(4):305–311. doi:10.1016/j.arcmed.2012.06.008
68. Zhu J-F, Cai L, Zhang X-W, et al. High plasma fibrinogen concentration and platelet count unfavorably impact survival in non-small cell lung cancer patients with brain metastases. Chin J Cancer. 2014;33(2):96–104. doi:10.5732/cjc.012.10307
69. Yamamoto M, Kurokawa Y, Miyazaki Y, et al. Usefulness of preoperative plasma fibrinogen versus other prognostic markers for predicting gastric cancer recurrence. World J Surg. 2016;40(8):1904–1909. doi:10.1007/s00268-016-3474-5
70. Fan S, Guan Y, Zhao G, An G. Association between plasma fibrinogen and survival in patients with small-cell lung carcinoma. Thorac Cancer. 2018;9(1):146–151. doi:10.1111/1759-7714.12556
71. Man Y-N, Wang Y-N, Hao J, et al. Pretreatment plasma D-dimer, fibrinogen, and platelet levels significantly impact prognosis in patients with epithelial ovarian cancer independently of venous thromboembolism. Int J Gynecol Cancer. 2015;25(1):24–32. doi:10.1097/IGC.0000000000000303
72. Erdem S, Amasyali AS, Aytac O, Onem K, Issever H, Sanli O. Increased preoperative levels of plasma fibrinogen and D dimer in patients with renal cell carcinoma is associated with poor survival and adverse tumor characteristics. Urol Oncol. 2014;32(7):1031–1040. doi:10.1016/j.urolonc.2014.03.013
73. Wang H, Gao J, Bai M, et al. The pretreatment platelet and plasma fibrinogen level correlate with tumor progression and metastasis in patients with pancreatic cancer. Platelets. 2014;25(5):382–387. doi:10.3109/09537104.2013.827782
74. Liu P, Zhu Y, Liu L. Elevated pretreatment plasma D-dimer levels and platelet counts predict poor prognosis in pancreatic adenocarcinoma. Onco Targets Ther. 2015;8:1335–1340. doi:10.2147/OTT.S82329
75. Guo Q, Zhang B, Dong X, et al. Elevated levels of plasma fibrinogen in patients with pancreatic cancer: possible role of a distant metastasis predictor. Pancreas. 2009;38(3):e75–e79. doi:10.1097/MPA.0b013e3181987d86
76. Kurahara H, Maemura K, Mataki Y, et al. Prognostication by inflammation-based score in patients with locally advanced pancreatic cancer treated with chemoradiotherapy. Pancreatology. 2015;15(6):688–693. doi:10.1016/j.pan.2015.09.015
77. Al-Mehdi AB, Tozawa K, Fisher AB, Shientag L, Lee A, Muschel RJ. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat Med. 2000;6(1):100–102. doi:10.1038/71429
78. Jurk K, Kehrel BE. Platelets: physiology and biochemistry. Semin Thromb Hemost. 2005;31(4):381–392. doi:10.1055/s-2005-916671
79. Jackson SP. Arterial thrombosis–insidious, unpredictable and deadly. Nat Med. 2011;17(11):1423–1436. doi:10.1038/nm.2515
80. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11(2):123–134. doi:10.1038/nrc3004
81. Karpatkin S, Pearlstein E, Ambrogio C, Coller BS. Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J Clin Invest. 1988;81(4):1012–1019. doi:10.1172/JCI113411
