")
Back to Journals » International Journal of General Medicine » Volume 15
A Review on the Diagnosis and Treatment of Proliferative Glomerulonephritis with Monoclonal Immunoglobulin Deposits
Received 21 September 2022
Accepted for publication 30 November 2022
Published 14 December 2022 Volume 2022:15 Pages 8577—8582
DOI https://doi.org/10.2147/IJGM.S386733
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Li Lin, Nan Chen
Department of Nephrology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, People’s Republic of China
Correspondence: Nan Chen, Department of Nephrology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, 197 Rui Jin Road II, Shanghai, 200025, People’s Republic of China, Tel +86 21 64370045, Fax +86 21 64370045, Email [email protected]
Abstract: Proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) is a new entity of monoclonal gammopathy of renal significance (MGRS) due to monoclonal immunoglobulin (mIg) deposits in the glomerulus leading to kidney injury. Patients with PGNMID typically present with proteinuria, haematuria, and abnormal renal function. Only ~30% of patients have a detectable clone in the blood, urine or bone marrow. Histologically, the membranoproliferative glomerulonephritis (MPGN) pattern of injury is the most common, with approximately 50% of patients demonstrating IgG3κ monoclonal deposition in the glomerulus. Approximately 20% of PGNMID patients progress to end-stage kidney disease (ES&Kgr;D), and recurrence with renal allograft is frequent. Treatment of PGNMID relies on targeting identifiable B-cell or plasma cell clones. Due to the relatively short history of research on this disease and incomplete understanding, it is easy to misdiagnose and miss diagnosis, and there are also objections on how to treat it. Therefore, we review the development of PGNMID understanding in the past 20 years and discuss this new entity from a holistic and progressive perspective.
Keywords: PGNMID, MPGN, treatment, RTX, DARA
Introduction
Monoclonal immunoglobulin diseases include light-chain deposition disease, light- and heavy-chain deposition disease, heavy-chain deposition disease, type I cryoglobulinemia, immune tubular glomerulopathy and fibrillar glomerulopathy. In recent years, the spectrum of monoclonal gammopathy has gradually expanded to include tissue deposits of mono-isotype Ig but lacks evidence for a circulating monoclonal Ig component.1 At present, there are still many problems worth exploring and solving for this disease. In this article, we discuss PGNMID in terms of history, clinicopathological manifestation and treatment.
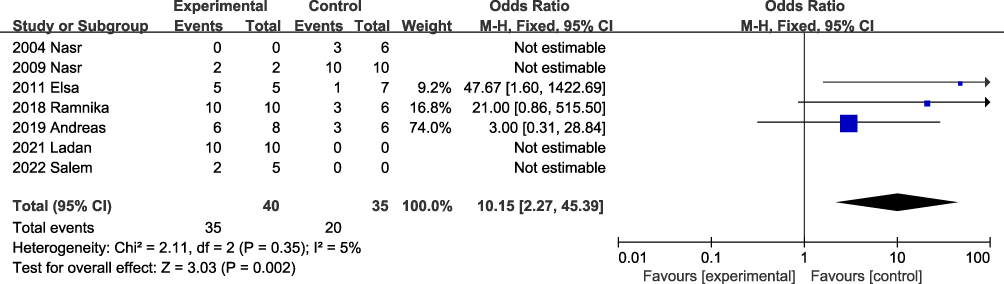 |
Figure 1 Meta-analysis of targeted drug therapy for PGNMID. Notes: Experimental (Targeted) drugs included RTX and/or proteasome inhibitors and/or CD38 monoclonal antibodies; Control (nontargeted) drugs include RAS inhibitors and/or immunosuppressants (CTX, CYA, MMF, FΚ, ASA and prednisone). |
History of PGNMID
Proliferative Glomerulonephritis with Monoclonal IgG Deposits (PGNMID-IgG)
As early as 1980s, scholars described PGNMID-like pathological changes.2 Alpers reported 11 cases of glomerulonephritis associated with immunoglobulin light chain or monoclonal immunoglobulin deposition, with MPGN as the main pathological manifestation, IgGκ present in mesangium and subendothelial deposition.2 The term PGNMID-IgG was first proposed by Nasr in 2004.1 Nasr’s team retrospectively identified 10 cases of unclassifiable immune-mediated glomerulonephritis with monoclonal IgG deposition among 4650 renal biopsies.1 The pathological manifestations under light microscopy were MPGN or proliferative glomerulonephritis (GN), and immunofluorescence showed the monoclonal expression of proteins composed of a single light-chain subtype (κ or λ) and a single γ subtype (two cases of IgG1κ, one of IgG1λ, one of IgG2λ, four of IgG3κ and one of IgG3λ).1 Electron microscopy revealed mesangial, subendothelial, and subepithelial granule-like electron-dense deposits, resembling immune complex-mediated GN. Three fixed regions of γ heavy chains (CH1, CH2 and CH3) were stained in all cases, suggesting the deposition of nondeleted immunoglobulin molecules. Renal insufficiency was present in 80% of the patients (mean serum creatinine was 2.8 mg/dL, mean urine protein level was 5.8 g/day), 44% presented with nephrotic syndrome, and 60% had microscopic haematuria. Monoclonal blood and urine species were detected in 50% of cases (3 IgGκ and 2 IgGλ), and no patient progressed to myeloma or lymphoma during the mean follow-up period of 12 months.
