")
Back to Journals » Clinical Epidemiology » Volume 13
A Search for Undiagnosed Charcot-Marie-Tooth Disease Among Patients Registered with Unspecified Polyneuropathy in the Danish National Patient Registry
Authors Vaeth S , Andersen H , Christensen R, Jensen UB
Received 18 November 2020
Accepted for publication 9 January 2021
Published 16 February 2021 Volume 2021:13 Pages 113—120
DOI https://doi.org/10.2147/CLEP.S292676
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Henrik Sørensen
Signe Vaeth,1 Henning Andersen,2 Rikke Christensen,1 Uffe Birk Jensen1
1Department of Clinical Genetics, Aarhus University Hospital, Aarhus N, Denmark; 2Department of Neurology, Aarhus University Hospital, Aarhus C, Denmark
Correspondence: Signe Vaeth
Department of Clinical Genetics, Aarhus University Hospital, Brendstrupgaardsvej 21 C, Skejby, Aarhus N, DK-8200, Denmark
Tel + 45 78455510
Fax +45 86783181
Email [email protected]
Purpose: In a recent study based on data from the Danish National Patients Registry (DNPR), we reported the prevalence of Charcot-Marie-Tooth disease (CMT) in Denmark to be 22.5 per 100.000. This prevalence is most likely a minimum estimate, as many cases of CMT may be misdiagnosed or remain undiagnosed due to the heterogeneous nature of the disorder. The aim of this study was to investigate the possible number of undiagnosed CMT cases among patients registered with unspecified polyneuropathy (UP) diagnoses in the DNPR.
Patients and Methods: From the DNPR we extracted data on all patients given an UP diagnosis in the period 1977 to 2012. We selected all patients diagnosed with a primary UP diagnosis before age 40 at a department of neurology, neurophysiology, clinical genetics or pediatrics, and excluded all patients with a specified polyneuropathy diagnosis or with diagnostic codes related to alcohol and diabetes mellitus. To assess the proportion of possible CMT patients, we performed medical record review in a random sample of patients diagnosed in the Central Denmark Region. To further investigate the possible overlap between UP and CMT in the DNPR, we performed a series of searches for ICD-8 and ICD-10 codes related to CMT.
Results: Between 1977 and 2012, 30.903 patients were diagnosed with UP without also being diagnosed with CMT. A total of 940 patients fulfilled the selection criteria. We found that 21.5% (95% CI 13.1%– 32.2%) of the cases in the random sample fulfilled our criteria for CMT. This estimate increases the prevalence of CMT in Denmark with 3.6 per 100,000 (95% CI 2.4%– 5.5%).
Conclusion: This study illustrates how hitherto undiagnosed CMT patients may be identified in the DNPR and further reports the number of possible CMT cases. Our results support the hypothesis that the true prevalence of CMT is higher than recently reported.
Keywords: registries, International Classification of Disease codes, epidemiology, hereditary neuropathy
Plain Language Summary
In this study we evaluate the large group of patients diagnosed with unspecified polyneuropathy (UP). We suspect that some of these patients may have a hereditary polyneuropathy called Charcot Marie Tooth disease, we therefore also suspect that the previously reported prevalence of Charcot Marie Tooth disease is underestimated. To investigate this, we collected data on all patients diagnosed with UP in the Danish National Patients Registry between 1977 and 2012. We further selected those UP patients who did not have diabetes- or alcohol-related diagnoses and who had received their diagnosis at a relevant clinical department. We then took a random sample from this selected group and reviewed their medical files to see how many fulfilled our criteria for CMT. We found that in total 30,903 patients were diagnosed with UP between 1977 and 2012, and 940 of these patients fulfilled our selection criteria (diagnosis at relevant clinical department and no alcohol or diabetes-related diagnosis). We found that 21.5% of random sample fulfilled our criteria for CMT. Our study shows that a considerable number of patients may have CMT but remain undiagnosed, and confirms that the previously reported prevalence of CMT is probably underestimated.
