")
Back to Journals » Clinical Ophthalmology » Volume 9
A single-arm, investigator-initiated study of the efficacy, safety, and tolerability of intravitreal aflibercept injection in subjects with exudative age-related macular degeneration previously treated with ranibizumab or bevacizumab (ASSESS study): 12-month analysis
Authors Singh R, Srivastava S, Ehlers J, Silva F, Bedi R, Schachat A, Kaiser P
Received 21 April 2015
Accepted for publication 2 June 2015
Published 22 September 2015 Volume 2015:9 Pages 1759—1766
DOI https://doi.org/10.2147/OPTH.S87043
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Rishi P Singh, Sunil K Srivastava, Justis P Ehlers, Fabiana Q Silva, Rumneek Bedi, Andrew P Schachat, Peter K Kaiser
Cole Eye Institute, Cleveland Clinic, Cleveland, OH, USA
Summary statement: In subjects with active exudative age-related macular degeneration, treating with a fixed intravitreal aflibercept injection dosing regimen for 12 months demonstrated improved anatomic and vision endpoints from baseline.
Purpose: Switching therapies in neovascular age-related macular degeneration (AMD) may offer an advantage for some patients. This study evaluates the efficacy of intravitreal aflibercept injection (IAI) in subjects previously treated with ranibizumab and/or bevacizumab.
Methods: Subjects (n=26) were given monthly 2 mg of IAI for 3 months, followed by 2 mg once in every 2 months for up to 12 months. The mean absolute change from baseline in central subfield thickness (CST) measured by optical coherence tomography and the mean change from baseline in best-corrected visual acuity (BCVA) early treatment in diabetic retinopathy study (ETDRS) letter score were obtained. Additionally, the percentage of subjects who gained or lost ≥15 letters of vision and the percentage of subjects who are 20/40 or better or 20/200 or worse were evaluated.
Results: There was a mean decrease in CST of -50.3 µm (P<0.001) and a mean increase in ETDRS BCVA of +9.2 letters (P<0.001). Twenty-seven percent of subjects experienced a ≥15-letter improvement in visual acuity, and no subject lost ≥3 lines of vision from baseline. Fifty percent of subjects were 20/40 or better, and 11.5% of subjects were 20/200 or worse at month 12.
Conclusion: Fixed IAI dosing regimen for 12 months demonstrated improved anatomic and vision endpoints in subjects with active exudative AMD.
Keywords: aflibercept, age-related macular degeneration, bevacizumab, ranibizumab, vascular endothelial growth factors
Corrigendum for this paper has been published
Introduction
Inhibitors of vascular endothelial growth factors (VEGFs) have provided significant therapeutic benefit in subjects suffering from exudative age-related macular degeneration (AMD).1–3 Aflibercept (Eylea; Regeneron Pharmaceuticals, Inc., Tarrytown, NY, USA) is a recombinant fusion protein consisting of key human VEGF receptor (VEGFR) extracellular domains from receptors 1 and 2 (VEGFR1 and VEGFR2) fused to the Fc domain of human IgG1.4 Aflibercept has several theoretical advantages over other pan-VEGF blockers: 1) it has a much higher binding affinity for VEGF-A (~0.5 pM dissociation constant for VEGF165 and VEGF121) than either bevacizumab or ranibizumab;5 2) it binds related growth factors such as placental growth factors 1 and 2 (PLGF1 and PLGF2), and VEGF-B, which may be advantageous in certain disease situations, including retinal neovascularization;6 and 3) the vitreous half-life of aflibercept (7.13 days) is longer than ranibizumab (4.75 days), but slightly shorter than bevacizumab (8.25 days).7
One of the limitations of the Phase III trials of intravitreal aflibercept injection (IAI) was the use of a fixed dosing scheme in the 1st year. The majority of retina specialists do not treat using a fixed dosing pattern used in the VIEW studies. Subjects are generally treated with as-needed dosing (pro re nata [PRN]) or on a treat-and-extend (TAE) paradigm. Studies such as the Comparison of Age-Related Macular Degeneration Treatment Trials (CATT) and the HARBOR trial have indicated that these treatment regimens are similar, but not fully equivalent to fixed dosing.8,9 More concerning are the results of the HORIZON AMD, SECURE, and SEVEN-UP studies, which demonstrated significant declines in visual acuity in subjects transitioned to PRN treatment from a fixed dosing scheme.10–12 Finally, there are little data for continuing fixed dosing in subsequent years and even less data on the outcome of switching patients from a PRN to fixed dosing regimen.
The ASSESS study is a 12-month, prospective, interventional, single-arm trial that transitioned patients from other anti-VEGF therapies to IAI using a fixed dosing regimen. The 6-month interim analysis of the ASSESS study demonstrated a mean decrease in central subfield thickness (CST) of 38.6 μm (P<0.001) and a mean increase in ETDRS best-corrected visual acuity (BCVA) of +5.9 letters (P<0.001).13 All the subjects were visually stable within the study, and the majority of subjects had either stable or improved anatomical optical coherence tomography (OCT) findings without any significant systemic or local adverse events (AEs). Herein, we report the 12-month results of the ASSESS study in subjects with active exudative AMD switched to fixed dosing IAI therapy.