82. Jain S, Zuka M, Liu J, et al. Platelet glycoprotein Ib alpha supports experimental lung metastasis. Proc Natl Acad Sci U S A. 2007;104(21):9024–9028. doi:10.1073/pnas.0700625104
83. Jain S, Russell S, Ware J. Platelet glycoprotein VI facilitates experimental lung metastasis in syngenic mouse models. J Thromb Haemost. 2009;7(10):1713–1717. doi:10.1111/j.1538-7836.2009.03559.x
84. Kim YJ, Borsig L, Varki NM, Varki A. P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci U S A. 1998;95(16):9325–9330. doi:10.1073/pnas.95.16.9325
85. Chen M, Geng JG. P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch Immunol Ther Exp (Warsz). 2006;54(2):75–84. doi:10.1007/s00005-006-0010-6
86. Wei M, Tai G, Gao Y, et al. Modified heparin inhibits P-selectin-mediated cell adhesion of human colon carcinoma cells to immobilized platelets under dynamic flow conditions. J Biol Chem. 2004;279(28):29202–29210. doi:10.1074/jbc.M312951200
87. McCarty OJ, Mousa SA, Bray PF, Konstantopoulos K. Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood. 2000;96(5):1789–1797. doi:10.1182/blood.V96.5.1789
88. Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28(3):403–412. doi:10.1161/ATVBAHA.107.150474
89. Da Q, Behymer M, Correa JI, Vijayan KV, Cruz MA. Platelet adhesion involves a novel interaction between vimentin and von Willebrand factor under high shear stress. Blood. 2014;123(17):2715–2721. doi:10.1182/blood-2013-10-530428
90. Hong S-H, Misek DE, Wang H, et al. Identification of a specific vimentin isoform that induces an antibody response in pancreatic cancer. Biomark Insights. 2006;1:175–183. doi:10.1177/117727190600100006
91. Palumbo JS, Talmage KE, Massari JV, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005;105(1):178–185. doi:10.1182/blood-2004-06-2272
92. Palumbo JS, Degen JL. Fibrinogen and tumor cell metastasis. Haemostasis. 2001;31 Suppl 1(Suppl 1):11–15.
93. Palumbo JS, Potter JM, Kaplan LS, Talmage K, Jackson DG, Degen JL. Spontaneous hematogenous and lymphatic metastasis, but not primary tumor growth or angiogenesis, is diminished in fibrinogen-deficient mice. Cancer Res. 2002;62(23):6966–6972.
94. Tsakadze NL, Zhao Z, D’Souza SE. Interactions of intercellular adhesion molecule-1 with fibrinogen. Trends Cardiovasc Med. 2002;12(3):101–108. doi:10.1016/S1050-1738(01)00157-8
95. Roland CL, Dineen SP, Toombs JE, et al. Tumor-derived intercellular adhesion molecule-1 mediates tumor-associated leukocyte infiltration in orthotopic pancreatic xenografts. Exp Biol Med (Maywood). 2010;235(2):263–270. doi:10.1258/ebm.2009.009215
96. Tempia-Caliera AA, Horvath LZ, Zimmermann A, et al. Adhesion molecules in human pancreatic cancer. J Surg Oncol. 2002;79(2):93–100. doi:10.1002/jso.10053
97. Shimoyama S, Gansauge F, Gansauge S, Kaminishi M, Beger HG. Basal expression and cytokine induction of intercellular adhesion molecule-1 in human pancreatic cancer cell lines. J Exp Clin Cancer Res. 1999;18(1):107–110.