Introduction
Charcot-Marie-Tooth disease (CMT) is characterized by great clinical and genetic heterogeneity, which challenges the diagnosis of cases with mild or atypical symptoms. CMT displays great variation in age of onset, severity and symptoms combined with a broad phenotypic overlap with other neurological disorders such as hereditary sensory and autonomic neuropathies (HSAN), distal hereditary motor neuropathy (dHMN), spinal muscular atrophy (SMA) and hereditary spastic paraplegia (HSP).1,2
The most common phenotype includes length dependent and slowly progressive muscle atrophy and weakness starting in the lower limbs, and later progressing to the upper limbs. Onset is often within the first two decades. Deformities of the feet and hands are common, tendon reflexes are usually reduced or absent, and many patients have sensory loss, although motor symptoms are predominant.3
Nagai et al have studied the prevalence of CMT among patients with bilateral cavovarus feet presenting between age 3 and 18 and found that 76% (116/148) were later diagnosed with CMT.4 Laurá et al reported foot deformities in 71% of a large cohort recruited into the Inherited Neuropathies Consortium5 Some patients develop additional symptoms such as scoliosis, hip dysplasia and hearing loss.6–8 In a study of associated features in CMT by Werheid et al, the authors found one or more additional symptom in 87% (43/49) of CMT cases, deafness, scoliosis and tremor were all reported in association with mutations in a large number of different CMT-related genes.9
Recently, we have performed a study on the prevalence of CMT in Denmark using data on patients registered with a CMT diagnosis in the Danish National Patients Registry (DNPR). The prevalence in 2012 was 22.5 per 100,000.10 The DNPR is a high-quality national registry,11 and the validity of the CMT diagnosis in the registry is high.12 However, likely the CMT population in the DNPR is incomplete as patients with atypical or mild symptoms may be misdiagnosed or go undiagnosed. Furthermore, due to diagnostic administrative procedures, probably some patients will not have been registered with a CMT diagnosis if they were already diagnosed with unspecified polyneuropathy, and then later reclassified as CMT.
The aim of the present study was to investigate whether the prevalence of CMT in Denmark is higher than previously reported by estimating the proportion of possible CMT cases among patients diagnosed with unspecified polyneuropathy (UP) diagnoses in the DNPR. The investigation was performed using two different study approaches: 1) A medical record review and 2) A combined search strategy for diagnoses related to CMT (foot and hand deformities, scoliosis and hearing loss).
Patients and Methods
Setting and Data Sources
In 2012 the population in Denmark was 5.6 million.13 Every resident has access to tax-supported healthcare provided by The Danish National Health Service, and every resident is registered in the Civil Personal Registry and is given a unique identification number (CPR number). The CPR number allows for unique matching of data between databases.14 Since 1977, information from all somatic hospital admissions in Denmark has been registered in the DNPR. Since 1995, outpatient contacts have been registered. The DNPR includes information on discharge diagnosis, diagnosis type, admission date, and hospital and department type. The International Classification of Diseases (ICD) version 8 (ICD-8) was used until 1993 and was then replaced by the ICD version 10 (ICD-10).11 Denmark is divided into 5 regions. The Central Denmark Region had 1.271.223 inhabitants in 2012.13
Data analyses were performed using STATA version 13.1.15 Exact 95% confidence intervals (CI) were calculated based on binomial distribution. A significance level of 5% was used for hypothesis testing.
The study was approved by The Danish Data Protection Agency (1–16-02-18-12) and the local ethics committee of the Central Denmark Region (1–10-72-331-12).
Identification of Study Populations
Using the DNPR, we extracted data from the period 1977 to 2012 on all patients diagnosed with the following diagnoses (collectively labelled “unspecified polyneuropathy” (UP) in this paper): Polyneuritis (ICD-8 35401), Idiopathic progressive neuropathy (ICD-10 G60.3), Other hereditary and idiopathic neuropathies (G60.8), Hereditary and idiopathic neuropathy, unspecified (G60.9), Other specified polyneuropathies (G62.8) and Polyneuropathy, unspecified (G62.9). We excluded all patients already diagnosed with CMT (ICD-8 33009 or ICD-10 DG60.0) or Refsums disease (ICD-10 DG60.1). Thereafter, we excluded the following patient groups: 1) Patients who had not been diagnosed with UP at a department of neurology, neurophysiology, clinical genetics or pediatrics, 2) patients without a primary UP diagnosis, 3) patients with their first neuropathy diagnosis after age 40 and 4) patients registered with a specified polyneuropathy diagnosis (eg, inflammatory polyneuropathy) or a diagnosis related to alcohol or diabetes mellitus as these conditions may be the cause of polyneuropathy. This selection process is illustrated in Figure 1, and a list of all exclusion diagnoses is presented in Table 1.