Materials and methods
Study design
The ASSESS study is a prospective, single-arm, investigator-initiated study. The study was performed at the Cole Eye Institute, Cleveland, OH, and received approval from the Cleveland Clinic Investigational Review Board. All study-related procedures were performed in accordance with good clinical practice (International Conference on Harmonization of Technical Requirements of Pharmaceuticals for Human Use E6), applicable Food and Drug Administration (FDA) regulations, and the Health Insurance Portability and Accountability Act. All subjects provided written informed consent for participation in the study.
Participants
Inclusion and exclusion criteria have previously been reported.13 Subjects were included in the study if they met the following criteria: 1) active subfoveal choroidal neovascularization (CNV) secondary to exudative AMD confirmed by fluorescein angiography; 2) E-ETDRS vision of 25–80 letters (Snellen equivalent of ~20/25 to 20/320); 3) at least one prior injection of 1.25 mg bevacizumab or 0.5 mg ranibizumab (Avastin and Lucentis, respectively; Genentech Inc., South San Francisco, CA, USA) within 3 months of enrollment; and 4) had an initial response on OCT defined as a decrease of retinal edema and/or subretinal fluid to anti-VEGF injections followed by recurrent increase in fluid on OCT (further defined as intraretinal, cystoid, subretinal fluid, or worsening pigment epithelial detachment [PED]) or the presence of a new hemorrhage on clinical examination. The study’s inclusion criteria intended to define a responder population to previous anti-VEGF treatment, which is in contrast to previous case series that focused on evaluating IAI in a nonresponding population.
The key exclusion criteria included any prior concomitant therapy with another investigational agent to treat neovascular AMD in the study eye, history of pars plana vitrectomy, trabeculectomy, surgery for retinal detachment, or any intraocular or periocular surgery in the study eye within 3 months of baseline, prior treatment with verteporfin photodynamic therapy (Visudyne, Valeant, New York, NY, USA) in the study eye, history of subfoveal laser photocoagulation, uncontrolled glaucoma, or uveitis. Prior systemic anti-VEGF therapy was only allowed up to 3 months prior to study enrollment. Additional systemic anti-VEGF therapy was not allowed during the study. Subjects who had a history of vascular diseases including diabetic retinopathy or previous retinal vein occlusion affecting the retina within the study eye were excluded from the study. Fellow eye treatment was allowed, and the interval of treatment was based on the investigator’s discretion.
Visits and assessments
Subjects were given 2 mg (0.05 mL) of IAI administered every month for the first 3 months, followed by 2 mg (0.05 mL) once every 2 months as per the drug label (Figure 1) for the next 9 months. Although imaging and clinical values were recorded at follow-up visits, treatment decisions were not based on these values, but on a fixed dosing regimen. Regeneron Pharmaceuticals, Inc. supplied the intravitreal aflibercept. The drug was administered using the standard aseptic intravitreal techniques as detailed in the package insert.14
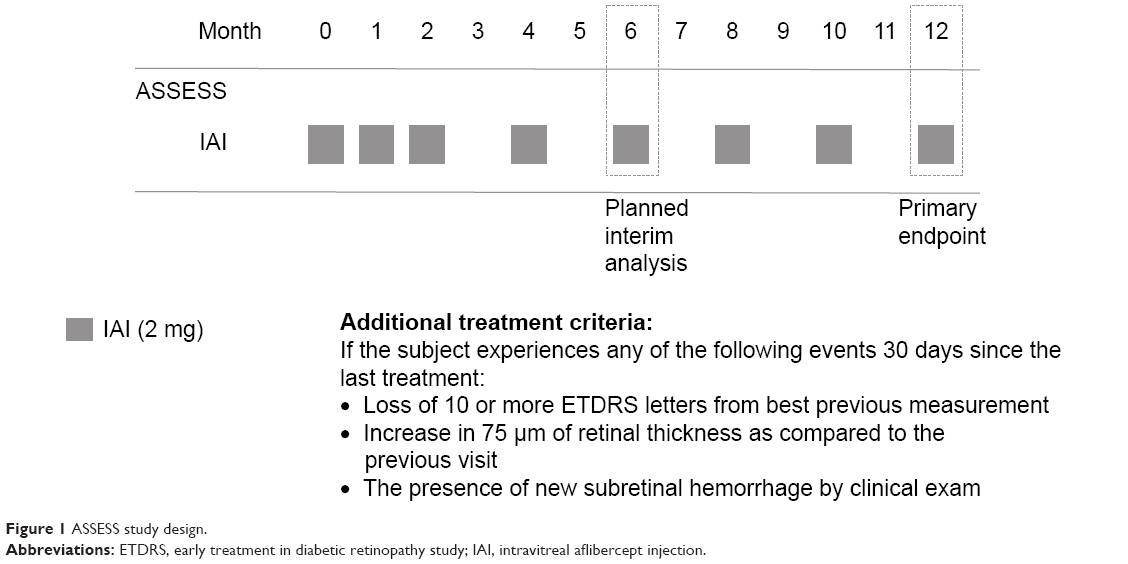 | Figure 1 ASSESS study design. |
After enrollment into the study, subjects were seen and treated according to the study schema as shown in Figure 1. Subjects were evaluated every month for the first 3 months, and then every 2 months thereafter until month 12. At each study visit, subjects were assessed using the E-ETDRS chart (M&S Systems) and protocol visual acuity measurement consisting of BCVA using a forced choice paradigm.15 Subjects also underwent a comprehensive eye examination at each visit. Spectral domain OCT scanning was performed on both eyes at every visit. The scanning protocol consisted of a series of high definition 6.0 mm linear scans centered on the fovea as well as a macular cube scan using the Cirrus SDOCT (Humphrey Zeiss, Inc., San Leandro, CA, USA; software version 6.0). Two independent graders scored the qualitative morphological patterns of the OCT scans. CST was measured using the automated software. Manual registration of pre-IAI, IAI, and post-IAI OCT images was done in cases of inaccurate automated registration by the Zeiss software before recording the CST.