In 2009, Nasr reported another 37 cases of PGNMID-IgG.3 The most common mIg was IgG3 (65.6%), the light-chain subtype was κ, and there was a lack of evidence of monoclonal B lymphocyte or plasma cell proliferation in bone marrow biopsies.3
In 2015, Nelson et al identified 60 patients with renal biopsy-proven glomerular monoclonal immunoglobulin deposition proliferative glomerulonephritis (MIPG) in the Mayo Clinic’s pathology database for retrospective analysis.4 The results showed that 90% of patients had IgG mIg deposition, 48% of patients showed an MPGN damage pattern, 20% of patients had mIg detected by serum immunofixation (SIFE), 21% of patients had an abnormal serum-free light-chain ratio (sFLCR), and 25% of patients bone marrow examination revealed mIg (10/40), including 6 plasma cell clones (5 IgG type, 1 IgA type), 3 chronic lymphocytic leukaemia (all IgG type), and 1 lymphoplasmacytic clone (IgM type).
Other Types of PGNMID
In 2007, Ramos et al reported a case of nonmalignant monoclonal immunoglobulinemia (IgM κ) with nephrotic syndrome and progressive renal failure. Light microscopy showed mesangial matrix expansion, focal mesangial cell hyperplasia, and fluorescence manifesting as monoclonal deposition of IgM and κ light chains in the glomerulus.5 In 2010, Κaneko et al reported a patient with monoclonal immunoglobulinemia (IgA κ) with haematuria. Light microscopy showed mesangial proliferative glomerulonephritis with intracapillary proliferation, necrosis and cell crescent formation. Immunofluorescence showed IgA1-κ monoclonal deposition.6
PGNMID-Light Chain (PGNMID-LC)
In 2020, Nasr proposed the concept of PGNMID-LC.7 The difference between PGNMID-LC and PGNMID-IgG is the higher probability of detecting pathogenic clones. Nasr reported 17 cases of PGNMID-LC, and these patients had haematological abnormalities, MGRS (71%) or multiple myeloma (29%). mIg was identified by blood and urine immunofixation electrophoresis in 65% and 73% of patients, respectively; 83% had abnormal serum-free light chains, and 88% had detectable clones in the bone marrow plasma cells. In light microscopy, most of the patients showed an MPGN injury pattern consisting of restricted light chains (71% κ) and C3.
Electron microscopy revealed subendothelial, mesangial and subepithelial granular deposits. Proteomic analysis of four patients with PGNMID-LC (κ) showed the expression of κ constant and variable domains but no Ig heavy-chain expression. The expression of alternative complement pathway components and the terminal complex was detected in three patients, but the expression of the classical complement pathway components was not detected. At a median follow-up of 70 months, 60% (6/10 patients) of the patients who received targeted plasma cell therapy achieved renal remission, whereas none of the 5 patients who received other treatments achieved remission.
