")
Back to Journals » Journal of Pain Research » Volume 16
A Review of the Therapeutic Targeting of SCN9A and Nav1.7 for Pain Relief in Current Human Clinical Trials
Authors Dormer A , Narayanan M, Schentag J, Achinko D, Norman E, Kerrigan J, Jay G, Heydorn W
Received 9 September 2022
Accepted for publication 14 March 2023
Published 4 May 2023 Volume 2023:16 Pages 1487—1498
DOI https://doi.org/10.2147/JPR.S388896
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Robert B. Raffa
Anton Dormer,1 Mahesh Narayanan,1 Jerome Schentag,1 Daniel Achinko,1 Elton Norman,1 James Kerrigan,2 Gary Jay,2 William Heydorn2
1Research and Development, Pepvax, Inc, Silver Spring, MD, USA; 2Research and Development, Navintus, Inc, Princeton, NJ, USA
Correspondence: Anton Dormer, Research and Development, PepVax, Inc, 8720 Georgia Ave #1000, Silver Spring, MD, 20910, USA, Email [email protected]
Introduction: There is a great need to find alternative treatments for chronic pain which have become a healthcare problem. We discuss current therapeutic targeting Nav1.7.
Areas Covered: Nav1.7 is a sodium ion channel protein that is associated with several human pain genetic syndromes. It has been found that mutations associated with Nav1.7 lead to the loss of the ability to perceive pain in individuals that are otherwise normal. Several therapeutic interventions are presently undergoing preclinical and research using the methodology of damping Nav1.7 expressions as a methodology to decrease the sensation of pain leading to analgesia.
Expert Opinion: It is our strong belief that there is a viable future in the targeting of protein of Nav1.7 for the relief of chronic pain in humans. The review will look at the genomics associated with SCN1A and proteomic of Nav1.7 as a foundation to explain the mechanism of the therapeutic interventions targeting Nav1.7, the human disease that are associated with Nav1.7, and the current development of treatment for chronic pain whether in preclinical or clinical trials targeting Nav1.7 expressions. The development of therapeutic antagonists targeting Nav1.7 could be a viable alternative to the current treatments which have led to the opioid crisis. Therefore, Nav1.7 targeted treatment has a major clinical significance that will have positive consequences as it relates to chronic pain interventions.
Keywords: SCN9A, Nav1.7, chronic pain, opioid crisis
Introduction
Neuronal sodium channels are coding by a family of genes, one of which is called sodium voltage-gated channel alpha subunit 9 (SCN9A).1 This gene encodes for sodium channels that are designed to assist in the transportation of positively charged sodium atoms (Na+ ions) into the cell. This action plays a vital role in the generation and the propagation of electrical signals along the neuron.2 A study which included 95 individuals was conducted of which from these persons 27 different tissues were examined to determine the SCN9A expression. The determination of SCN9A protein coding gene tissue-specificity was analyzed using RNA-seq. It was found that SCN9A was expressed in all 27 different tissues at variant degree. The highest expression of SCN9A in non-neuronal tissues was in testis, placenta, and colon.3–5 SCN9A consists of 180,803 nucleotides which encodes a 1988 amino acid Nav1.7 protein.6
Nav1.7 is the sodium channel expression protein of the SCN9A gene.7 The pain signals that are transmitted to the brain are facilitated by Nav1.7 nociceptors preferentially expressed on the neurons that are part of the peripheral nervous system.8 The peripheral nervous system (PNS) is connected to the central nervous system (CNS) which consists of the brain and the spinal cord.9 Nociceptors, which are part of the PNS, consist of cells that have the ability to detect many different types of sensation including pain, smell, and taste.10 Neuronal structures consisting of nociceptors are designed primarily in the transmission of pain signals.11 It is within the cell bodies that the centers of nociceptors are found. Those centers are located within the dorsal root ganglion of the spinal cord.12 The nerve fibers that extend from those cells’ bodies come from all over the human body receiving sensory information from the periphery that will be transmitted to the brain.12 SCN9A which is the gene that encodes for the voltage-gated Nav1.7 sodium channel have a major role in the proper functioning of the nociceptive signaling.13 Mutations within SCN9A gene have been associated with several diseases including congenital insensitivity to pain,14 erythromelalgia,15 paroxysmal extreme pain disorder,16 and small fiber neuropathy.17
Health Conditions Related to Genetic Changes
Congenital Insensitivity to Pain
The inability of an individual from birth to have a perception of physician pain is a condition called congenital insensitivity to pain.18 Those individuals that are suffering from this condition when injured anywhere in and on their body will never feel the pain.19 Persons who have congenital insensitivity to pain do have their ability to distinguish between temperature differences and between dull and sharp stimuli. Many individuals show injuries such as lip and tongue injuries due to the individuals biting those areas during their first 4 years of life. These patients have many bruises and cuts that go unnoticed. They even have fractures that go undiagnosed, leading to additional health challenges.20 The lack of awareness of pain does lead to bruises, bones that are broken, wounds, and many other health issues will not be checked.21 Other notable challenges include children self-biting their lips and their fingers (Klein, CJ et al, 2012). Also, many experience multiple injuries related to burns due to their insensitivity to pain (Klein et al, 2012). The life expectancy of those with congenital insensitivity to pain is severely reduced in comparison to the normal population.21 The condition of congenital insensitivity to pain is one that is extremely rare. Only 80 cases have been documented, with approximately 300 cases reported worldwide within the medical literature.22