98. Weyrich AS, Zimmerman GA. Platelets: signaling cells in the immune continuum. Trends Immunol. 2004;25(9):489–495. doi:10.1016/j.it.2004.07.003
99. Lam FW, Vijayan KV, Rumbaut RE. Platelets and their interactions with other immune cells. Compr Physiol. 2015;5(3):1265–1280. doi:10.1002/cphy.c140074
100. Kopp H-G, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009;69(19):7775–7783. doi:10.1158/0008-5472.CAN-09-2123
101. Placke T, Örgel M, Schaller M, et al. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012;72(2):440–448. doi:10.1158/0008-5472.CAN-11-1872
102. Placke T, Kopp H-G, Salih HR. Modulation of natural killer cell anti-tumor reactivity by platelets. J Innate Immun. 2011;3(4):374–382. doi:10.1159/000323936
103. Rachidi S, Metelli A, Riesenberg B, et al. Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci Immunol. 2017;2(11):eaai7911. doi:10.1126/sciimmunol.aai7911
104. Maugeri N, Campana L, Gavina M, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12(12):2074–2088. doi:10.1111/jth.12710
105. Cools-Lartigue J, Spicer J, McDonald B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123(8):3446–3458. doi:10.1172/JCI67484
106. Boone BA, Orlichenko L, Schapiro NE, et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 2015;22(6):326–334. doi:10.1038/cgt.2015.21
107. Ahrens I, Chen Y-C, Topcic D, et al. HMGB1 binds to activated platelets via the receptor for advanced glycation end products and is present in platelet rich human coronary artery thrombi. Thromb Haemost. 2015;114(11):994–1003. doi:10.1160/TH14-12-1073
108. Chung HW, Lim J-B, Jang S, Lee KJ, Park KH, Song SY. Serum high mobility group box-1 is a powerful diagnostic and prognostic biomarker for pancreatic ductal adenocarcinoma. Cancer Sci. 2012;103(9):1714–1721. doi:10.1111/j.1349-7006.2012.02358.x
109. Wittwer C, Boeck S, Heinemann V, et al. Circulating nucleosomes and immunogenic cell death markers HMGB1, sRAGE and DNAse in patients with advanced pancreatic cancer undergoing chemotherapy. Int J Cancer. 2013;133(11):2619–2630. doi:10.1002/ijc.28294
110. Zheng S, Shen J, Jiao Y, et al. Platelets and fibrinogen facilitate each other in protecting tumor cells from natural killer cytotoxicity. Cancer Sci. 2009;100(5):859–865. doi:10.1111/j.1349-7006.2009.01115.x
111. Mastoraki A, Mastoraki S, Schizas D, et al. Facing the challenge of venous thromboembolism prevention in patients undergoing major abdominal surgical procedures for gastrointestinal cancer. World J Gastrointest Oncol. 2018;10(10):328–335. doi:10.4251/wjgo.v10.i10.328
112. Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. 2004;104(2):397–401. doi:10.1182/blood-2004-02-0434
113. Amirkhosravi A, Mousa SA, Amaya M, et al. Inhibition of tumor cell-induced platelet aggregation and lung metastasis by the oral GpIIb/IIIa antagonist XV454. Thromb Haemost. 2003;90(09):549–554. doi:10.1160/TH03-02-0102
114. Zhang W, Dang S, Hong T, et al. A humanized single-chain antibody against beta 3 integrin inhibits pulmonary metastasis by preferentially fragmenting activated platelets in the tumor microenvironment. Blood. 2012;120(14):2889–2898. doi:10.1182/blood-2012-04-425207
115. Bakewell SJ, Nestor P, Prasad S, et al. Platelet and osteoclast beta3 integrins are critical for bone metastasis. Proc Natl Acad Sci U S A. 2003;100(24):14205–14210. doi:10.1073/pnas.2234372100
116. Erpenbeck L, Nieswandt B, Schön M, Pozgajova M, Schön MP. Inhibition of platelet GPIb alpha and promotion of melanoma metastasis. J Invest Dermatol. 2010;130(2):576–586. doi:10.1038/jid.2009.278
117. Lavergne M, Janus-Bell E, Schaff M, Gachet C, Mangin PH. Platelet integrins in tumor metastasis: do they represent a therapeutic target? Cancers (Basel). 2017;9(10):133. doi:10.3390/cancers9100133
118. Qi C-L, Wei B, Ye J, et al. P-selectin-mediated platelet adhesion promotes the metastasis of murine melanoma cells. PLoS One. 2014;9(3):e91320. doi:10.1371/journal.pone.0091320
119. Qi C, Wei B, Zhou W, et al. P-selectin-mediated platelet adhesion promotes tumor growth. Oncotarget. 2015;6(9):6584–6596. doi:10.18632/oncotarget.3164
120. Xu XR, Yousef GM, Ni H. Cancer and platelet crosstalk: opportunities and challenges for aspirin and other antiplatelet agents. Blood. 2018;131(16):1777–1789.