PGNMID in Kidney Allografts
In 2011, Nasr et al reported that PGNMID-IgG recurred in kidney allografts.8 Similar to in MGRS diseases, relapse after kidney transplantation in PGNMID patients is extremely common, with a relapse rate of 89%; 86% of the patients experience relapse within 3–4 months after transplantation.9 The transplant kidney biopsies revealed that mesangial proliferative glomerulonephritis is the most common type; 89% of the cases were renal tissue mIg of IG3, and 20% of the patients had detectable serum paraprotein.9,10 The median survival time after transplant was 92 months, with nearly 50% of patients experiencing transplant failure within 3 years of diagnosis.9–11
Pathogenesis
The pathophysiology of PGNMID remains unclear. As in other types of MGRS, nephrotoxicity likely arises primarily from the mIg or subunits, those may relate to antigen-binding antibody or with special conformation to promote mIg deposition.12,13 IgG3 has a greater predominance in PGNMID, and IgG3 usually constitutes only 4–8% of total serum IgG. IgG3 has a large molecular mass (170 kDa) and is easily restricted by the glomerular filtration barrier.14 IgG3 easily self-aggregates through the Fc–Fc interaction and has the largest C1q immobilization capacity. These two characteristics promote IgG3 deposition in the glomerulus and activate the classical complement pathway.14–16 Sirac et al found that changes in some amino acids, which may reduce the stability and folding of immunoglobulins, lead to oversecretion of unstable immunoglobulins or immunoglobulin fragments in an animal model of monoclonal immunoglobulin-associated kidney disease.17 Therefore, the diagnosis of mIg in renal deposits requires not only the demonstration of a single (restricted) heavy- and light-chain isotype, but also evidence of homogeneous variable domains either by empirical approaches such as immunoelectron microscopy or by determination of amino acid sequences by mass spectrometry assays.14,17,18 In addition, spontaneous remission of PGNMID patients that occurs at a young age or after viral infection may suggest that pathogenesis also involves induction of B-cell repertoires by viral or other antigenic stimulation mechanisms, producing low clonal nephrotoxic IgG3.19–22
Clinical Characteristics
PGNMID is common in Caucasians, with no sex difference, accounting for 0.17%–3.7% of renal biopsies.16,23 In patients over 20 years of age, PGNMID accounts for 0.8% of renal biopsies. The incidence of PGNMID is 7-fold lower than that of light-chain amyloidosis and 2-fold lower than that of MIDD.1 The average age of onset is 55 years, and the disease can occur at any age.24
The clinical manifestations of PGNMID are nonspecific; nearly half of the patients present with nephrotic syndrome, 60% have oedema, and 80% have haematuria, sometimes visible to the naked eye.24 Two-thirds of patients have renal insufficiency, the median estimated glomerular filtration rate (eGFR) at baseline is 36 mL/min/1.73 m2, and <10% require dialysis at diagnosis. Rapidly progressive glomerulonephritis is uncommon. Hypocomplementemia is uncommon, with low serum levels of C3 and/or C4 in 20% of patients and a negative cryoglobulin test.19
The positive detection rate of mIg was lower for PGNMID than for MIDD.14,15 Under conditions in which immunofixation electrophoresis and free light-chain detection are feasible, nephrotoxic mIg is found in the circulation in ~30% of cases. Monoclonal findings on bone marrow examination are similarly low. Bhutani reported that only 25% of patients had abnormal bone marrow clones, of which 5 patients (50%) had plasma cell clones detected by immunohistochemistry and flow cytometry, and the rest were CD20+ clones. Peripheral blood flow lymphocyte detection revealed B-cell clones in one patient (1/9).4 Although patients with PGNMID are less likely to develop haematological malignancies, the presence of CLL, rare B-cell lymphomas or smouldering MM necessitates long-term monitoring.
Diagnosis and Differential Diagnosis
Early diagnosis of PGNMID is key to improving prognosis, and diagnosis and treatment are based on comprehensive clinical evaluations, complete renal biopsies, and in-depth immunological and haematological examinations.