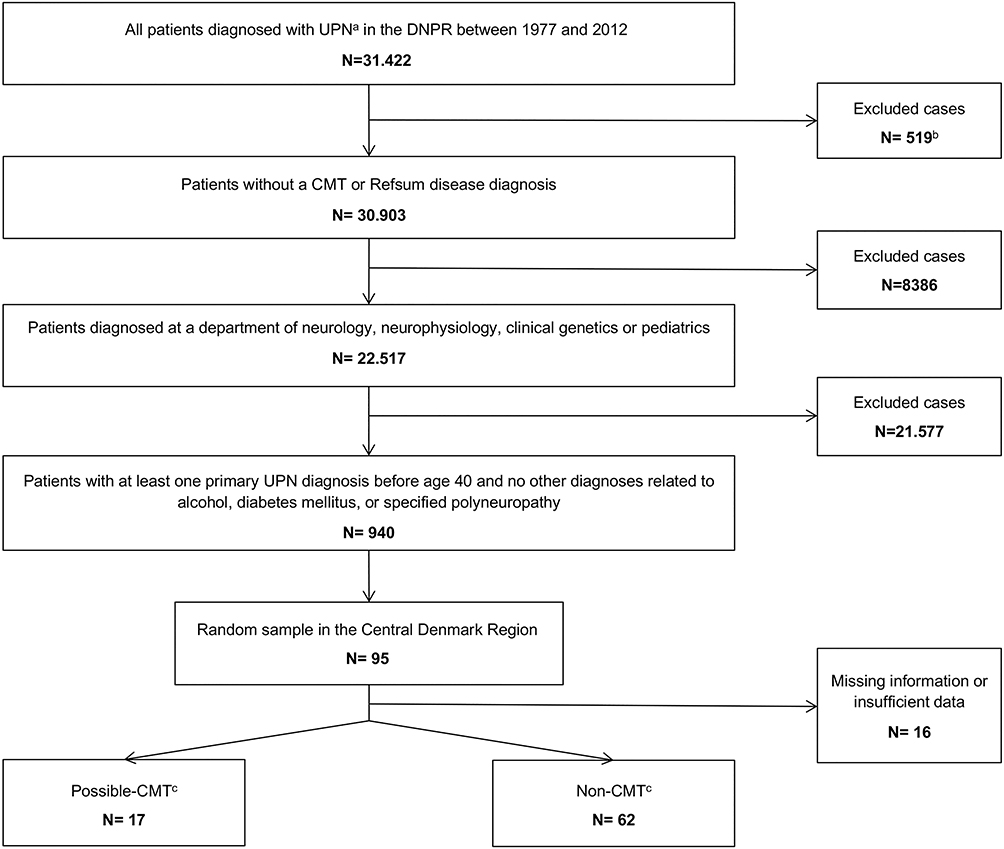 |
Figure 1 Flowchart of the selection process and review of the medical records. Abbreviations: UP, unspecified polyneuropathy; CMT, Charcot-Marie-Tooth disease. Notes: aDiagnostic codes ICD-8 35401, ICD-10 G60.3, G60.8, G60.9, G62.8 and G62.9; b516 CMT diagnoses and 3 Refsum disease diagnoses; cAccording to medical record review. |
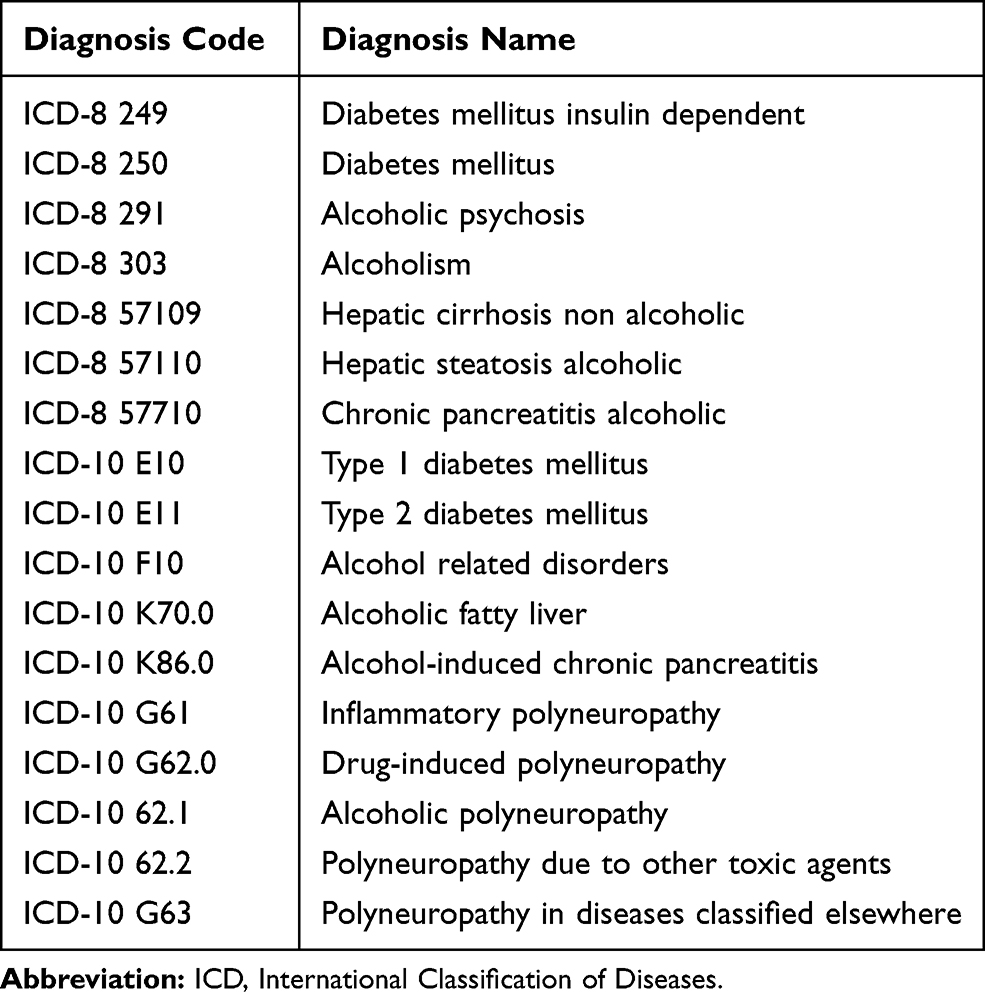 |
Table 1 Diagnoses Used as Exclusion Criteria |
From the final cohort, we selected those who had been given their first UP diagnosis in the Central Denmark Region and generated a random sample by randomly selecting 20 patients for every 5 calendar years. The random sample was then used in the medical record review.
