")
Back to Journals » OncoTargets and Therapy » Volume 9
AA-PMe, a novel asiatic acid derivative, induces apoptosis and suppresses proliferation, migration, and invasion of gastric cancer cells
Authors Jing Y, Wang G, Ge Y, Xu M, Tang S, Gong Z
Received 21 October 2015
Accepted for publication 16 January 2016
Published 17 March 2016 Volume 2016:9 Pages 1605—1621
DOI https://doi.org/10.2147/OTT.S98849
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Yue Jing,1 Gang Wang,1 Ying Ge,1 Minjie Xu,1 Shuainan Tang,1 Zhunan Gong1,2
1Center for New Drug Research and Development, 2Jiangsu Key Laboratory for Molecular and Medical Biotechnology, College of Life Sciences, Nanjing Normal University, Nanjing, People’s Republic of China
Abstract: Asiatic acid (AA; 2α,3β,23-trihydroxyurs-12-ene-28-oic acid) is widely used for medicinal purposes in many Asian countries due to its various bioactivities. A series of AA derivatives has been synthesized in attempts to improve its therapeutic potencies. Herein we investigated the anti-tumor activities of N-(2α,3β,23-acetoxyurs-12-en-28-oyl)-l-proline methyl ester (AA-PMe), a novel AA derivative. AA-PMe exhibited a stronger anti-cancer activity than its parent compound AA. AA-PMe inhibited the proliferation of SGC7901 and HGC27 human gastric cancer cells in a dose-dependent manner but had no significant toxicity in human gastric mucosa epithelial cells (GES-1). AA-PMe induced cell cycle arrest in G0/G1 phase and blocked G1-S transition, which correlated well with marked decreases in levels of cyclin D1, cyclin-dependent kinase CKD4, and phosphorylated retinoblastoma protein, and increase in cyclin-dependent kinase inhibitor P15. Further, AA-PMe induced apoptosis of human gastric cancer cells by affecting Bcl-2, Bax, c-Myc, and caspase-3. Moreover, AA-PMe suppressed the migration and invasion of human gastric cancer cells (SGC7901 and HGC27) cells by downregulating the expression of MMP-2 and MMP-9. Overall, this study investigated the potential anti-cancer activities of AA-PMe including inducing apoptosis and suppressing proliferation, migration and invasion of gastric cancer cells, as well as the underlying mechanisms, suggesting that AA-PMe is a promising anti-cancer drug candidate in gastric cancer therapy.
Keywords: Asiatic acid derivatives, gastric cancer cells, anti-tumor effect, cytotoxicity, apoptosis, cell cycle arrest, migration, invasion, mobility
Introduction
Gastric cancer has a high mortality rate. Among all the malignant tumors, gastric cancer is the second leading cause of global cancer-related death,1 especially in East Asian countries such as the People’s Republic of China, Japan, and Korea.2,3 The development of gastric cancer is a multi-step process, starting from normal mucosa and developing to chronic gastritis, then to precancerous lesions including gastric atrophy, intestinal metaplasia and dysplasia, and eventually to invasive cancer.4 Gastric cancer is also frequently associated with lymph node metastasis, peritoneal dissemination, and hematogenous metastasis.5 It is believed that alterations of various regulators (eg, Bcl-2 and c-Myc), tumor suppressor genes (eg, Bax), DNA repair genes, cell cycle regulators (eg, cyclin D1, CDK4, CDK6, and P15), cell adhesion molecules (eg, Matrix metalloproteinase [MMP]-2 and MMP-9), and growth factor/receptor system are collectively involved in the onset and progression of gastric cancer.6–8 Rapid proliferation, migration, invasion, and multidrug resistance of tumor cells are responsible for poor prognosis of gastric cancer.9 At present, surgery and chemotherapy10,11 are the two main ways to treat gastric cancer. However, the existing chemotherapeutic drugs for gastric cancer lack robust efficacy and come with many side effects. As a result, current research has begun to focus on drugs that are more effective at preventing and treating gastric cancer as well as other malignant tumors with much lower side effect.
Asiatic acid (AA; 2α,3β,23-trihydroxyurs-12-ene-28-oic acid) is a pentacyclic triterpenoid widely distributed in plant kingdom. AA has been reported to have a variety of pharmacological effects, including anti-inflammatory,12,13 antioxidative,14 anti-tumor,15,16 neuroprotective,17,18 and wound healing19,20 activities. In addition, AA and its derivatives have been shown to have anti-tumor activities in various cancer cell lines.21,22 They are also inhibitors of glycogen phosphorylases23,24 and have hepatoprotective effect.25–27
We have synthesized a series of novel AA derivatives with amino acid conjugates and compared their bioactivities. Among them, N-(2α,3β,23-acetoxyurs-12-en-28-oyl)-L-proline methyl ester (AA-PMe) exhibited the strongest anti-tumor activities in most of the cancer cell lines used. In the present study, we sought to elucidate the effects of AA-PMe on the malignant phenotype of SGC7901 and HGC27 gastric cancer cells. Our result indicated that AA-PMe could efficiently inhibit proliferation and induce apoptosis of the gastric cancer cells via arresting cell cycle in G1/S phase. Moreover, AA-PMe could suppress migration and invasion of gastric cancer cells.
Materials and methods
Materials and reagents
AA was extracted from Centella asiatica (L.) and AA-PMe was synthesized in our laboratory.28 The structures of AA and AA-PMe are shown in Figure 1. Proline was purchased from Aladdin (Shanghai, China). DMEM, RPMI 1640, and fetal bovine serum (FBS) were purchased from Gibco (Thermo Fisher Scientific, Grand Island, USA). Rabbit anti-caspase 3, anti-c-Myc, anti-phosphorylated Rb (Ser807/811), anti-p15, anti-MMP-2 and anti-MMP-9 polyclonal antibodies and mouse anti-cyclin D1, anti-CDK4, and anti-CDK6 monoclonal antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). Rabbit anti-Bax and anti-Bcl-2 polyclonal antibodies and the horseradish peroxidase (HRP)-conjugated anti-rabbit IgG secondary antibody were purchased from ABCam (Cambridge, MA, USA). The HRP-conjugated anti-mouse IgG secondary antibody was purchased from Boster (Pleasanton, CA, USA). Annexin V-FITC/propidium iodide (PI) double staining apoptosis detection kit, cell cycle kit, and cell counting kit-8 (CCK8) were purchased from Vazyme Co. Ltd (Nanjing, Jiangsu, People’s Republic of China). All other reagents and chemicals were purchased from Sigma (St Louis, MO, USA). All animal experiments in this investigation were conducted in compliance with the Guide outlining the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996) and was approved by the Animal Care and Use Committee of Nanjing Normal University, China (Permit Number: 2090658).
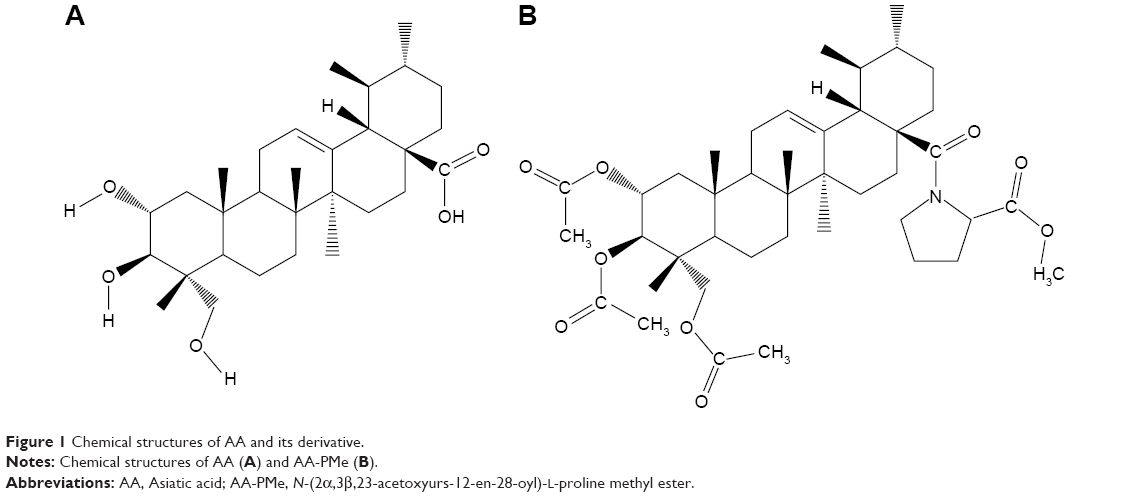 | Figure 1 Chemical structures of AA and its derivative. |
Cell culture
The human gastric mucosa epithelial GES-1, gastric cancer cell lines SGC7901 and HGC27, were purchased from the Cell Bank of the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Science, and cultured in DMEM and RPMI 1640 media, respectively, supplemented with 10% FBS in a humidified atmosphere containing 5% CO2 in air at 37°C. This study was approved by the Ethics Committee of Nanjing Normal University, and conformed with the provisions of the Declaration of Helsinki.