Study endpoints
The primary study endpoint was the mean absolute change from baseline in CST (defined as the average thickness within the central 1 mm subfield) at month 12. Secondary outcomes included the mean change from baseline in BCVA score, change from baseline in macular volume, and cube average thickness by SDOCT, the percentage of subjects who gained greater than 15 letters of vision, the percentage of subjects who lost greater than 15 letters of vision, the percentage of subjects who were 20/40 or better, and the percentage of subjects who were 20/200 or worse. The incidence of ocular and nonocular AEs and serious AEs were also documented.
Safety analysis
Safety was monitored through the collection and summary of ocular and nonocular AEs, systemic AEs (SAEs), and ocular assessments. The occurrence of any severe postoperative complications after the first IAI was recorded, including infection, inflammation, arteriothrombolic events, and death. Nondirective questioning was performed by the study coordinator to prompt AE reports from subjects at each study visit. All AEs and SAEs, whether volunteered by the subject, discovered by the study site personnel during questioning, or detected by examination, laboratory testing, or other means, were recorded in the case report forms.
Statistical analysis
Measures were summarized using means, standard deviations, median, and range. Normality of measures was evaluated using the Shapiro–Wilk test. Since the data were normally distributed, comparisons with and between groups were performed using two-sided paired t-tests. Where appropriate, sensitivity analyses using nonparametric Wilcoxon signed rank tests were also performed. Analyses were performed using SAS software version 9.2 (SAS Institute, Inc., Cary, NC, USA). A significance level of 0.05 was assumed for all tests.
Results
Twenty-six subjects were included in the study (Table 1).
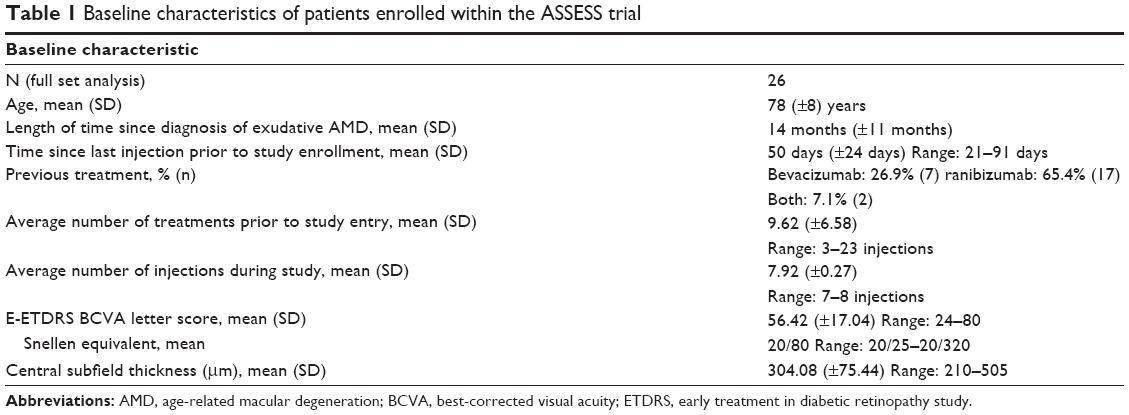 | Table 1 Baseline characteristics of patients enrolled within the ASSESS trial |
The average age of the subject was 78 years, ranging from 69 to 90 years. The average length of time since diagnosis and first treatment of exudative AMD was 14±11 months (range 3–35 months). Twenty-seven percent (seven subjects) were previously treated with bevacizumab, 65.4% (17 subjects) were previously treated with ranibizumab, and 7.1% of subjects were treated with both drugs. The average number of anti-VEGF treatments prior to study entry was 9.6 injections (range 3–23 injections), and all the subjects were mainly treated with PRN. This is accounted by the fact that these subjects were referred by outside physicians, without complete documentation of how many previous anti-VEGF injections were given to the patient prior to study entry. The average number of injections during the study was 7.92 (range 7–8 injections). Classic CNV was the initial lesion subtype in 30.77% of subjects, whereas 69.23% of patients had occult CNV at study enrollment. No patients with retinal angiomatous proliferation were enrolled.