121. Zelenay S, van der Veen AG, Böttcher JP, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162(6):1257–1270. doi:10.1016/j.cell.2015.08.015
122. Alsubaie H, Leggett C, Lambert P, et al. Diagnosis of VTE postdischarge for major abdominal and pelvic oncologic surgery: implications for a change in practice. Can J Surg. 2015;58(5):305–311. doi:10.1503/cjs.012314
123. Cui G, Wang X, Yao W, Li H. Incidence of postoperative venous thromboembolism after laparoscopic versus open colorectal cancer surgery: a meta-analysis. Surg Laparosc Endosc Percutan Tech. 2013;23(2):128–134. doi:10.1097/SLE.0b013e3182827cef
124. Cheung HY, Chung CC, Yau KK, et al. Risk of deep vein thrombosis following laparoscopic rectosigmoid cancer resection in chinese patients. Asian J Surg. 2008;31(2):63–68. doi:10.1016/S1015-9584(08)60060-3
125. Gil-Bernabé AM, Lucotti S, Muschel RJ. Coagulation and metastasis: what does the experimental literature tell us? Br J Haematol. 2013;162(4):433–441. doi:10.1111/bjh.12381
126. Cuzick J, Thorat MA, Bosetti C, et al. Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann Oncol. 2015;26(1):47–57. doi:10.1093/annonc/mdu225
127. Jiang M-J, Dai -J-J, Gu D-N, Huang Q, Tian L. Aspirin in pancreatic cancer: chemopreventive effects and therapeutic potentials. Biochim Biophys Acta. 2016;1866(2):163–176. doi:10.1016/j.bbcan.2016.08.002
128. Streicher SA, Yu H, Lu L, Kidd MS, Risch HA. Case-control study of aspirin use and risk of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2014;23(7):1254–1263. doi:10.1158/1055-9965.EPI-13-1284
129. Risch HA, Lu L, Streicher SA, et al. Aspirin use and reduced risk of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2017;26(1):68–74. doi:10.1158/1055-9965.EPI-16-0508
130. Cui X-J, He Q, Zhang J-M, Fan H-J, Wen Z-F, Qin Y-R. High-dose aspirin consumption contributes to decreased risk for pancreatic cancer in a systematic review and meta-analysis. Pancreas. 2014;43(1):135–140. doi:10.1097/MPA.0b013e3182a8d41f
131. Everhart J, Wright D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA. 1995;273(20):1605–1609. doi:10.1001/jama.1995.03520440059037
132. Yue W, Yang CS, DiPaola RS, Tan X-L. Repurposing of metformin and aspirin by targeting AMPK-mTOR and inflammation for pancreatic cancer prevention and treatment. Cancer Prev Res (Phila). 2014;7(4):388–397. doi:10.1158/1940-6207.CAPR-13-0337
133. Yue W, Zheng X, Lin Y, et al. Metformin combined with aspirin significantly inhibit pancreatic cancer cell growth in vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and Bcl-2. Oncotarget. 2015;6(25):21208–21224. doi:10.18632/oncotarget.4126
134. Mezouar S, Darbousset R, Dignat-George F, Panicot-Dubois L, Dubois C. Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int J Cancer. 2015;136(2):462–475. doi:10.1002/ijc.28997
135. Mitrugno A, Sylman JL, Ngo ATP, et al. Aspirin therapy reduces the ability of platelets to promote colon and pancreatic cancer cell proliferation: implications for the oncoprotein c-MYC. Am J Physiol Cell Physiol. 2017;312(2):C176–C189. doi:10.1152/ajpcell.00196.2016
136. Chen H, Lan X, Liu M, Zhou B, Wang B, Chen P. Direct TGF-β1 signaling between activated platelets and pancreatic cancer cells primes cisplatin insensitivity. Cell Biol Int. 2013;37(5):478–484. doi:10.1002/cbin.10067
137. Green D, Hull RD, Brant R, Pineo GF. Lower mortality in cancer patients treated with low-molecular-weight versus standard heparin. Lancet (London, England). 1992;339(8807):1476. doi:10.1016/0140-6736(92)92064-m