Measurement of cell viability with CCK8 assay
Cell counting kit-8 was used to examine cell viability. Briefly, gastric cancer cells (5×103/well) were seeded in 96-well plates. Then AA and AA-PMe were added at a series of concentrations (1–100 μM) and incubated for 24, 48, or 72 hours, followed by addition of 10 μL CCK8 and incubated for another 1 hour. The absorbance was measured at 450 nm using a microplate reader (Thermo Fisher Scientific, Grand Island, USA).
Cell count using trypan blue dye exclusion
SGC7901 cells were seeded in a six-well plate (105 cells/well) and cultured overnight. Then AA and AA-PMe were added at a series of concentrations and incubated for 24, 48, or 72 hours. After that, cells were harvested and mixed with Trypan blue solution (0.14% in Hank’s balanced salt solution; Gibco BRL Co. Ltd., USA). The colored (dead) and dye-excluding (live) cells were counted using a hemocytometer.
Cell cycle analysis by flow cytometry
Cells were seeded in a six-well plate (1.5×106 cells/well) and treated with AA-PMe or AA (1–50 μM) for 24 hours. Then the cells were harvested, fixed with 75% ethanol for 2 hours at 4°C, washed with PBS, and stained with PI (5 mg/L) containing RNase A (1 g/L), sodium citrate (1 g/L), and 0.5% Triton X-100 for 30 minutes in the dark. Then the cells were analyzed by a FACS Vantage cell sorter (BD Bioscience, Franklin Lakes, NJ, USA) equipped with a 488 nm argon laser. Cell distribution in different phases of the cell cycle was estimated using ModFit LT™ software.
Annexin V-FITC/PI assay
SGC7901 and HGC27 cells were seeded in six-well plates and cultured overnight to reach 80% confluence. Apoptosis was induced by 24 hours of treatment with AA or AA-PMe at a series of concentrations (1, 5, 10, 25, and 50 μM). Apoptosis was analyzed using an Annexin V-FITC and PI double-staining apoptosis detection kit by following the manufacturer’s instruction. Briefly, after treatment, cells were harvested, washed with PBS, centrifuged, and resuspended in 195 μL Annexin V-FITC binding buffer. The cells were incubated with 5 μL Annexin V-FITC solution for 10 minutes at room temperature in the dark, then the solution was removed and 190 μL Annexin V-FITC binding buffer and 10 μL PI were added and incubated for another 5 minutes. Cell suspension was analyzed by FACS with CellQuest version 3.3 software.
Wound-healing assay
Cells were seeded into six-well plates and grew into monolayer, and a plastic pipette tip was used to scratch across the center of wells to generate a 1 mm-wide wound area and washed twice with PBS to remove detached cells. The photomicrographs of initial wounds were taken using Canon Power Shot A640 digital camera. Then, cells were treated with AA or AA-PMe (1, 5, 10, 30, and 50 μM) in FBS-free medium for 6, 12, or 24 hours, and the photomicrographs of wounds at different time points were taken under a phase-contrast microscope and analyzed.
Invasion assay
A 24-well plate with transwells (Costar, Corning, NY, USA) precoated with matrigel (BD Biosciences) was used to analyze cell invasion. Briefly, 600 μL of culture medium containing 10% FBS with or without AA or AA-PMe was added to the lower chamber as chemoattractant. Cells were placed in the upper chamber in 200 μL medium containing 1% FBS at a density of 1×105 cells/well; 24 hours later, the nonmigrated cells were removed with cotton swabs, and the cells migrated to the lower surface of the filters were fixed with 90% alcohol for 30 minutes at room temperature and stained with 0.1% crystal violet for visualization. Images were captured under a phase-contrast microscope and the invasive cells were quantified by manual counting. A total of 33% acetic acid was then used to decolor invasive cells for 10 minutes. The destaining solution was collected and the absorbance was measured at 570 nm using a microplate reader (Thermo FC, USA). The inhibition of invasion was calculated as follows:
|
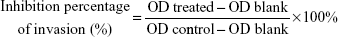
Protein extraction and Western blot analysis
Cells were lysed in RIPA buffer (50 mM Tris, pH 8, 150 mM NaCl, 0.1% SDS, 0.2% EDTA, 1% Triton X-100, 1% sodium deoxycholate, and 0.01% PMSF) supplemented with protease and phosphatase inhibitors cocktail (Roche, Basel, Switzerland) by 30 minutes of incubation on ice and centrifuged at 12,000 rpm for 15 minutes at 4°C to remove nuclei and cell debris (Eppendorf centrifuge 5804R, Eppendorf, Germany). The protein concentration of the extracts was determined using a Bicinchoninic Acid Protein Assay Kit. Then proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (10% gels) and then were transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Massachusetts, USA) using a semidry transfer system (Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5% skimmed milk in Tris-buffered saline containing 0.05% Tween-20 at room temperature for 1 hour, followed by incubation with primary antibodies overnight at 4°C and HRP-conjugated secondary antibody for 1 hour at room temperature. Proteins were visualized by enhanced chemiluminescence detection system (Tanon-3500R, Tanon Co. Ltd, Shanghai, China).
Real-time reverse transcription PCR
Total RNA was extracted with Trizol Reagent (Invitrogen, Carlsbad, CA, USA) and quantified using a spectrophotometer (Nano Drop ND-1000, Thermo Fisher Scientific, Grand Island, USA). Reverse transcription was performed using a cDNA synthesis kit (Invitrogen). Quantitative RT-PCR was performed by following the manufacturer’s protocol (Takara, Takara, Tokyo, Japan). The sequences of primers used are listed in Table 1. The target mRNA was quantified in comparison with GAPDH mRNA. The data were calculated with ΔΔCt method.
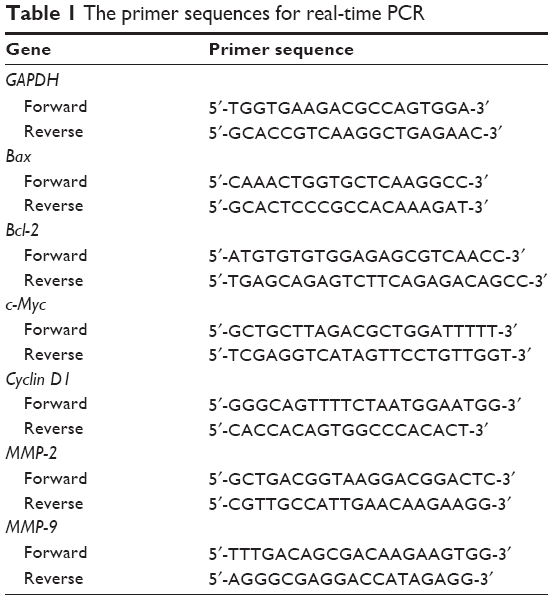 | Table 1 The primer sequences for real-time PCR |
Statistical analysis
All the data were presented as mean ± standard deviation (SD) of at least triplicate measurements. The data shown are representatives of at least two independent experiments with similar results. Statistical analysis was performed using SPSS software (version 13.0). The significant differences between groups were evaluated by one-way analysis of variance (ANOVA). Statistical significance was defined as P<0.05.
Results
AA-PMe reduces the viability of gastric cancer cells
The anti-cancer effect of AA-PMe and its parent compound AA was compared by exposing tumor cells at a series of concentrations and time points. The CCK-8 analysis results showed that both AA-PMe and AA reduced the viability of SGC7901 and HGC27 cells in a concentration- and time-dependent manner (Figure 2). The half maximal inhibitory concentration (IC50) of AA-PMe for SGC7901 cells was 4.48 μM, which was much lower than that of AA (36.9 μM) (Table 2 and Figure 2A). We also evaluated cell viability using a trypan blue exclusion assay. It was shown that AA-PMe could inhibit cell survival at a low concentration of 5 μM (Figure 2B), whereas AA inhibited cell survival only at a high concentration of 50 μM (Figure 2C). Similar results were also obtained in HGC27 cells. AA-PMe exerted a stronger inhibition than AA in HGC27 cells with an IC50 of 7.92 μM and 61.23 μM for AA-PMe and AA, respectively (Figure 2D–F).