Prior to study entry, average CST was 316 μm, and average VA was 20/63. The mean baseline E-ETDRS BCVA letter score upon entry was 56.4 letters (Snellen equivalent ~20/80). The mean baseline CST on study entry was 304.1 μm (range 210–505 μm). Thirty-five percent of subjects were 20/40 or better, and 23.1% of subjects were 20/200 or worse at baseline.
The primary outcome was the mean change in CST at month 12, which improved from 304.1 to 253.7 μm at month 12 (−50.3 μm) and was significantly improved from baseline in the study eye at all visits (M12: P<0.001) (Figure 2). The mean BCVA score improved from 56.4 at baseline to 65.6 at month 12 (+9.2 letters) (Figure 3). There was also a significant improvement in BCVA in all follow-up visits except month 1 (M12: P<0.001). The average macular volume improved from 10.3 mm3 at baseline to 9.9 mm3 at month 12 (−0.4 mm3 reduction from baseline). There were statistically significant changes from baseline at all follow-up visits (M12: P=0.038). The mean cube average thickness improved from 286.2 μm at baseline to 274.3 μm at month 12 (−11.9 μm reduction from baseline) and was statistically significant at all follow-up time periods (M12: P=0.038).
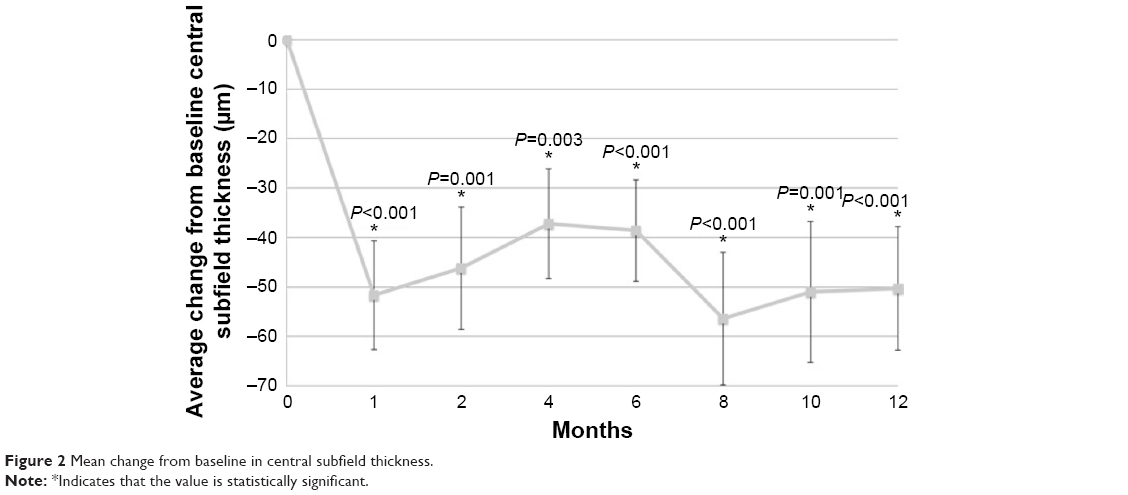 | Figure 2 Mean change from baseline in central subfield thickness. |
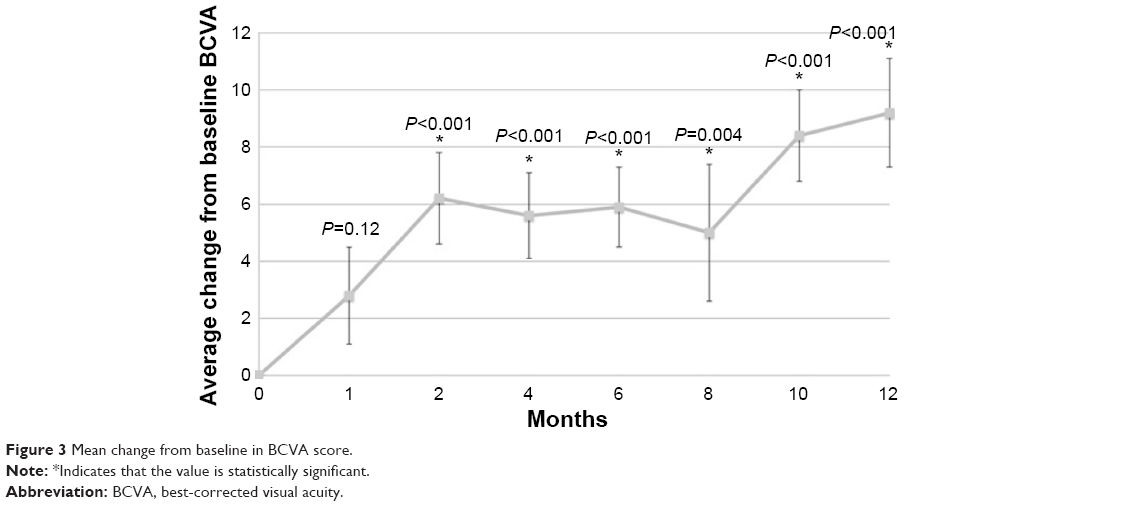 | Figure 3 Mean change from baseline in BCVA score. |
At 12 months, 26.9% of subjects gained at least 15 letters of visual acuity (Figure 4). One-hundred percent of subjects were considered visually stable (defined as the number of subjects who gained, were stable, or lost up to two lines of acuity). Fifty percent of subjects were 20/40 or better, and 11.5% of patients were 20/200 or worse at month 12, both improvements from baseline.