The pathological diagnostic criteria for PGNMID are as follows:19 (1) Light microscope: proliferative or membranous proliferative GN (or atypical membranous GN); (2) Immunofluorescence or immunohistochemistry: glomerular monotypic deposits (glomerular positivity for a single immunoglobulin class, a single IgG subclass in the case of IgG PGNMID and a single light-chain isotype), usually IgG3; rarely IgG1, IgG2, IgA, IgM, LC only; (3) Electron microscopy: predominantly granular electron-dense deposits in mesangial, subendothelial and/or subepithelial locations and (4) Clinical: absence of clinical or laboratory evidence of cryoglobulinaemia.
When the pathological manifestation indicates PGNMID, a thorough examination by immunology and haematology is required to determine whether there is pathological plasma cell or lymphocyte disease.25 Testing includes blood examinations, urine immunoelectrophoresis, haematuria immunofixation electrophoresis, blood-free light-chain tests, bone marrow histology examinations, and bone marrow flow examinations.25 If all of the above test results are negative, immunophenotyping of circulating lymphocytes by flow cytometry, CT scan of the chest, abdomen and pelvis, and PET-CT should be considered for the detection of indolent B-cell lymphoma.19
PGNMID must be differential diagnosis from fibrillary glomerulopathy, cryoglobulinemia type 1, membranous glomerulonephritis with IgGκ deposition, light-chain amyloidosis, light- and heavy-chain deposition disease, lupus nephritis, atypical anti-GBM nephritis with IgG deposition and idiopathic MPGN.
Treatment and Prognosis
The specific treatment required for PGNMID is currently unclear, and the data are based on small sample data. In Nasr’s cohort study, 5 of 9 patients treated with RAS blockade alone achieved remission, including 2 achieving complete remission (proteinuria<500 mg/d).3 Therefore, in patients without nephrotic syndrome and stable CΚD stage 1–2 (especially those who are frail) and in patients with PGNMID associated with viral infection, the use of RAS inhibitors to reduce proteinuria may be a reasonable treatment option. Intense immunotherapy appears to be inappropriate for patients with stage 5 CΚD who are not candidates for kidney transplantation.16
In Nasr’s cohort study, 18 patients received immunosuppressive therapy (steroid, MMF, CTX, n=12), cyclophosphamide combined with RTX (n=4) or antineoplastic drugs (bortezomib or thalidomide; n=2). At the 30-month follow-up appointment, one-third of patients had achieved renal remission (4 complete responses), one-third had stable disease, 22% had experienced progression to ESRD, and 16% had died.3 Several recent studies have shown that treatment with targeted drugs, such as rituximab, proteasome inhibitors, and antiplasmacytomab, increases renal remission.26–28 Gumber et al provided new insights into the management of PGNMID using targeted clonal drug therapy.27 Treatment targeting either identified latent clones (n=4) or empirically putative clones (n=12) had an overall response rate of 88%, with 6 patients (38%) achieving complete responses.27 Daratumumab is an anti-CD38 human IgG1κ monoclonal antibody. Zand et al reported the cases of 10 patients with PGNMID (mIg detected in only 1 patient) treated with daratumumab. At the 12-month follow-up, all patients had achieved partial remission, and four achieved complete remission.28
Targeted cloning therapy improves renal response rates and renal prognosis in PGNMID patients with detectable mIg.14 Although empirical chemotherapy may show some benefit for those with no detectable clones, it remains challenging.29,30 Through a literature search (PubMed), 7 studies, including over 5 cases with PGNMID treatment, were selected, and a meta-analysis was conducted to determine whether targeted therapy was beneficial (Figure 1).1,3,16,26–28,31 Forty patients were treated with targeted drugs (single drug or combination) and 35 did not receive targeted drug treatment. The efficacy rate of targeted drug therapy was 10.15 times (95% CI 2.27, 45.39) that of no targeted therapy.
Outlook
PGNMID is the least understood type of MGRS disease. In the nearly 20 years since its discovery, the “veil” has been gradually lifted. The disease diagnosis relies on renal biopsy, and clonal found in circulation or bone marrow provides the basis for treatment. Integrative molecular biological methods such as immunoelectron microscopy and mass spectrometry will be used to explore the characteristics and mechanisms of mIg deposition in the kidneys, formulate treatment plans in conjunction with multidisciplinary consultation, and compare B-cell targeting drugs (rituximab) and plasma cell targets. Further investigation of the therapeutic effect of bortezomib/anti-CD38 monoclonal antibody will help to improve the diagnosis and treatment system.