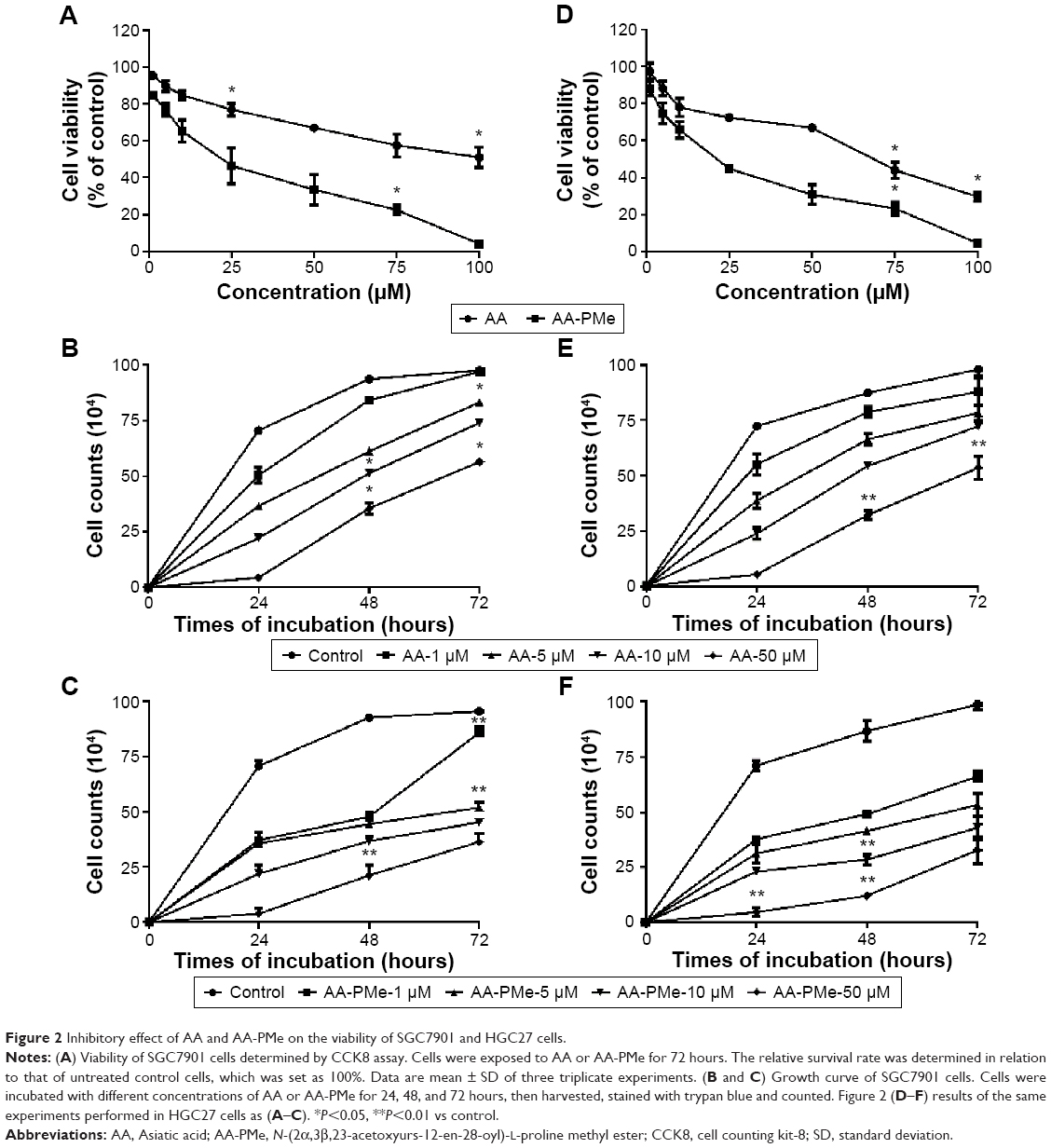 | Figure 2 Inhibitory effect of AA and AA-PMe on the viability of SGC7901 and HGC27 cells. |
 | Table 2 Comparison of anti-tumor activities of AA and AA-PMe in SGC7901 and HGC27 cells |
We also examined the toxicity of AA-PMe and AA on human gastric mucosa epithelial GES-1 cells. Both AA-PMe and AA had no significant effect on viability of GES-1 cells, except for a slight inhibition at high concentration of 100 μM (Table 2 and Figure 3).
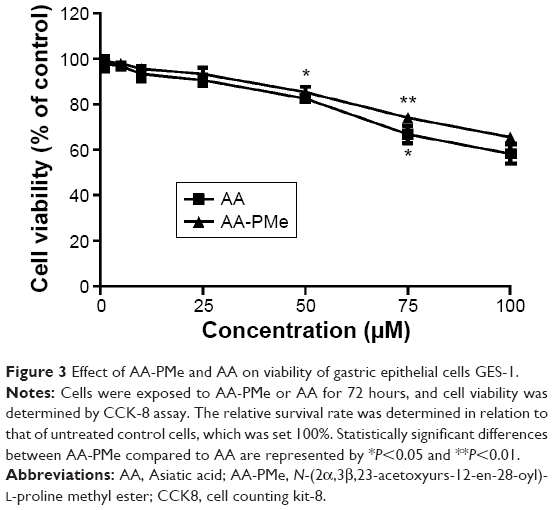 | Figure 3 Effect of AA-PMe and AA on viability of gastric epithelial cells GES-1. |
AA-PMe arrests cell cycle of gastric cancer cells in G0/G1 phase
We examined the cell cycle after treatment with AA-PMe and AA. We found that both AA-PMe and AA concentration-dependently induced cell cycle arrest in G0/G1 phase (Figure 4). Again, AA-PMe exhibited a stronger activity than AA. After a 24-hours exposure, the proportion of cells in G0/G1 phase treated by 25 μM AA-PMe increased from 51.31% to 77.7% in SGC7901 cells (Figure 4A and C), and from 52.12% to 77.26% in HGC27 cells (Figure 4B and D). AA increased the proportion of cells in G0/G1 phase from 50.81% to 65.08% in SGC7901 cells (Figure 4A and C) and from 54.66% to 65.63% in HGC27 cells (Figure 4B and D).
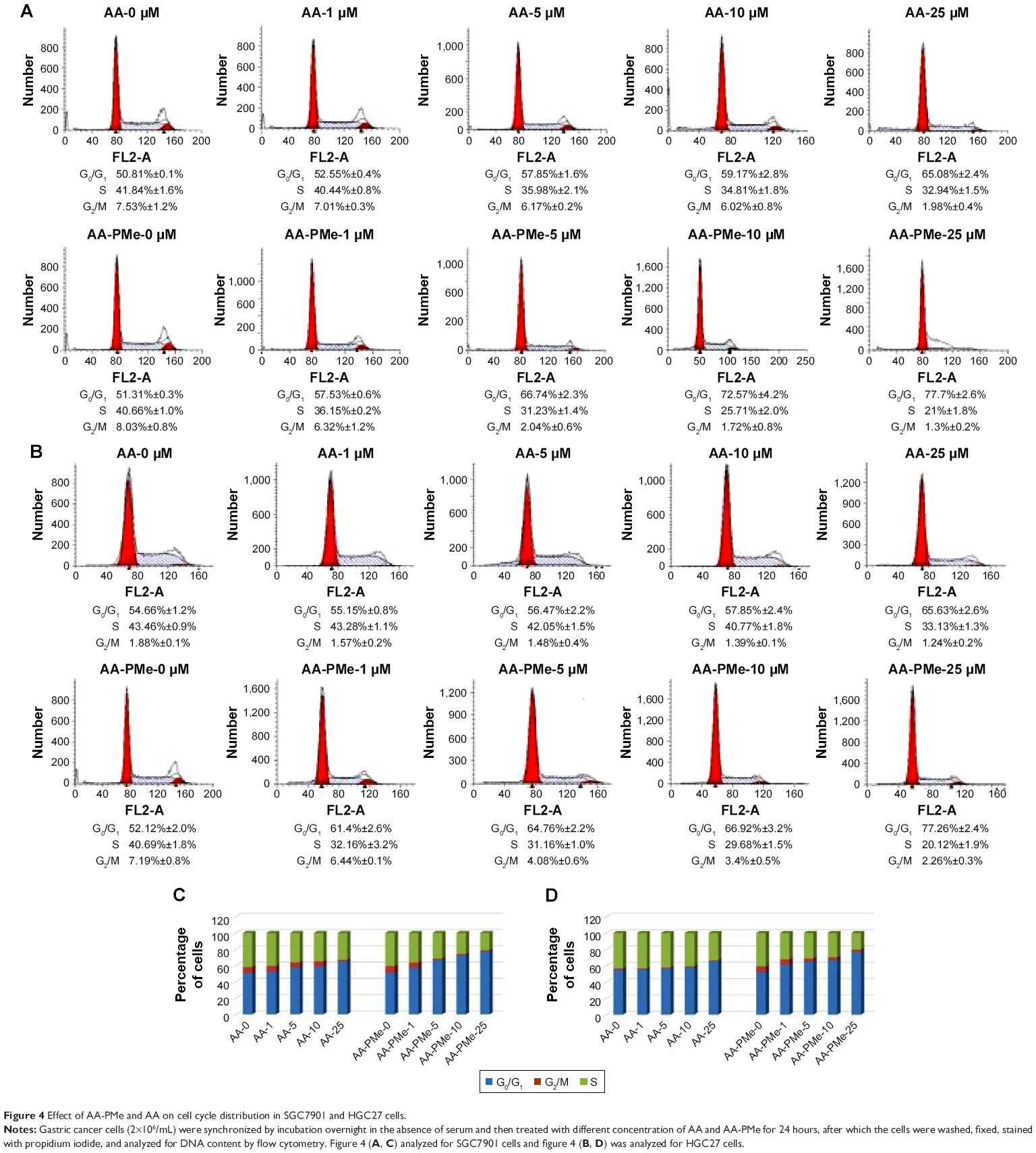 | Figure 4 Effect of AA-PMe and AA on cell cycle distribution in SGC7901 and HGC27 cells. |
We next observed the influence of AA-PMe and AA on cell cycle regulators including cyclin D1, CDK4, CDK6, p15, and phosphorylated Rb. We found that AA-PMe remarkably decreased the expression of cyclin D1, CDK4, and phosphorylated Rb, and increased the amount of p15, but failed to affect the CDK6 level or only slightly decreased it. In contrast, AA only had effects on these cell cycle regulators at high concentration of 50 μM (Figure 5).