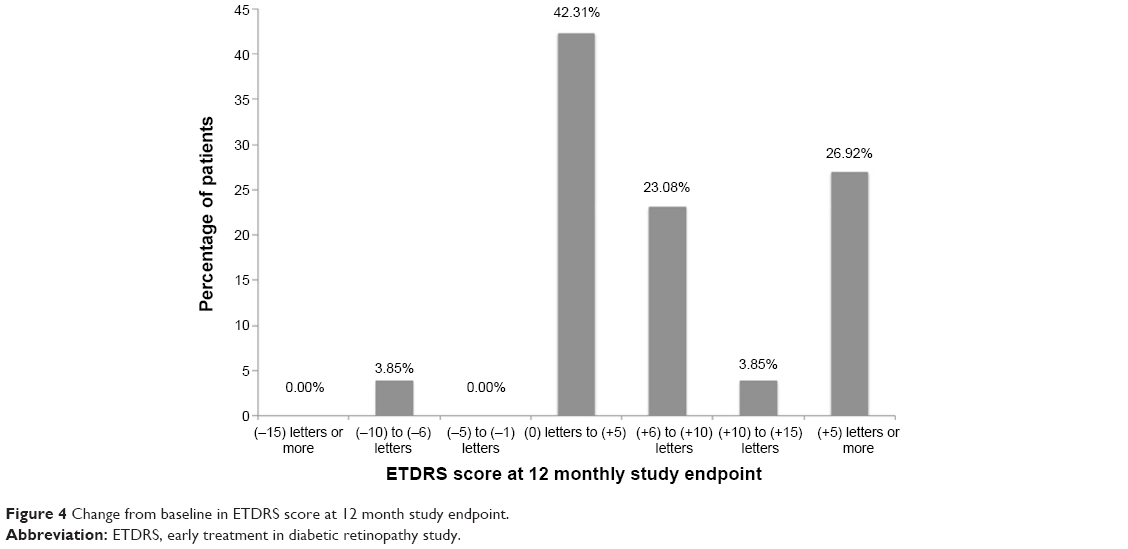 | Figure 4 Change from baseline in ETDRS score at 12 month study endpoint. |
Qualitative OCT grading of registered scans was performed to observe anatomical changes, specifically for the presence of subretinal fluid, intraretinal fluid (or cysts), and PED. A PED was defined as a highly reflective elevated band of retinal pigment epithelium overlying a less reflective cavity, which is almost optically empty. Two independent OCT graders performed all morphologic analysis and defined the changes from baseline as improved, stable, or worse for each OCT characteristic. Any disagreement between graders was given to a third grader for adjudication.
A total of 45.5% patients manifested intraretinal fluid, and 63.6% presented with subretinal fluid at baseline. At 12 months, 22.7% of study eyes exhibited intraretinal fluid, and 50% of study eyes exhibited subretinal fluid. At month 12, 30.8% of study eyes exhibited an anatomical improvement in subretinal fluid, 46.2% demonstrated anatomically stable subretinal fluid in comparison to their baseline visit, and 23.1% had worsened subretinal fluid (Figure 5). In comparison to baseline, 34.6% of subjects had anatomically improved intraretinal fluid, 53.9% were anatomically stable, and 11.5% had worsened intraretinal fluid. Improvement in PED was seen in 15.4% of study eyes, 69.2% were anatomically stable, and 15.4% of PEDs were anatomically worse compared to baseline. Figure 6 is a case representation of a patient within the trial.