The combined search strategy for diagnoses related CMT was performed on 1) the entire group of UP patients without a CMT diagnosis, and 2) the final study population (see Figure 1). To compare the findings with those in patients a CMT diagnosis, we performed the combined search on a cohort of patients diagnosed with CMT obtained by extracted data on all patients registered with a CMT diagnosis (ICD-10 DG60.0 Hereditary motor and sensory neuropathy and ICD-8 33009 Atrophia mm. neuropathica, Charcot-Marie-Tooth) from the DNPR between 1977 and 2012.
Medical Record Review
Medical records were identified and located using CPR numbers. Medical records were accessed without written informed consent from the patients; this was approved by the local ethics committee of the Central Denmark Region and The Danish Data Protection Agency. All cases were reviewed by SV and undetermined cases were reviewed together with HA. Cases were classified as “possible-CMT” or “non-CMT” using a list of seven criteria developed for validation of CMT diagnoses in a recent study.12 These criteria were based on the European CMT Consortium criteria,16,17 and are shown in Table 2. A case was labeled possible-CMT if 1) at least 4 of the 7 criteria were fulfilled, and 2) one of the fulfilled criteria was criterion 1, 2 or 3. The proportion of possible-CMT cases in the random sample was calculated and applied to the final study population to assess the volume of patients with UP who may have undiagnosed CMT.
 |
Table 2 Criteria for CMT Validation |
Combined Search Strategy for Diagnoses Related to CMT