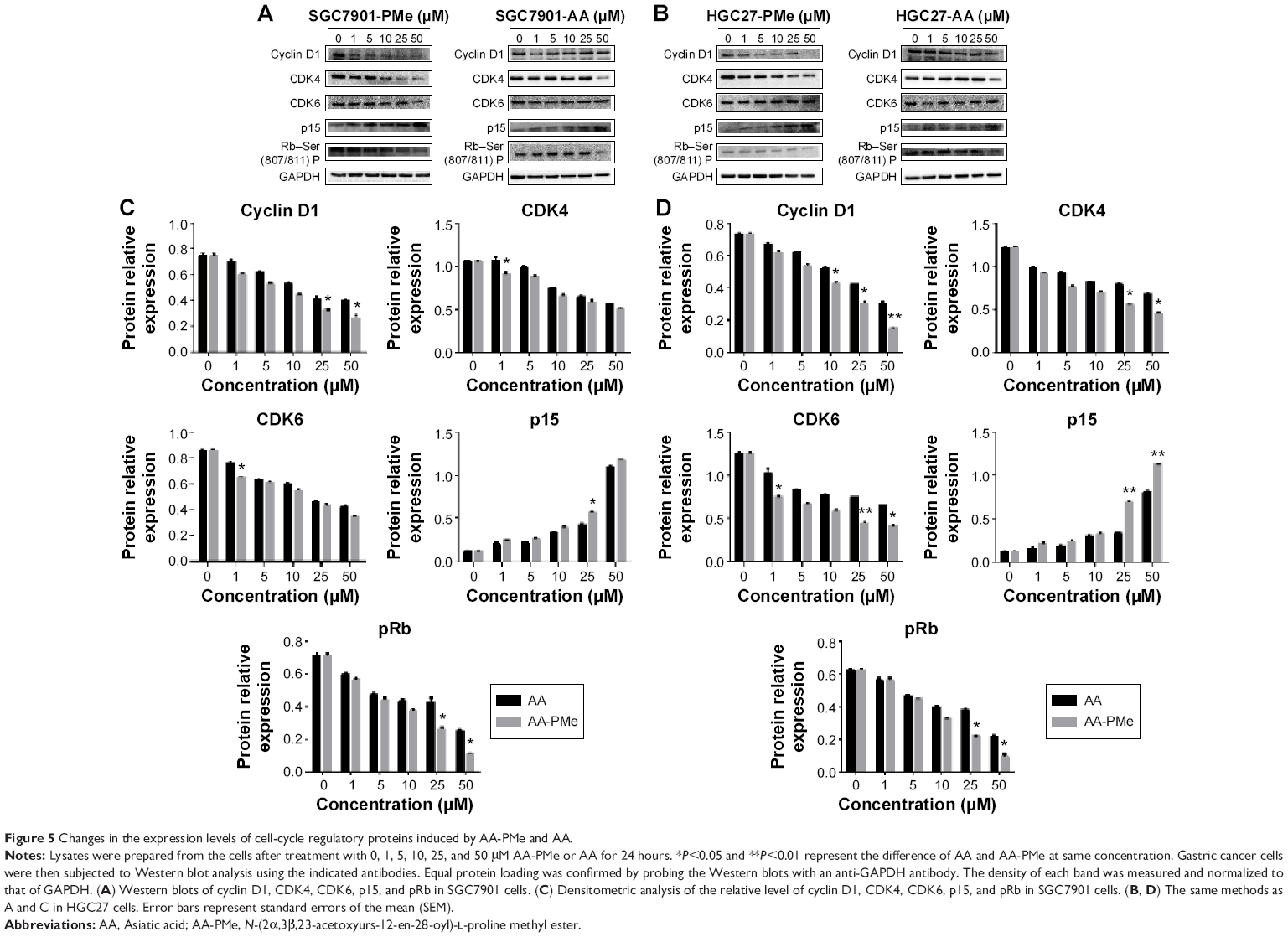 | Figure 5 Changes in the expression levels of cell-cycle regulatory proteins induced by AA-PMe and AA. |
AA-PMe induces apoptosis of gastric cancer cells
We used Annexin V-FITC/PI double staining to analyze AA-PMe-induced apoptosis. SGC7901 and HGC27 cells were treated with AA-PMe and AA (0–50 μM) for 24 hours. As shown in Figure 6, the fluorescent microscopic images show that both AA-PMe and AA induced significant cell apoptosis in SGC7901 and HGC27 cells in a concentration-dependent manner. The green light shows the cells at early stage of apoptosis, while the red light shows those at late stage of apoptosis. Consistently, AA-PMe exhibited a stronger activity than AA. The apoptotic cells induced by 10 μM AA-PMe were much more than those induced by 10 μM AA in both SGC7901 and HGC27 cells.
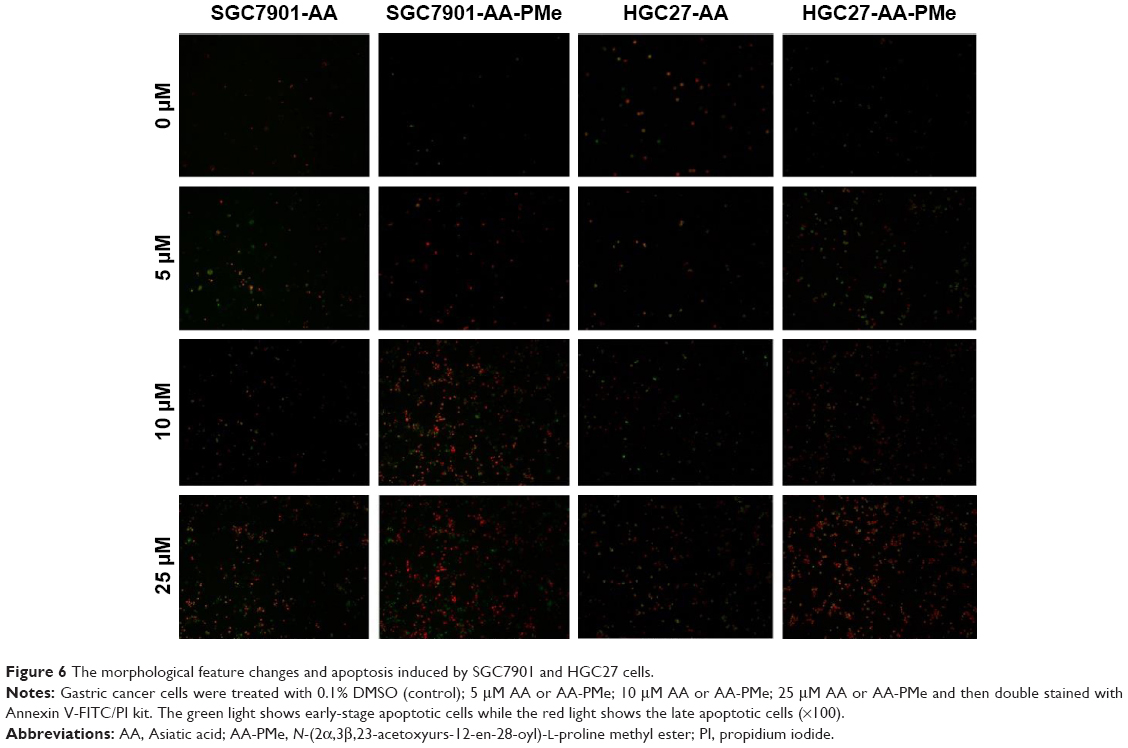 | Figure 6 The morphological feature changes and apoptosis induced by SGC7901 and HGC27 cells. |
To further quantify the apoptosis of SGC7901 and HGC27 cells induced by AA-PMe and AA, we performed Annexin V-FITC/PI double-staining assay by flow cytometric analysis, which detects different stages of apoptosis. The results indicated that AA-PMe had a stronger capability to induce apoptosis than AA. A total of 50 μM AA-PMe induced an apoptosis rate of 65.4% in SGC7901 cells and 59.02% in HGC27 cells after 24 hours of treatment. In contrast, 50 μM AA induced an apoptosis rate of 42.68% in SGC7901 cells and 54.6% in HGC27 cells (Figure 7).