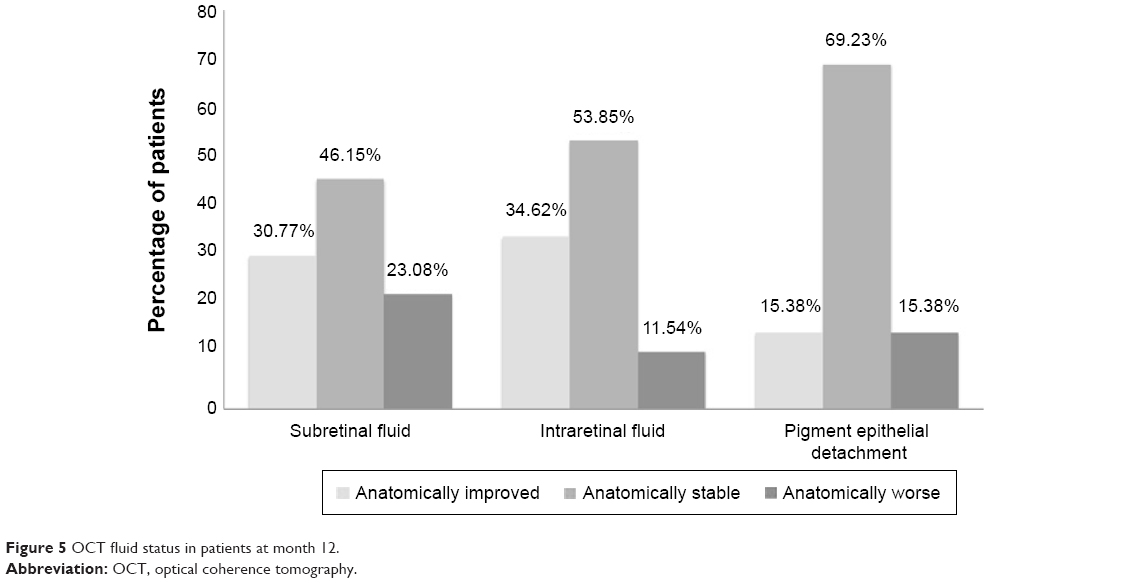 | Figure 5 OCT fluid status in patients at month 12. |
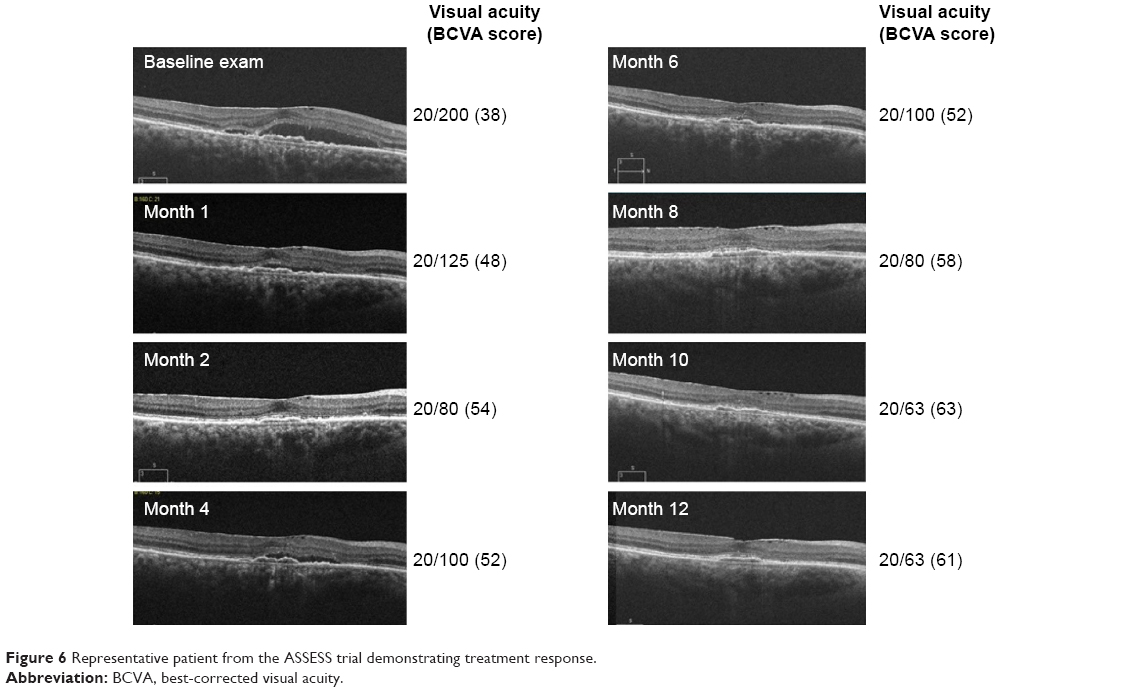 | Figure 6 Representative patient from the ASSESS trial demonstrating treatment response. |
One serious ocular AE (3.85%) occurred at month 8 of the study, and was not thought to be related to the study medication. The patient began noticing a myopic shift, and vision loss due to cataract progression was identified. The patient underwent uncomplicated phacoemulsification and lens implant placement with significant improvement in visual acuity. No other serious ocular or SAEs were identified during the 12-month duration of this study including Anti-platelet Trialist Collaborative-defined events.