We performed seven different searches using data from the DNPR, in which we counted the number of patients registered with a diagnosis related to hearing loss, limb deformities or scoliosis in different combinations (see Tables 3 and 4). The search was performed on the following three study populations: 1) patients diagnosed with UP who did not also have a CMT diagnosis, 2) the final study population (see Figures 1) and 3) a cohort of patients registered with a CMT diagnosis in the DNPR. We chose to restrict the hearing loss diagnoses to those given before the age of 40, to reduce the amount of cases with age-related hearing loss.
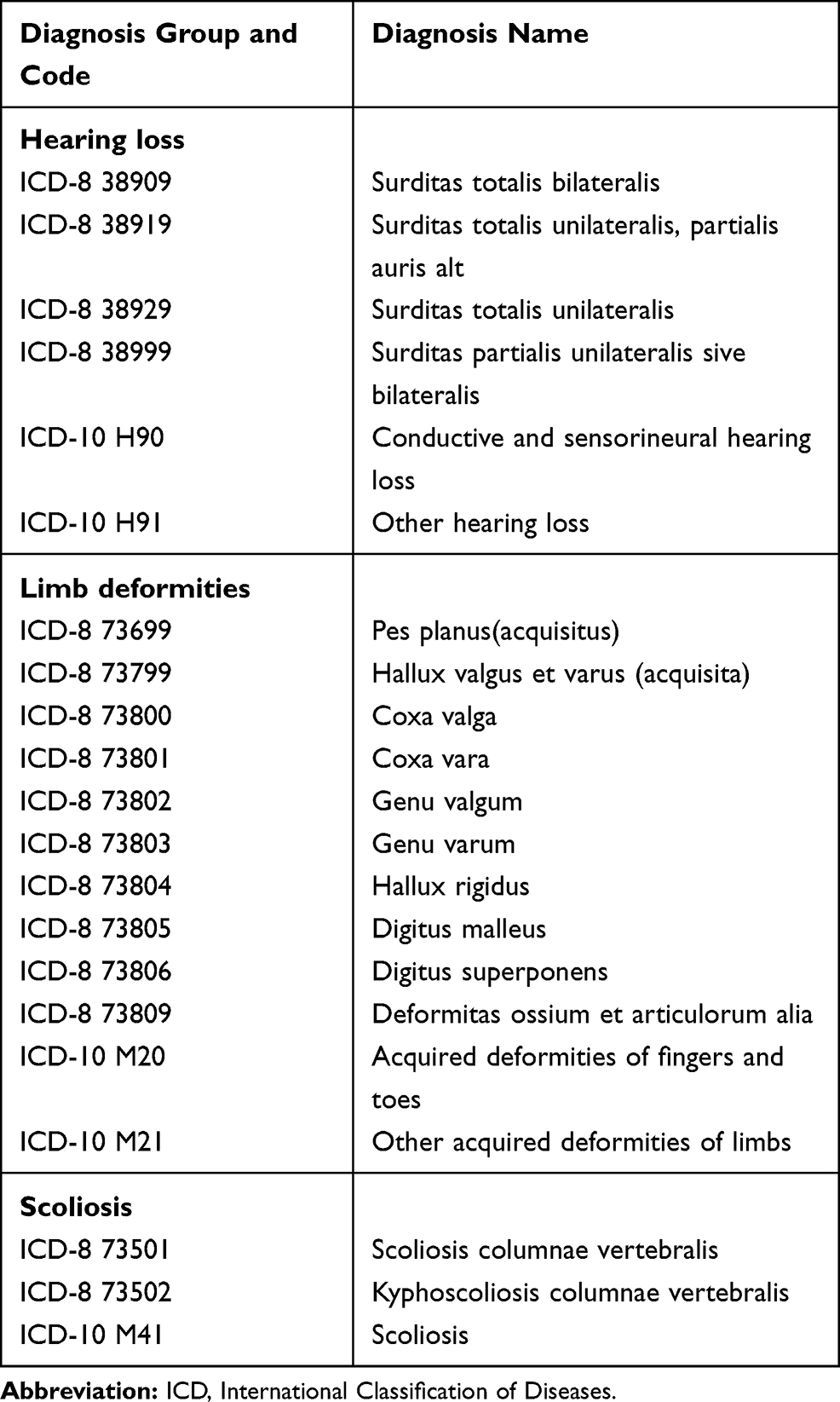 |
Table 3 Diagnosis Used in the Combined Search Strategy |
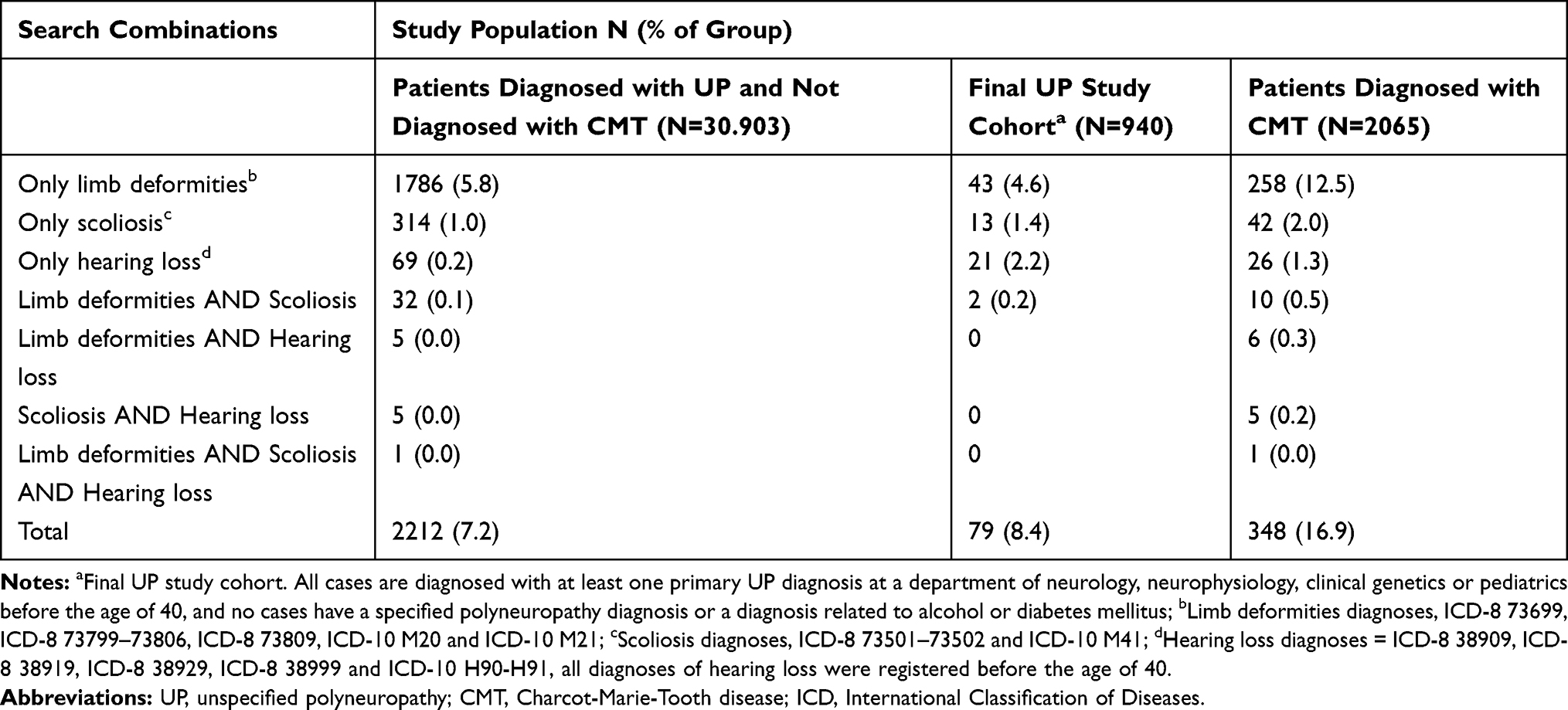 |
Table 4 Distribution of Cases in Three Study Populations from the Danish National Patients Registry According to Different Search Combinations on Diagnoses of Limb Deformities, Scoliosis and Hearing Loss |
Results
In total, 31.422 patients have been given an UP diagnosis in the DNPR between 1977 and 2012; hereof 516 were also diagnosed with CMT and 3 were diagnosed with Refsums disease. Of the remaining 30.903 cases, a total of 22.517 were diagnosed at a department of neurology, neurophysiology, clinical genetics or pediatrics, hereof 39.5% (95% CI 38.9–40.2) were female, and 60.5% (95% CI 59.8–61.1) were male. The vast majority was diagnosed with G62.9. After performing all exclusion steps, the final study cohort consisted of 940 cases (Figure 1).