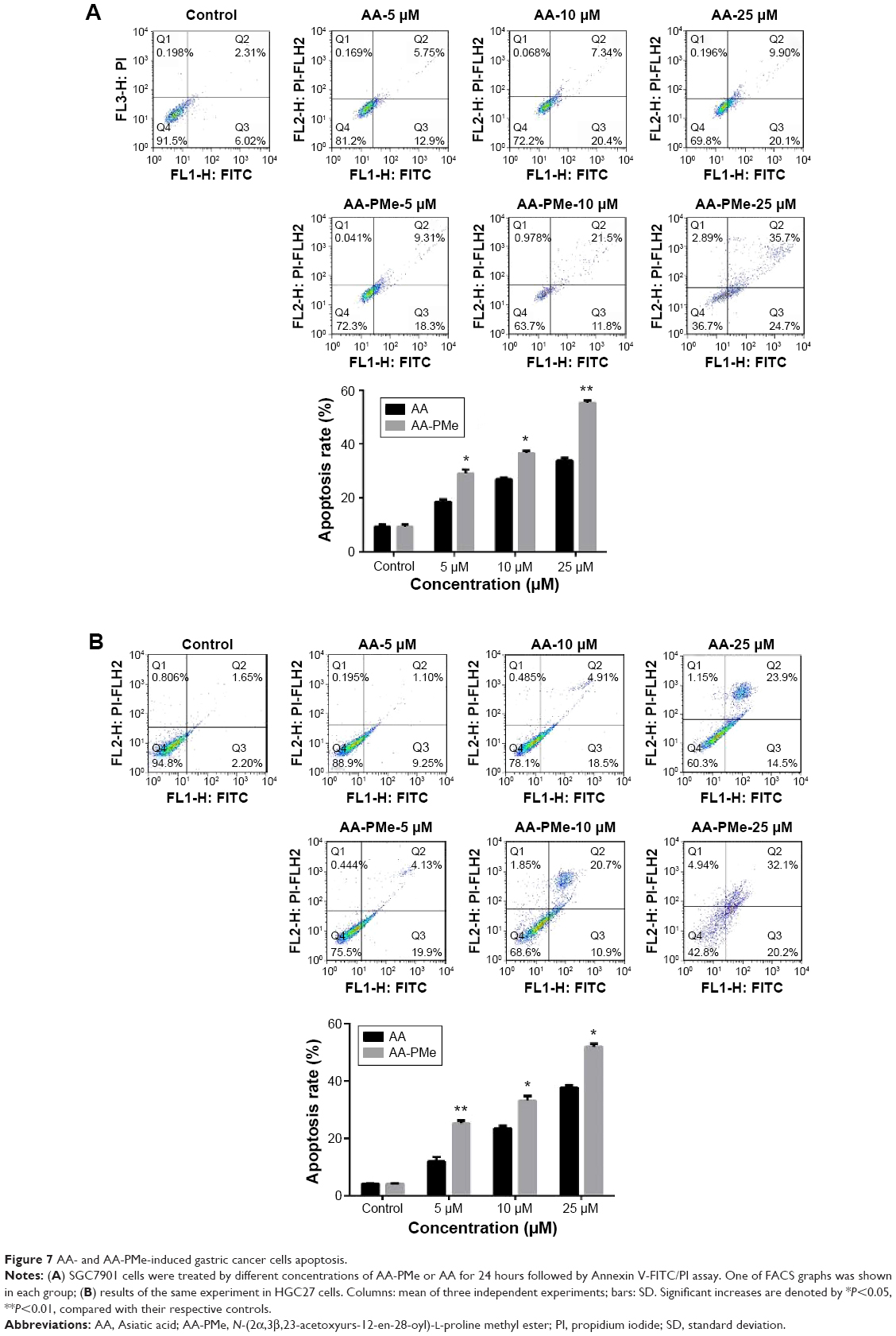 | Figure 7 AA- and AA-PMe-induced gastric cancer cells apoptosis. |
We next analyzed the effects of AA-PMe and AA on apoptosis-related regulators including Bcl-2, caspase 3, c-Myc, and Bax in SGC7901 and HGC27 cells. Results showed that AA-PMe treatment downregulated the expression of Bcl-2, caspase 3, and c-Myc, and increased the accumulation of proapoptotic protein Bax in a concentration-dependent manner in both SGC7901 and HGC27 cells (Figure 8). AA exhibited weaker activities in affecting these signaling events. Overall, these results clearly showed that AA-PMe could decrease viability and induce apoptosis in SGC7901 and HGC27 cells through altering the expression of pro-apoptotic and anti-apoptotic molecules.
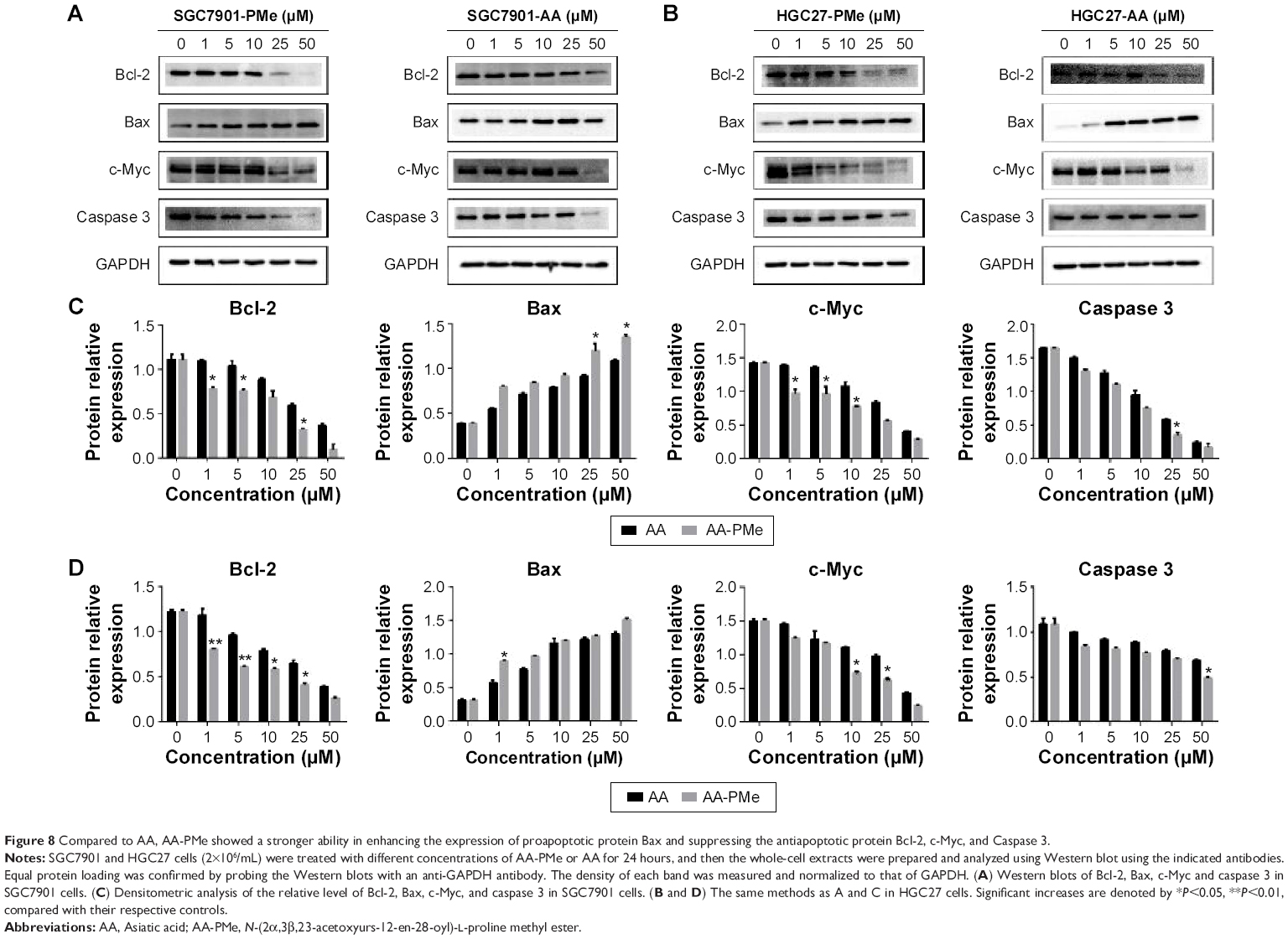 | Figure 8 Compared to AA, AA-PMe showed a stronger ability in enhancing the expression of proapoptotic protein Bax and suppressing the antiapoptotic protein Bcl-2, c-Myc, and Caspase 3. |
AA-PMe inhibits migration and invasion of gastric cancer cells
Cancer cell migration and invasion are critical events for tumor growth and metastasis.29 So we next studied the effect of AA-PMe and AA on the migration and invasion of SGC7901 and HGC27 cells using wound-healing and transwell assays. As shown in Figure 9, AA-PMe dramatically inhibited the migration of both SGC7901 and HGC27 cells in a concentration- and time-dependent manner in wound-healing assay. Similarly, AA-PMe also significantly suppressed the invasion of both SGC7901 and HGC27 cells through matrigel in a concentration-dependent manner. Again, AA-PMe showed a much stronger activity than AA in these experiments (Figure 10). It is well known that MMP-2 and MMP-9 could degrade the intercellular mesenchyme and promote tumor migration. Expressions of MMP-2 and MMP-9 were detected following treatment with AA-PMe for 24 hours. Western blot results showed that AA-PMe downregulated the expression of both MMP-2 and MMP-9 (Figure 11).