Summary
With the recognition of PGNMID, its diagnosis rate has gradually increased, and the application of biological agents targeting clones has greatly improved the diagnosis and prognosis of PGNMID patients.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Nasr SH, Markowitz GS, Stokes MB., et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65(1):85–96. doi:10.1111/j.1523-1755.2004.00365.x
2. Alpers CE, Tu WH, Hopper J, Biava CG. Single light chain subclass (kappa chain) immunoglobulin deposition in glomerulonephritis. Hum Pathol. 1985;16(3):294–304. doi:10.1016/s0046-8177(85)80017-4
3. Nasr SH, Satoskar A, Markowitz GS, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20(9):2055–2064. doi:10.1681/ASN.2009010110
4. Bhutani G, Nasr SH, Said SM, et al. Hematologic characteristics of proliferative glomerulonephritides with nonorganized monoclonal immunoglobulin deposits. Mayo Clin Proc. 2015;90(5):587–596. doi:10.1016/j.mayocp.2015.01.024
5. Ramos R, Poveda R, Sarra J, Domingo A, Carreras L, Grinyo JM. Renal involvement in non-malignant IgM gammopathy. Nephrol Dial Transplant. 2007;22(2):627–630. doi:10.1093/ndt/gfl709
6. Kaneko S, Usui J, Narimatsu Y, et al. Renal involvement of monoclonal immunoglobulin deposition disease associated with an unusual monoclonal immunoglobulin A glycan profile. Clin Exp Nephrol. 2010;14(4):389–395. doi:10.1007/s10157-010-0285-0
7. Nasr SH, Larsen CP, Sirac C, et al. Light chain only variant of proliferative glomerulonephritis with monoclonal immunoglobulin deposits is associated with a high detection rate of the pathogenic plasma cell clone. Kidney Int. 2020;97(3):589–601. doi:10.1016/j.kint.2019.10.025
8. Nasr SH, Sethi S, Cornell LD, et al. Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol. 2011;6(1):122–132. doi:10.2215/CJN.05750710
9. Said SM, Cosio FG, Valeri AM, et al. Proliferative glomerulonephritis with monoclonal immunoglobulin G deposits is associated with high rate of early recurrence in the allograft. Kidney Int. 2018;94(1):159–169. doi:10.1016/j.kint.2018.01.028
10. Kamal J, Khairallah P, Crew RJ, et al. Clinicopathologic Assessment of Monoclonal Immunoglobulin-associated Renal Disease in the Kidney Allograft: a Retrospective Study and Review of the Literature. Transplantation. 2020;104(7):1341–1349. doi:10.1097/TP.0000000000003010
11. Wen J, Wang W, Xu F, et al. Clinicopathological analysis of proliferative glomerulonephritis with monoclonal IgG deposits in 5 renal allografts. BMC Nephrol. 2012;19(1):173. doi:10.1186/s12882-018-0969-3
12. Leung N, Bridoux F, Batuman V, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. 2019;15(1):45–59. doi:10.1038/s41581-018-0077-4
13. Motwani SS, Herlitz L, Monga D, Jhaveri KD, Lam AQ; American Society of Nephrology Onco-Nephrology F. Paraprotein-Related Kidney Disease: glomerular Diseases Associated with Paraproteinemias. Clin J Am Soc Nephrol. 2016;11(12):2260–2272. doi:10.2215/CJN.02980316
14. Aucouturier P, D’Agati VD, Ronco P, Fresh A. Perspective on monoclonal gammopathies of renal significance. Kidney Int Rep. 2021;6(8):2059–2065. doi:10.1016/j.ekir.2021.04.026
15. Hogan JJ, Alexander MP, Leung N. Dysproteinemia and the Kidney: core Curriculum 2019. Am J Kidney Dis. 2019;74(6):822–836. doi:10.1053/j.ajkd.2019.04.029
16. Kousios A, Duncan N, Tam FWK, et al. Proliferative glomerulonephritis with monoclonal Ig deposits (PGNMID): diagnostic and treatment challenges for the nephrologist! Kidney Int. 2019;95(2):467–468. doi:10.1016/j.kint.2018.10.016