Medical Record Review
In the final study cohort (n=940), a total of 202 cases were diagnosed in the Central Denmark Region. The random sample consisted of 95 cases and we were able to locate 85 medical records. All except one of the 10 missing medical records were dated earlier than 1994. Six records did not hold sufficient data to allow a conclusion. In total, 79 medical records were successfully assessed, hereof 17 fulfilled our criteria for CMT corresponding to 21.5% (95% CI 13.1%-32.2%) of the random sample. Among the 17 cases that fulfilled our CMT criteria, 1 had a genetically confirmed hereditary neuropathy with liability to pressure palsy (HNPP), 13 had a neurophysiological analysis result consistent with CMT and 1 had a nerve biopsy consistent with CMT. When applying the percentage of possible CMT to the study cohort of 940 cases, this results in approximately 200 patients with possible undiagnosed CMT in Denmark, and corresponds to an estimated increase in prevalence of 3.6 per 100000 (95% CI 2.4%–5.5%).
Based on this estimate and the results from our previous study,10 the prevalence of CMT in Denmark is 26.1 (22.5+3.6) per 100000 (95% CI 24.1%–28.0%).
Combined Search Strategy for Diagnoses Related to CMT
Our search results are presented in Table 4. A positive finding in one or more of the searches was recorded in 7.2% (95% CI 6.9%–7.5%) of the total UP cohort (2212/30.903) and in 8.4% (95% CI 6.7%–10.4%) of the final study cohort (79/940) compared to a significantly higher percentage, 16.9% (95% CI 15.3%–18.5%), the CMT cohort (348/2065). In all three cohorts, the search for limb deformities alone gave the highest number of positive findings. “Hearing loss diagnosed before age 40” was the only category with a significant difference between the two UP cohorts (p<0.0001); 2.2% in the selected cohort compared to 0.2% in the total UP cohort.
Discussion
The main finding of this study is that 21.5% (95% CI 13.1%–32.2%) of a selected cohort of patients diagnosed with UP may have undiagnosed CMT. This supports our hypothesis; that using only CMT diagnoses from the DNPR will lead to an underestimation of the prevalence of CMT in Denmark. Our finding is supported by a study from 1981 by Dyck et al. They performed an intensive evaluation of 205 patients with undiagnosed polyneuropathy, and found that 42% could be diagnosed with inherited neuropathies.18
Our study revealed only a very small difference between the number of CMT-related diagnoses (limb deformities, scoliosis and hearing loss) in the selected and the unselected UP cohort. Based on the strict selection criteria used in the selection of UP cases, we expected to find a larger difference. This finding suggests that limb deformities, scoliosis and hearing loss are not useful to identify undiagnosed CMT patients in the DNPR. Alternatively, the findings could indicate that the selection process of UP cases did not have the effect we expected - to increase the concentration of possible CMT cases.
A number of strengths and limitations should be considered when interpreting our results. Firstly, the DNPR is a very high-quality national registry containing prospectively collected data.11 The use of a national database minimizes selection bias, however, misclassification of patients will cause information bias.19 An unknown number of cases registered with the diagnoses used for selection in this study, may be misclassified, and therefore lead to a false selection. Likewise, an unknown number of true cases will not have been selected due to missing diagnosis. We have reported a high PPV of CMT diagnoses in DNPR (PPV=88.5).12 According to an extensive review by Schmidt et al,19 only two other diagnoses used in the present study (or closely related to) have been validated: diabetes mellitus (PPV= 96.9)20 and alcoholic cirrhosis (PPV=77.7).21 We do not know the completeness of the remaining diagnoses. Furthermore, in a patient with polyneuropathy a secondary diagnosis of limb deformities, scoliosis or hearing loss might go unregistered as a separate diagnosis in the medical record.