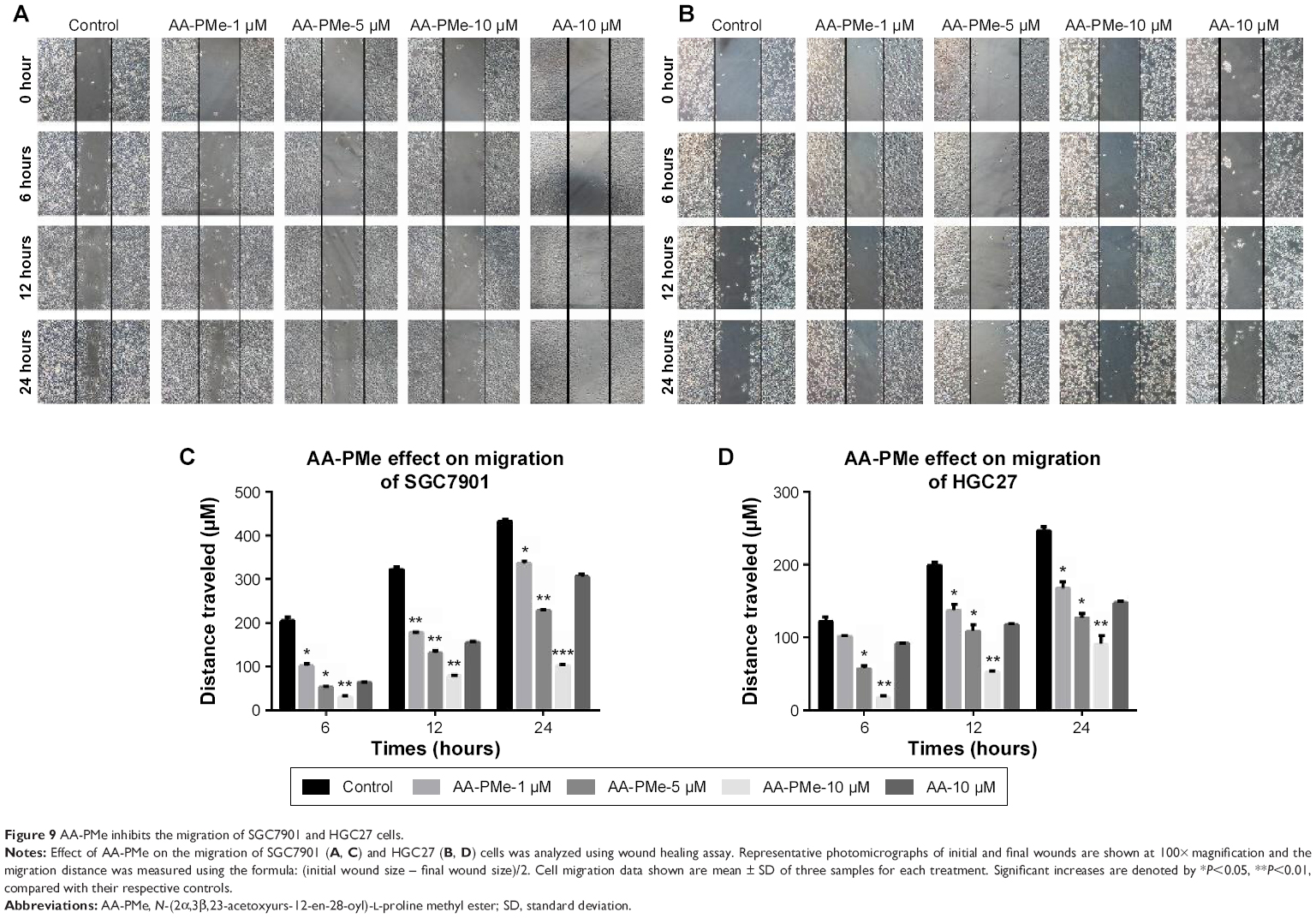 | Figure 9 AA-PMe inhibits the migration of SGC7901 and HGC27 cells. |
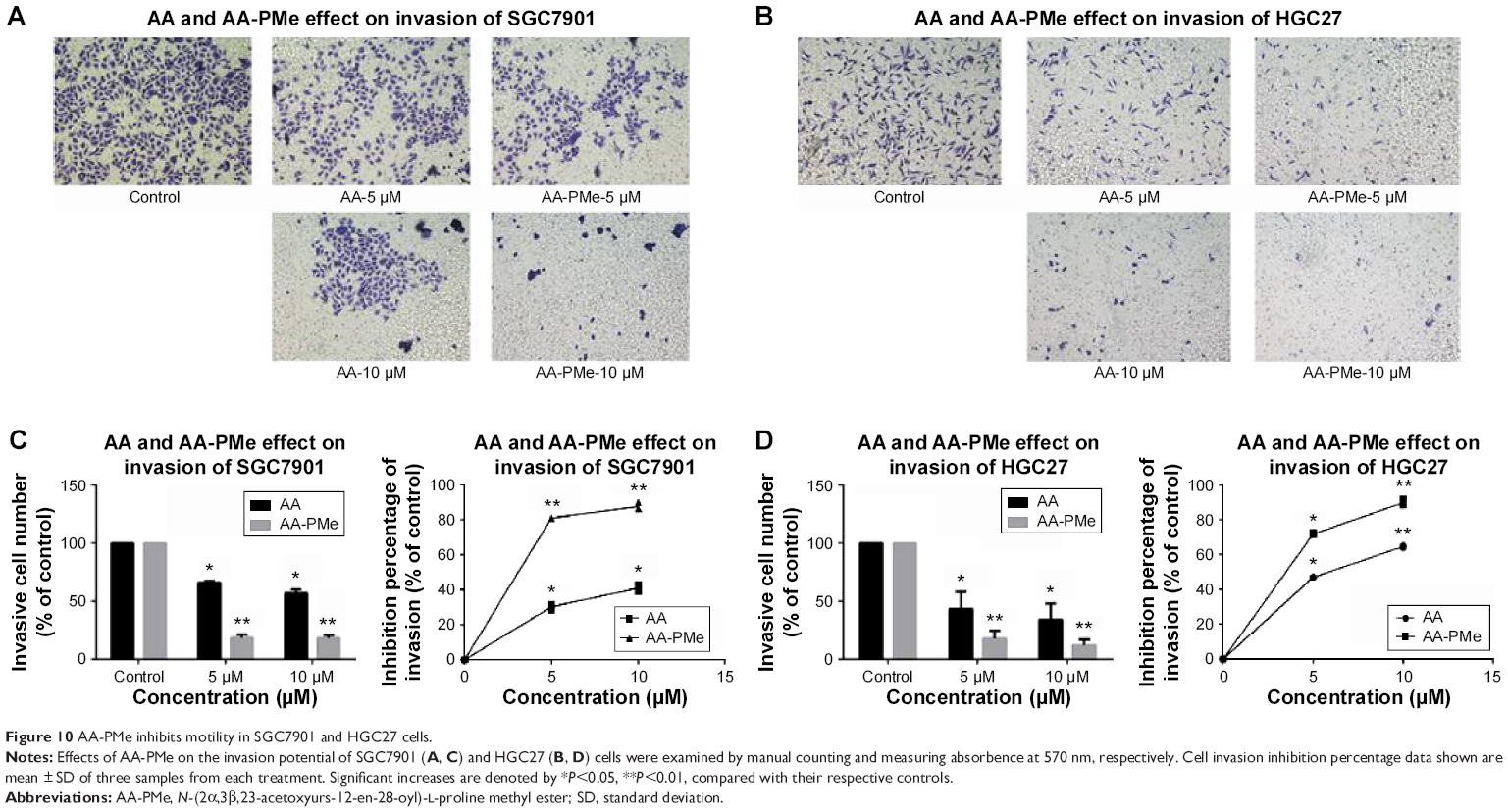 | Figure 10 AA-PMe inhibits motility in SGC7901 and HGC27 cells. |
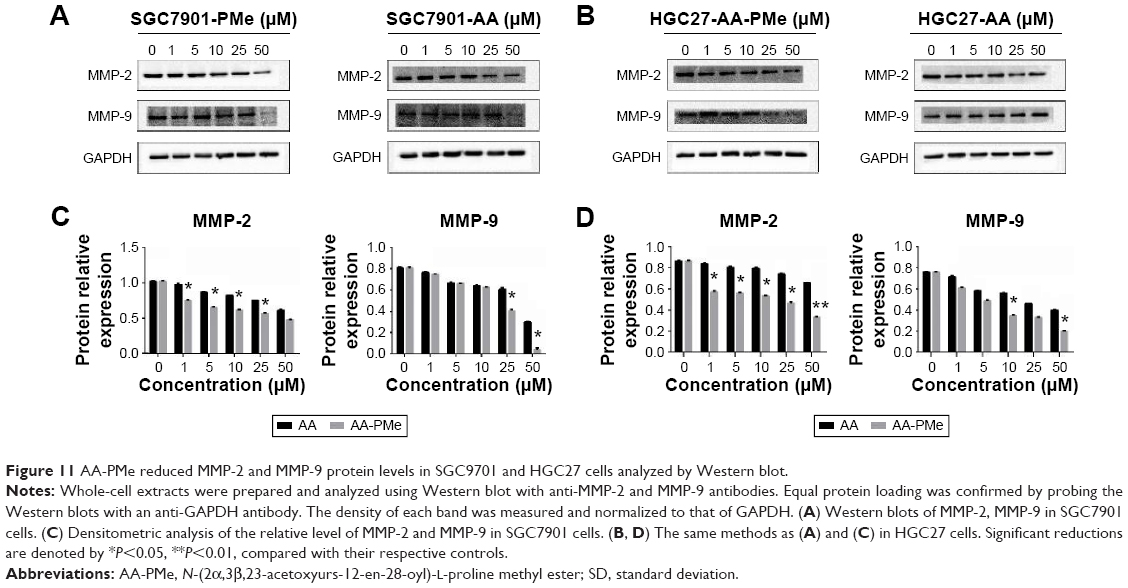 | Figure 11 AA-PMe reduced MMP-2 and MMP-9 protein levels in SGC9701 and HGC27 cells analyzed by Western blot. |
AA-PMe modulates gene expression
To determine whether AA-PMe affects the transcription in SGC7901 and HGC27 cells, the mRNA expression of Bax, Bcl-2, cyclin D1, c-Myc, MMP-2, and MMP-9 was examined. As shown in Figure 12, AA-PMe treatment increased the expression of proapoptotic Bax gene and suppressed the expression of the rest – Bcl-2, cyclin D1, c-Myc, MMP-2, and MMP-9 genes at high concentration used, suggesting that AA-PMe modulates the expression of these genes at transcriptional level.