17. Sirac C, Herrera GA, Sanders PW, et al. Animal models of monoclonal immunoglobulin-related renal diseases. Nat Rev Nephrol. 2018;14(4):246–264. doi:10.1038/nrneph.2018.8
18. Nasr SH, Fidler ME, Said SM, Koepplin JW, Altamirano-Alonso JM, Leung N. Immunofluorescence staining for immunoglobulin heavy chain/light chain on kidney biopsies is a valuable ancillary technique for the diagnosis of monoclonal gammopathy-associated kidney diseases. Kidney Int. 2021;100(1):155–170. doi:10.1016/j.kint.2021.02.038
19. Bridoux F, Javaugue V, Nasr SH, Leung N. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits: a nephrologist perspective. Nephrol Dial Transplant. 2021;36(2):208–215. doi:10.1093/ndt/gfz176
20. Fujita E, Shimizu A, Kaneko T, et al. Proliferative glomerulonephritis with monoclonal immunoglobulin G3kappa deposits in association with parvovirus B19 infection. Hum Pathol. 2012;43(12):2326–2333. doi:10.1016/j.humpath.2012.04.004
21. Miller P, Xiao AY, Kung VL, et al. Progression of proliferative glomerulonephritis with monoclonal IgG deposits in pediatric patients. Pediatr Nephrol. 2021;36(4):927–937. doi:10.1007/s00467-020-04763-5
22. Xu ZG, Li WL, Wang X, et al. Proliferative glomerulonephritis with monoclonal immunoglobulin G deposits in a young woman: a case report. World J Clin Cases. 2021;9(10):2357–2366. doi:10.12998/wjcc.v9.i10.2357
23. Gowda KK, Nada R, Ramachandran R, et al. Proliferative glomerulonephritis with monoclonal immunoglobulin deposition disease: the utility of routine staining with immunoglobulin light chains. Indian J Nephrol. 2015;25(6):344–348. doi:10.4103/0971-4065.151354
24. Li M, Xu G. An update of proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Clin Kidney J. 2022;15(6):1041–1048. doi:10.1093/ckj/sfab269
25. Rovin BH, Adler SG, Barratt J, et al. Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney Int. 2021;100(4):753–779. doi:10.1016/j.kint.2021.05.015
26. Guiard E, Karras A, Plaisier E, et al. Patterns of noncryoglobulinemic glomerulonephritis with monoclonal Ig deposits: correlation with IgG subclass and response to rituximab. Clin J Am Soc Nephrol. 2011;6(7):1609–1616. doi:10.2215/CJN.10611110
27. Gumber R, Cohen JB, Palmer MB, et al. A clone-directed approach may improve diagnosis and treatment of proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Kidney Int. 2018;94(1):199–205. doi:10.1016/j.kint.2018.02.020
28. Zand L, Rajkumar SV, Leung N, Sethi S, El Ters M, Fervenza FC. Safety and Efficacy of Daratumumab in Patients with Proliferative GN with Monoclonal Immunoglobulin Deposits. J Am Soc Nephrol. 2021;32(5):1163–1173. doi:10.1681/ASN.2020101541
29. van Kruijsdijk RCM, Abrahams AC, Nguyen TQ, Minnema MC, Jacobs JFM, Limper M. Clone-directed therapy for proliferative glomerulonephritis with monoclonal immunoglobulin depositions: is it always necessary?: two case reports and literature review. J Nephrol. 2020;33(3):611–617. doi:10.1007/s40620-020-00723-2
30. Buxeda A, Said SM, Nasr SH, Leung N, El Ters M, Cosio FG. Recurrent Proliferative Glomerulonephritis With Monoclonal Immunoglobulin Deposits in Kidney Allografts Treated With Anti-CD20 Antibodies. Transplantation. 2019;103(7):1477–1485. doi:10.1097/TP.0000000000002577
31. Almaani S, Parikh SV, Satoskar AA, et al. Daratumumab in Patients With Bortezomib-Refractory Proliferative Glomerulonephritis With Monoclonal Immunoglobulin Deposits. Kidney Int Rep. 2021;6(8):2203–2206. doi:10.1016/j.ekir.2021.05.008
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.