Secondly, our medical record review only provides the proportion of possible CMT cases in a strictly selected UP cohort. If additional medical record reviews had been performed in random samples from the unselected cohort and, eg, in a cohort without age-restriction, we would have achieved a more complete picture of the proportion of possibly undiagnosed CMT patients in the DNPR. Likewise, it could have been of interest to perform a review of the medical records within each of the diagnoses collectively termed UP (35401, G60.3, G60.8, G60.9, G62.8, and G62.9), as some may contain a higher proportion of possible-CMT patients than others. Furthermore, we have chosen to exclude all cases with “specified polyneuropathy” such as inflammatory polyneuropathy. In some cases, CMT cannot clearly be separated from inflammatory polyneuropathy; and undiagnosed CMT cases could be found in this group of patients also.22,23 The same could be the case for some patients diagnosed with acquired or toxic polyneuropathy, as several studies describe cases of undiagnosed or asymptomatic CMT in patients with drug-induced polyneuropathy.24–26 Thirdly, the list of criteria used in the medical record review were based on the classical CMT phenotype, hence atypical CMT cases and mildly affected cases will remain unidentified using these criteria. This is problematic, as especially these subgroups of CMT are at a higher risk of remaining undiagnosed or misdiagnosed. The criteria in this study are designed to ensure high specificity; therefore, patients with undiagnosed CMT with medical files that do not fulfill our selected criteria will be missed by this study. Fourthly, in some cases the medical record did not hold sufficient data. Most missing cases were old, and probably lost over time; however, other reasons for loss of data may have caused bias. Lastly, limb deformities, especially pes cavus and hammer toes, belong to the classical CMT phenotype.1 The list of diagnoses in our “limb deformities” group contains a very broad range of conditions, whereof not all are as common in CMT. It may have been better to separate this group into smaller entities, eg, feet, hands, hip, etc., or in groups according to how commonly they are observed.
Conclusion
In a selected group of patients diagnosed with an unspecified polyneuropathy diagnosis in the DNPR, we found that 21.5% could have undiagnosed CMT. This corresponds to an increase in the prevalence of CMT in Denmark with 3.6 per 100,000 (95% CI 2.4%-5.5%): from 22.5 per 100,000 (95% CI 21.2%-23.7%) as reported previously10 to 26.1 per 100,000 (95% CI 24.1%-28.0%). This study supports the hypothesis, that many patients with CMT are undiagnosed. As we expected, the estimates of prevalence and incidence of CMT in Denmark, as reported in an earlier study, are likely to be underestimated.
Acknowledgments
We are grateful to the following for assistance in medical records retrieval: Anita Højholt, Department of Neurology, Aarhus University Hospital, Birgitte Forsom Sandal, Department of Neurology, Holstebro Regional Hospital West Jutland, Friedrich Emanuel Schlesinger, Department of Neurology, Viborg Regional Hospital Central Jutland, Hatice Tankisi, Department of Neurophysiology, Aarhus University Hospital, Lene Vels Pedersen, Department of Pediatrics, Herning Regional Hospital West Jutland and Thomas Helweg Thelle, Department of Pediatrics, Viborg Regional Hospital Central Jutland. We also wish to thank Michael Væth from The Department of Public Health, Section for Biostatistics at Aarhus University for technical assistance in management of register data and review of the statistical analyses. This work has been supported by a grant from Innovation Fund Denmark, Aarhus University Hospital and Aarhus University.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol. 2013;9(10):562–571. doi:10.1038/nrneurol.2013.179
2. Pipis M, Rossor AM, Laura M, Reilly MM. Next-generation sequencing in Charcot-Marie-Tooth disease: opportunities and challenges. Nat Rev Neurol. 2019;15(11):644–656. doi:10.1038/s41582-019-0254-5
3. Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103(2):259–280. doi:10.1093/brain/103.2.259
4. Nagai MK, Chan G, Guille JT, Kumar SJ, Scavina M, Mackenzie WG. Prevalence of Charcot-Marie-Tooth disease in patients who have bilateral cavovarus feet. J Pediatr Orthop. 2006;26(4):438–443. doi:10.1097/01.bpo.0000226278.16449.c4
5. Laura M, Singh D, Ramdharry G, et al. Prevalence and orthopedic management of foot and ankle deformities in Charcot-Marie-Tooth disease. Muscle Nerve. 2018;57(2):255–259. doi:10.1002/mus.25724
6. Yagerman SE, Cross MB, Green DW, Scher DM. Pediatric orthopedic conditions in Charcot-Marie-Tooth disease: a literature review. Curr Opin Pediatr. 2012;24(1):50–56. doi:10.1097/MOP.0b013e32834e9051
7. Vallat JM, Mathis S, Funalot B. The various Charcot-Marie-Tooth diseases. Curr Opin Neurol. 2013;26(5):473–480. doi:10.1097/WCO.0b013e328364c04b
8. Lerat J, Magdelaine C, Lunati A, et al. Implication of the SH3TC2 gene in Charcot-Marie-Tooth disease associated with deafness and/or scoliosis: illustration with four new pathogenic variants. J Neurol Sci. 2019;406:116376. doi:10.1016/j.jns.2019.06.027
9. Werheid F, Azzedine H, Zwerenz E, et al. Underestimated associated features in CMT neuropathies: clinical indicators for the causative gene? Brain Behav. 2016;6(4):e00451. doi:10.1002/brb3.451
10. Vaeth S, Vaeth M, Andersen H, Christensen R, Jensen UB. Charcot-Marie-Tooth disease in Denmark: a nationwide register-based study of mortality, prevalence and incidence. BMJ Open. 2017;7(11):e018048. doi:10.1136/bmjopen-2017-018048
11. Lynge E, Sandegaard JL, Rebolj M. The Danish national patient register. Scand J Public Health. 2011;39(7 Suppl):30–33. doi:10.1177/1403494811401482
12. Vaeth S, Jensen UB, Christensen R, Andersen H. Validation of diagnostic codes for Charcot-Marie-Tooth disease in the Danish national patient registry. Clin Epidemiol. 2016;8:783–787. doi:10.2147/CLEP.S115565