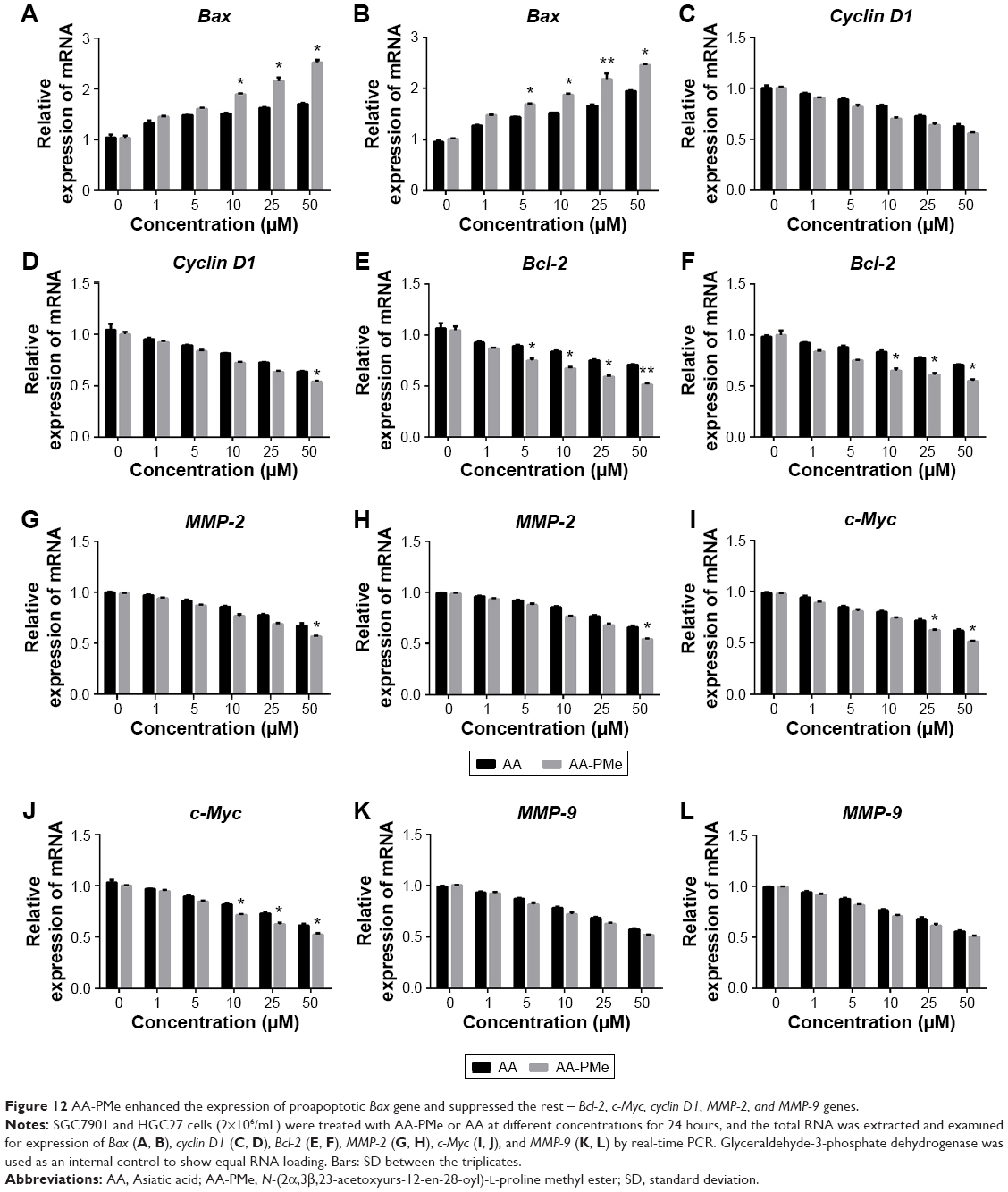 | Figure 12 AA-PMe enhanced the expression of proapoptotic Bax gene and suppressed the rest – Bcl-2, c-Myc, cyclin D1, MMP-2, and MMP-9 genes. |
Discussion
In this paper, we have studied the anti-tumor activities of a novel AA derivative, AA-PMe, in human gastric cancer cells, which has been proved to have the highest anti-cancer efficacy among all the AA derivatives in numerous cancer cell lines, suggesting that the increased anti-cancer activity of AA derivatives may result not only from increased water solubility but also from the optimization of hydrophilic–lipophilic balance to a great extent.30,31 Moreover, it suggests that proline methyl ester and acetoxy groups could lead to an increased activity jointly when put together.
Our studies showed that AA-PMe had stronger anti-cancer activities than its parent compound AA in gastric cancer cells. AA-PMe remarkably inhibited the proliferation of gastric cancer cells by arresting cell cycle in G0/G1 phase via affecting cell cycle regulators including cyclin D1, CDK4, p15, and phosphorylated Rb, and induced cell apoptosis by regulating apoptosis-related signaling molecules including Bcl-2, caspase 3, c-Myc, and Bax. Moreover, AA-PMe also suppressed the migration and invasion of gastric cancer cells by downregulating the expression of MMP-2 and MMP-9, two critical proteins in cell migration and invasion. Furthermore, we have found that AA-PMe regulated the expression of these related genes including Bax, Bcl-2, cyclin D1, c-Myc, MMP-2, and MMP-9 at transcriptional level. In contrast, AA-PMe exhibited very weak toxicity in normal human embryonic kidney 293 cells, suggesting that AA-PMe may have some selectivity for tumor cells over normal cells, which is very important for an anti-cancer drug candidate.
Cell cycle arrest induced by AA-PMe might be the mechanism underlying the anti-proliferation effect of AA-PMe. AA-PMe increased the percentage of tumor cells in G0/G1 phase but decreased the proportion of cells in S phase, suggesting that AA-PMe might induce cell cycle arrest into G0/G1 phase and block the entry of cells into DNA synthesis phase. The cell cycle progression is driven by CDK family proteins, which are positively regulated by cyclin proteins and negatively regulated by CDKI. During the G1 phase, cyclin D engages with CDK4 and/or CDK6 to form a complex, resulting in the activation of CDKs. The activated CDKs can phosphorylate Rb protein, which subsequently disrupts the binding of Rb to E2F, leading to E2F activation and the gene transcriptions necessary for G1–S transition and cell cycle progression.32,33 In our study, AA-PMe treatment decreased the levels of CDK4, cyclin D, and phosphorylated Rb proteins but increased the levels of p15, a specific inhibitor of CDK4 and CDK6, thereby regulating cell cycle progression.
Then, we elucidated the mechanism underlying AA-PMe-induced apoptosis in gastric cancer cells. Apoptosis is essential for the normal development and maintenance of tissue homeostasis. On the other hand, apoptosis is also involved in pathologic conditions especially cancers. Bcl-2 family proteins cytochrome c and caspases are well-known apoptosis-inducing signaling pathways.34 Induction of cancer cell apoptosis has become a promising direction for anti-cancer drug development.35 Apoptosis is directly mediated by caspase-3, a family member of cysteine proteases,36,37 which is activated by the formation of apoptosome,38 relying on the release of cytochrome c from mitochondria to cytosol regulated by Bax and Bcl-2. The proapoptotic protein Bax favors cytochrome c release by forming pores in the outer mitochondrial membrane, while the antiapototic protein Bcl-2 prevents the opening of mitochondrial transition pore by binding to Bax.39,40 In our study, we found that AA-PMe could induce apoptosis of gastric cancer cells by decreasing the levels of Bcl-2, caspase 3, and c-Myc proteins and by increasing the levels of Bax.
We also investigated the antimigration/invasion activities of AA-PMe as well as the underlying mechanisms. Cell migration and invasion results demonstrated that AA-PMe efficiently decrease the migration and invasion of SGC7901 and HGC27 cells. MMPs are critical molecules favoring tumor invasion and metastasis by degrading extracellular matrix surrounding the tumor, especially the basement membrane.41 Higher expression levels of MMP-2 and MMP-9 have been correlated with increased tumor metastasis and poorer prognosis.42 Our study indicated that AA-PMe downregulated MMP-2 and MMP-9 levels, which may at least partially explain the antiinvasion activity of AA-PMe.