138. Wahrenbrock M, Borsig L, Le D, Varki N, Varki A. Selectin-mucin interactions as a probable molecular explanation for the association of Trousseau syndrome with mucinous adenocarcinomas. J Clin Invest. 2003;112(6):853–862. doi:10.1172/JCI18882
139. Klerk CPW, Smorenburg SM, Otten H-M, et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol. 2005;23(10):2130–2135. doi:10.1200/JCO.2005.03.134
140. Mousa SA, Mohamed S. Inhibition of endothelial cell tube formation by the low molecular weight heparin, tinzaparin, is mediated by tissue factor pathway inhibitor. Thromb Haemost. 2004;92(09):627–633. doi:10.1160/TH04-02-0069
141. Stevenson JL, Varki A, Borsig L. Heparin attenuates metastasis mainly due to inhibition of P- and L-selectin, but non-anticoagulant heparins can have additional effects. Thromb Res. 2007;120(Suppl):S107–s111. doi:10.1016/S0049-3848(07)70138-X
142. Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM, Varki A. Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci U S A. 2001;98(6):3352–3357. doi:10.1073/pnas.061615598
143. Yoshitomi Y, Nakanishi H, Kusano Y, et al. Inhibition of experimental lung metastases of Lewis lung carcinoma cells by chemically modified heparin with reduced anticoagulant activity. Cancer Lett. 2004;207(2):165–174. doi:10.1016/j.canlet.2003.11.037
144. Maraveyas A, Ettelaie C, Echrish H, et al. Weight-adjusted dalteparin for prevention of vascular thromboembolism in advanced pancreatic cancer patients decreases serum tissue factor and serum-mediated induction of cancer cell invasion. Blood Coagul Fibrinolysis. 2010;21(5):452–458. doi:10.1097/MBC.0b013e328338dc49
145. Sudha T, Yalcin M, Lin H-Y, et al. Suppression of pancreatic cancer by sulfated non-anticoagulant low molecular weight heparin. Cancer Lett. 2014;350(1–2):25–33. doi:10.1016/j.canlet.2014.04.016
146. Sudha T, Phillips P, Kanaan C, Linhardt RJ, Borsig L, Mousa SA. Inhibitory effect of non-anticoagulant heparin (S-NACH) on pancreatic cancer cell adhesion and metastasis in human umbilical cord vessel segment and in mouse model. Clin Exp Metastasis. 2012;29(5):431–439. doi:10.1007/s10585-012-9461-9
147. Agnelli G, Gussoni G, Bianchini C, et al. Nadroparin for the prevention of thromboembolic events in ambulatory patients with metastatic or locally advanced solid cancer receiving chemotherapy: a randomised, placebo-controlled, double-blind study. Lancet Oncol. 2009;10(10):943–949. doi:10.1016/S1470-2045(09)70232-3
148. von Delius S, Ayvaz M, Wagenpfeil S, Eckel F, Schmid RM, Lersch C. Effect of low-molecular-weight heparin on survival in patients with advanced pancreatic adenocarcinoma. Thromb Haemost. 2007;98(08):434–439. doi:10.1160/TH07-01-0004
149. Icli F, Akbulut H, Utkan G, et al. Low molecular weight heparin (LMWH) increases the efficacy of cisplatinum plus gemcitabine combination in advanced pancreatic cancer. J Surg Oncol. 2007;95(6):507–512. doi:10.1002/jso.20728
150. Pelzer U, Opitz B, Deutschinoff G, et al. Efficacy of prophylactic low-molecular weight heparin for ambulatory patients with advanced pancreatic cancer: outcomes from the CONKO-004 trial. J Clin Oncol. 2015;33(18):2028–2034. doi:10.1200/JCO.2014.55.1481
151. Sideras K, Schaefer PL, Okuno SH, et al. Low-molecular-weight heparin in patients with advanced cancer: a Phase 3 clinical trial. Mayo Clin Proc. 2006;81(6):758–767. doi:10.4065/81.6.758
152. Di Nisio M, Porreca E, Candeloro M, De Tursi M, Russi I, Rutjes AW. Primary prophylaxis for venous thromboembolism in ambulatory cancer patients receiving chemotherapy. Cochrane Database Syst Rev. 2016;12(12):CD008500. doi:10.1002/14651858.CD008500.pub4
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.