13. Statistics Denmark. http://www.statistikbanken.dk/.
14. Pedersen CB. The Danish civil registration system. Scand J Public Health. 2011;39(7 Suppl):22–25. doi:10.1177/1403494810387965
15. StataCorp. Stata: Release 13. Statistical Software. College Station, TX: StataCorp LP. [computer program]; 2013.
16. Haites NE, Nelis E, Van Broeckhoven C. 3rd workshop of the European CMT consortium: 54th ENMC International Workshop on genotype/phenotype correlations in Charcot-Marie-Tooth type 1 and hereditary neuropathy with liability to pressure palsies 28–30 November 1997, Naarden, The Netherlands. Neuromuscul Disord. 1998;8(8):591–603. doi:10.1016/S0960-8966(98)00067-4
17. De Jonghe P, Timmerman V, Van Broeckhoven C. 2nd Workshop of the European CMT Consortium: 53rd ENMC International Workshop on Classification and Diagnostic Guidelines for Charcot-Marie-Tooth Type 2 (CMT2-HMSN II) and Distal Hereditary Motor Neuropathy (distal HMN-Spinal CMT) 26–28 September 1997, Naarden, The Netherlands. Neuromuscul Disord. 1998;8(6):426–431.
18. Dyck PJ, Oviatt KF, Lambert EH. Intensive evaluation of referred unclassified neuropathies yields improved diagnosis. Ann Neurol. 1981;10(3):222–226. doi:10.1002/ana.410100304
19. Schmidt M, Schmidt SA, Sandegaard JL, Ehrenstein V, Pedersen L, Sorensen HT. The Danish national patient registry: a review of content, data quality, and research potential. Clin Epidemiol. 2015;7:449–490. doi:10.2147/CLEP.S91125
20. Thomsen RW, Hundborg HH, Lervang HH, Johnsen SP, Sorensen HT, Schonheyder HC. Diabetes and outcome of community-acquired pneumococcal bacteremia: a 10-year population-based cohort study. Diabetes Care. 2004;27(1):70–76. doi:10.2337/diacare.27.1.70
21. Jepsen P, Vilstrup H, Sorensen HT. Alcoholic cirrhosis in Denmark - population-based incidence, prevalence, and hospitalization rates between 1988 and 2005: a descriptive cohort study. BMC Gastroenterol. 2008;8:3. doi:10.1186/1471-230X-8-3
22. Rajabally YA, Adams D, Latour P, Attarian S. Hereditary and inflammatory neuropathies: a review of reported associations, mimics and misdiagnoses. J Neurol Neurosurg Psychiatry. 2016;87(10):1051–1060. doi:10.1136/jnnp-2015-310835
23. Campagnolo M, Taioli F, Cacciavillani M, et al. Sporadic hereditary neuropathies misdiagnosed as chronic inflammatory demyelinating polyradiculoneuropathy: pitfalls and red flags. J Peripher Nerv Syst. 2020;25(1):19–26. doi:10.1111/jns.12362
24. Nakamura T, Hashiguchi A, Suzuki S, Uozumi K, Tokunaga S, Takashima H. Vincristine exacerbates asymptomatic Charcot-Marie-tooth disease with a novel EGR2 mutation. Neurogenetics. 2012;13(1):77–82. doi:10.1007/s10048-012-0313-1
25. Aghajan Y, Yoon JM, Crawford JR. Severe vincristine-induced polyneuropathy in a teenager with anaplastic medulloblastoma and undiagnosed Charcot-Marie-Tooth disease. BMJ Case Rep. 2017;2017. doi:10.1136/bcr-2016-218981
26. Beutler AS, Kulkarni AA, Kanwar R, et al. Sequencing of Charcot-Marie-Tooth disease genes in a toxic polyneuropathy. Ann Neurol. 2014;76(5):727–737. doi:10.1002/ana.24265
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.