Conclusion
Our studies demonstrated that AA-PMe, a novel derivative of AA, has stronger anti-cancer activities in human gastric cancer cells than its parent compound AA. However, AA-PMe almost has no toxicity in normal human gastric mucosa epithelial cells. AA-PMe inhibited the proliferation of gastric cancer cells by blocking G1–S phase transition and arresting cell cycle in G0/G1 phase via affecting cell cycle regulators. AA-PMe induced cell apoptosis by enhancing the expression of proapoptotic protein and suppressing the expression of antiapoptotic proteins. Moreover, AA-PMe also suppressed the migration and invasion of gastric cancer cells by downregulating the expression of MMP-2 and MMP-9. Our study suggests that AA-PMe is a promising anti-cancer drug candidate in gastric cancer therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Li L, Ying XJ, Sun TT, et al. Overview of methodological quality of systematic reviews about gastric cancer risk and protective factors. Asian Pac J Cancer Prev. 2012;13(5):2069–2079. | ||
Alberts SR, Cervantes A, van de Velde CJ. Gastric cancer: epidemiology, pathology and treatment. Ann Oncol. 2003;14(Suppl 2):ii31–ii36. | ||
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. | ||
Correa P. Human gastric carcinogenesis: a multistep and multifactorial process – First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52(24):6735–6740. | ||
Boyle P, Ferlay J. Cancer incidence and mortality in Europe, 2004. Ann Oncol. 2005;16(3):481–488. | ||
Wu MS, Shun CT, Wang HP, et al. Genetic alterations in gastric cancer: relation to histological subtypes, tumor stage, and Helicobacter pylori infection. Gastroenterology. 1997;112(5):1457–1465. | ||
Yasui W, Yokozaki H, Fujimoto J, Naka K, Kuniyasu H, Tahara E. Genetic and epigenetic alterations in multistep carcinogenesis of the stomach. J Gastroenterol. 2000;35(Suppl 12):111–115. | ||
Werner M, Becker KF, Keller G, Hofler H. Gastric adenocarcinoma: pathomorphology and molecular pathology. J Cancer Res Clin Oncol. 2001;127(4):207–216. | ||
Lochhead P, El-Omar EM. Gastric cancer. Br Med Bull. 2008;85:87–100. | ||
Huang JY, Xu YY, Sun Z, et al. Comparison different methods of intraoperative and intraperitoneal chemotherapy for patients with gastric cancer: a meta-analysis. Asian Pac J Cancer Prev. 2012;13(9):4379–4385. | ||
Shi WT, Wei L, Xiang J, et al. Chinese patients with gastric cancer need targeted adjuvant chemotherapy schemes. Asian Pac J Cancer Prev. 2012;13(10):5263–5272. | ||
Won JH, Shin JS, Park HJ, et al. Anti-inflammatory effects of madecassic acid via the suppression of NF-kappaB pathway in LPS-induced RAW 264.7 macrophage cells. Planta Med. 2010;76(3):251–257. | ||
Yun KJ, Kim JY, Kim JB, et al. Inhibition of LPS-induced NO and PGE2 production by asiatic acid via NF-kappa B inactivation in RAW 264.7 macrophages: possible involvement of the IKK and MAPK pathways. Int Immunopharmacol. 2008;8(3):431–441. | ||
Pittella F, Dutra RC, Junior DD, Lopes MT, Barbosa NR. Antioxidant and cytotoxic activities of Centella asiatica (L) Urb. Int J Mol Sci. 2009;10(9):3713–3721. | ||
Hsu YL, Kuo PL, Lin LT, Lin CC. Asiatic acid, a triterpene, induces apoptosis and cell cycle arrest through activation of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways in human breast cancer cells. J Pharmacol Exp Ther. 2005;313(1):333–344. | ||
Park BC, Bosire KO, Lee ES, Lee YS, Kim JA. Asiatic acid induces apoptosis in SK-MEL-2 human melanoma cells. Cancer Lett. 2005;218(1):81–90. | ||
Krishnamurthy RG, Senut MC, Zemke D, et al. Asiatic acid, a pentacyclic triterpene from Centella asiatica, is neuroprotective in a mouse model of focal cerebral ischemia. J Neurosci Res. 2009;87(11):2541–2550. | ||
Soumyanath A, Zhong YP, Gold SA, et al. Centella asiatica accelerates nerve regeneration upon oral administration and contains multiple active fractions increasing neurite elongation in-vitro. J Pharm Pharmacol. 2005;57(9):1221–1229. | ||
Cheng CL, Guo JS, Luk J, Koo MW. The healing effects of Centella extract and asiaticoside on acetic acid induced gastric ulcers in rats. Life Sci. 2004;74(18):2237–2249. | ||
Kimura Y, Sumiyoshi M, Samukawa K, Satake N, Sakanaka M. Facilitating action of asiaticoside at low doses on burn wound repair and its mechanism. Eur J Pharmacol. 2008;584(2–3):415–423. | ||
Zhao LX, Tian MZ, Jin LJ, et al. Synthesis and characterization of derivatives of Asiatic acid and primary study on anti-cancer activity. Chin J Org Chem. 2011;31(5):646–652. | ||
Meng YQ, Li YY, Li FQ, et al. Synthesis and antitumor activity evaluation of new asiatic acid derivatives. J Asian Nat Prod Res. 2012;14(9):844–855. | ||
Zhang LY, Chen J, Gong YC, et al. Synthesis and biological evaluation of Asiatic acid derivatives as inhibitors of glycogen phosphorylases. Chem Biodivers. 2009;6(6):864–874. | ||
Wen X, Sun H, Liu J, et al. Naturally occurring pentacyclic triterpenes as inhibitors of glycogen phosphorylase: synthesis, structure-activity relationships, and X-ray crystallographic studies. J Med Chem. 2008;51(12):3540–3554. | ||
Jeong BS, Lee MK, Kim YC, Lee ES. Modification of C2 functional group on asiatic acid and the evaluation of hepatoprotective effects. Arch Pharm Res. 2007;30(3):282–289. | ||
Jeong BS, Kim YC, Lee ES. Modification of C2,3,23,28 functional groups on asiatic acid and evaluation of hepatoprotective effects. Bull Korean Chem Soc. 2007;28(6):977–982. | ||
Zhao LX, Park HG, Jew SS, et al. Modification of C11, C28, C2,3,23 or C2,23,28 functional groups on Asiatic acid and evaluation of hepatoprotective effects. Bull Korean Chem Soc. 2007;28(6):970–976. | ||
Jing Y, Wang G, Ge Y, Xu M, Gong Z. Synthesis, anti-tumor and anti-angiogenic activity evaluations of asiatic acid amino acid derivatives. Molecules. 2015;20(4):7309–7324. | ||
van Moorselaar RJ, Voest EE. Angiogenesis in prostate cancer: its role in disease progression and possible therapeutic approaches. Mol Cell Endocrinol. 2002;197(1–2):239–250. | ||
McKeage MJ, Maharaj L, Berners-Price SJ. Mechanisms of cytotoxicity and antitumor activity of gold(I) phosphine complexes: the possible role of mitochondria. Coord Chem Rev. 2002;232(1–2):127–135. | ||
Seiler N. Pharmacological aspects of cytotoxic polyamine analogs and derivatives for cancer therapy. Pharmacol Ther. 2005;107(1):99–119. | ||
Shapiro GI, Harper JW. Anticancer drug targets: cell cycle and checkpoint control. J Clin Invest. 1999;104(12):1645–1653. | ||
Israels ED, Israels LG. The cell cycle. Oncologist. 2000;5(6):510–513. | ||
Antonsson B, Martinou JC. The Bcl-2 protein family. Exp Cell Res. 2000;256(1):50–57. | ||
Kornblau SM. The role of apoptosis in the pathogenesis, prognosis, and therapy of hematologic malignancies. Leukemia. 1998;12(Suppl 1):S41–S46. | ||
Polverino AJ, Patterson SD. Selective activation of caspases during apoptotic induction in HL-60 cells. Effects of a tetrapeptide inhibitor. J Biol Chem. 1997;272(11):7013–7021. | ||
Thornberry NA. Caspases: key mediators of apoptosis. Chem Biol. 1998;5(5):R97–R103. | ||
Bratton SB, Walker G, Srinivasula SM, et al. Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 2001;20(5):998–1009. | ||
Antonsson B. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277(5324):370–372. | ||
Zamzami N, Marchetti P, Castedo M, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182(2):367–377. | ||
Hua H, Li M, Luo T, Yin Y, Jiang Y. Matrix metalloproteinases in tumorigenesis: an evolving paradigm. Cell Mol Life Sci. 2011;68(23):3853–3868. | ||
Lin CY, Tsai PH, Kandaswami CC, et al. Matrix metalloproteinase-9 cooperates with transcription factor Snail to induce epithelial-mesenchymal transition. Cancer Sci. 2011;102(4):815–827. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.