Discussion
This study is the first prospective study examining the effect of a fixed dosing paradigm with intravitreal aflibercept on subjects transitioned from other anti-VEGF agents who were treated earlier in a PRN fashion. We found a statistically significant improvement from baseline in the primary outcome of CST, as well as all secondary outcomes including BCVA, average thickness, and total macular volume. This 12-month interim analysis demonstrated a mean decrease in CST of 50.3 μm (P<0.001) and a mean increase in E-ETDRS visual acuity of +9.2 letters (P<0.001). All subjects were considered visually stable within the study, and the majority of subjects had either stable or improved anatomical OCT findings with regard to subretinal fluid, intraretinal fluid, and PEDs. No significant AEs was identified.
These results indicate that the majority of subjects demonstrated improved visual and anatomical outcomes after switching from other anti-VEGF agents and treated with a fixed dosing scheme. The population selected for this study utilized subjects who were considered anti-VEGF responders albeit requiring frequent treatment (average of more than nine anti-VEGF injections prior to enrollment). These subjects were not necessarily “treatment failures” but were more consistent with the average subjects who could experience multiple improvements and declines in their clinical course. The 12-month interim analysis of the ASSESS study supports the conversion of patients from TAE and PRN regimens from other anti-VEGF agents to a fixed dosing pattern of every 8 weeks with IAI.
There have been numerous reports on the conversion of patients to IAI from other therapies.16–19 The drawbacks of such studies include their retrospective nature, lack of standardized treatment interval, and a lack of protocol visual or anatomic analyses. The ASSESS study tried to avoid some of these potential biases; however, there are still drawbacks to our study, including the small sample size and only 12 months of follow-up. This study does have significant strengths including its prospective nature, strict entry criteria, and rigorous evaluation of anatomical and visual outcomes outweigh these drawbacks.
Tachyphylaxis has been suggested in subjects treated for exudative AMD, and some studies have shown better resolution of fluid after switching anti-VEGF treatments as was done in this study.16–19 One may argue that patients repeatedly treated with ranibizumab and bevacizumab may demonstrate tachyphylaxis over time. However, this study attempted to alleviate the bias of tachyphylaxis as an explanation for the improved outcomes by including patients responding to previous anti-VEGF agents.
The pharmacodynamics of aflibercept is different than other intravitreal anti-VEGF treatments. In vitro studies of aflibercept demonstrate a binding affinity 94 times that of ranibizumab and 119 times that of bevacizumab.5 Additionally, the sustained benefits of aflibercept in this 12-month study could also be supported by mathematical models predicting that a single IAI 2.0 mg injection could last for 48–83 days in comparison to only 30 days with an intravitreal ranibizumab injection.20 The primary analysis of this 12-month study demonstrates a majority of patients with stable or improved anatomical outcomes. A similar outcome was also seen in a post hoc analysis of the combined VIEW trials.21 This analysis showed that the anatomical benefits of aflibercept at 1 year in comparison to other anti-VEGF treatments were observed; 38% of patients on ranibizumab had persistent fluid for 1 year in comparison to 27.6% of patients in the 2.0 mg every 4 weeks arm and 32.3% in the IAI 2.0 mg every 8 weeks (following three initial monthly doses) arm.21
Additionally, the fixed dosing regimen every 8 weeks with IAI, as per the FDA label, within this study could be another factor contributing to the apparently sustained long-term benefits. While TAE is the most popular dosing frequency of retina specialists, one may argue that using the fixed dosing regimen presented in this study might decrease the burden to both the patient and the physician by taking the added work out of calculating TAE intervals, reducing the need for monthly assessments for PRN treatment potentially by reducing the consequence of extending the intervals too long, which may result in an AE such as a submacular hemorrhage, and by reducing the need for repeated examinations and diagnostic testings. Moreover, the results of the HORIZON AMD, SECURE, and SEVEN-UP studies demonstrate declines in visual acuity in subjects transitioned to PRN treatment from a fixed dosing scheme and, therefore, a fixed dosing regimen may return the vision and anatomy to the best possible levels.10–12
The aim of this study was to determine the outcomes following the switch from other anti-VEGF therapies to a fixed IAI dosing regimen following the FDA drug label. These subjects were not necessarily “treatment failures” but were more consistent with average subjects within clinical practice who are being treated in a nonfixed dosing pattern. Results of this study demonstrated that 100% of subjects remained visually stable and 96.2% gained vision after switching to this fixed IAI dosing regimen. It is possible the subjects could have also remained stable had they continued their original treatment paradigm albeit transitioned to a fixed regimen. However, it is our assumption that the primary result seen in the ASSESS study is due to a combination of exposure to a different anti-VEGF drug, with theoretical benefits over their previous treatments, and a switch to a fixed pattern of treatment.
Funding
Support provided by a research trial grant from Regeneron, Inc. and an unrestricted grant from research to prevent blindness.
Disclosure
RPS: Thrombogenics (consultant), Alcon (consultant), Regeneron (consultant); SKS: Bausch and Lomb (consultant), Allergan (contract research), Bioptigen (royalty payment), Synergetics (royalty payment); JPE: Bioptigen (royalty payment), Thrombogenics (consultant, contract research), Genentech (contract research), Leica (consultant), Zeiss (consultant), Synergetics (royalty payment); RB: None; APS: None; PKK: Thrombogenics (consultant), Alcon (consultant), Novartis (consultant), Allegro (consultant). The authors report no other conflicts of interest in this work.
References
Rosenfeld PJ, Brown DM, Heier JS, et al; MARINA Study Group. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–1431. | ||
ANCHOR Study Group. Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: two-year results of the ANCHOR study. Ophthalmology. 2009;116(1):57–65. | ||
Busbee BG, Ho AC, Brown DM, et al; HARBOR Study Group. Twelve-month efficacy and safety of 0.5 mg or 2.0 mg ranibizumab in patients with subfoveal neovascular age-related macular degeneration. Ophthalmology. 2013;120:1046–1056. | ||
Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. | ||
Papadopoulos N, Martin J, Ruan Q, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15:171–185. | ||
Rakic JM, Lambert V, Devy L, et al. Placental growth factor, a member of the VEGF family, contributes to the development of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3186–3193. | ||
Stewart MW. What are the half-lives of ranibizumab and aflibercept (VEGF Trap-eye) in human eyes? Calculations with a mathematical model. Eye Rep. 2011;1.1:e5. | ||
CATT Research Group, Martin DF, Maguire MG, et al. Ranibizumab and Bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011;364:1897–1908. | ||
Martin DF, Maguire MG, Fine SL, et al; Comparison of Age-Related Macular Degeneration Treatment Trials (CATT) Research Group. Ranibizumab and bevacizumab for neovascular age-related macular degeneration: two-year results. Ophthalmology. 2012;119: 1388–1398. | ||
Rofagha S, Bhisitkul RB, Boyer DS, Sadda SR, Zhang K; SEVEN-UP Study Group. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multi-center cohort study (SEVEN-UP). Ophthalmology. 2013;120:1–8. | ||
Silva R, Axer-Siegel R, Eldem B, et al; SECURE Study Group. The SECURE study: long-term safety of ranibizumab 0.5 mg in neovascular age-related macular degeneration. Ophthalmology. 2013;120:130–139. | ||
Singer MA, Awh CC, Sadda S, et al. HORIZON: an open-label extension trial of ranibizumab for choroidal neovascularization secondary to age-related macular degeneration. Ophthalmology. 2012;119:1175–1183. | ||
Singh RP, Srivastava S, Ehlers JP, Bedi R, Schachat AP, Kaiser PK. A single-arm, investigator-initiated study of the efficacy, safety and tolerability of intravitreal aflibercept injection in subjects with exudative age-related macular degeneration, previously treated with ranibizumab or bevacizumab: 6-month interim analysis. Br J Ophthalmol. 2014;98(suppl 1):i22–i27. | ||
Eylea [package insert]. Tarrytown, NY: Regeneron Pharmaceuticals, Inc; 2011. Available from: http://www.regeneron.com/Eylea/eylea-fpi.pdf | ||
Beck RW, Moke PS, Turpin AH, et al. A computerized method of visual acuity testing: adaptation of the early treatment of diabetic retinopathy study testing protocol. Am J Ophthalmol. 2003;135(2):194–205. | ||
Cho H, Shah CP, Weber M, Heier JS. Aflibercept for exudative AMD with persistent fluid on ranibizumab and/or bevacizumab. Br J Ophthalmol. 2013;0:1–4. | ||
Bakall B, Folk JC, Boldt HC, et al. Aflibercept therapy for exudative age-related macular degeneration. Am J Ophthalmol. 2013;156:15–22. | ||
Ho VY, Yeh S, Olsen TW, et al. Short-term outcomes of aflibercept for neovascular age-related macular degeneration in eyes previously treated with other vascular endothelial growth factor inhibitors. Am J Ophthalmol. 2013;156:23–28. | ||
Yonekawa Y, Andreoli C, Miller JB, et al. Conversion to aflibercept for chronic refractory or recurrent neovascular age-related macular degeneration. Am J Ophthalmol. 2013;156:29–35. | ||
Stewart MW, Rosenfeld PJ. Predicted biological activity of intravitreal VEGF Trap. Br J Ophthalmol. 2008;92(5):667–668. | ||
Heier JS, Brown DM, Chong V, et al; VIEW 1 and VIEW 2 Study Groups. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology. 2012;119:2537–2548. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.