This condition can be considered a form of peripheral neuropathy since it affects the peripheral nervous system23 due to the disconnection of the transmission of touch, small, and pain sensations through the spinal cord to the brain.24 The cause of this disease is the production of an SCN9A gene that has undergone a nonsense mutation.25 This leads to the SCN9A gene product to produce nonfunctional alpha subunits that cannot be incorporated in the proper Nav1.7 sodium channel configuration. The formation of sodium channels that are needed for the proper propagation of pain sensory information from the peripheral nervous system to the spinal cord towards the brain.6 In the absence of Nav1.7 sodium channels, there is an insensitivity to pain that is caused since there is an impairment of pain signal transmission from the periphery through the spinal cord to the brain.6 Congenital insensitivity to pain is a genetic disease that follows an autosomal recessive pattern. The parents are not affected by the condition but are carriers of the mutated gene which they both pass on to their child.18
Erythromelalgia
Another disease condition that can arise from SCN9A genetic abnormalities is erythromelalgia. One of the key characteristics of this condition is the consistent pain, swelling throughout the body mostly within the hands and feet, and redness.26,27 The condition is episodic and is, in many cases, triggered when there is an increase in body temperature. There are many different reasons that could lead to a spike in temperature including exercise and even entering a warm room.28 A triggering episode can also occur with the ingesting alcohol or with spicy foods.29 Simple activities such as walking and the wearing of shoes can stimulate pain episodes when patients wear warm socks30 and shoes31 that are tight. Gloves can cause pain episodes in those with this condition.31 These painful episodes prevent individuals suffering with this condition from being able to go to school or to have regular employment.32
In many cases, the condition will begin to show signs and symptoms during childhood. Those with a milder manifestation of the condition may have their first painful episode later in their life.33 The hands and feet of those with erythromelalgia become adults; the disease progresses to the point that their hands and feet are constantly red. The affected area could be extended to cover their entire arms and legs, face and shoulders.26 Like congenital insensitivity to pain, erythromelalgia is a form of peripheral neuropathy.31 The prevalence of erythromelalgia is estimated to be approximately 1 to 2 individuals per 100,000 individuals in the general population have either moderate or severe erythromelalgia.34 The mutations of the SCN9A gene which causes erythromelalgia codes for the protein Nav1.7 sodium channels which open more easily when stimulated and then stays open longer than a normal Nav1.7. Thus, there is an increase into the Nav1.7 nociceptor of sodium ions. The increase in the flow of the sodium ion allows for the increase in pain signal transmission that leads to erythromelalgia signs and symptoms.35–40
The reasons for why the pain episodes occur mostly within the hands and feet with this condition is not known.1 A mutation of the SCN9A gene is estimated to be the cause of 15% of the erythromelalgia cases. The other 85% of the cases is thought to be caused due to non-genetic means or may be caused by yet unidentified mutation in one or more genes.41 Autosomal dominant patterns seem to be the genetic pattern associated with this condition. It takes only one copy of the gene which is altered from an affected parent to lead to the disorder.31
Paroxysmal Extreme Pain Disorder
Flushing (warmth), skin redness, and severe pain attacks in different parts of the body represent the characteristic manifestation of paroxysmal extreme pain disorder.42 The pain site is associated with the flushing that is found manifested with infants.42 Pain attacks can last for hours in some cases, but in this consideration, the pain episode usually lasts for a few seconds to a few minutes.16 Pain attacks can occur during infancy. The pain is usually triggered during infancy beginning with a movement of the bowel, and the pain is concentrated mostly around the rectum.43 Children with this condition may develop constipation in an attempt to prevent pain attack.44 The attacks of pain that occur with these young children may be also associated with seizures,45 slowed heartbeat2 and apnea.46 As the children get older, the pain location seems to change, switching from the lower portion of the body to affecting mostly the face and the head with the pain being focused on the eyes and the jaws.
As a person with paroxysmal extreme pain disorder ages, the location of pain changes. Pain attacks switch from affecting the lower body to affecting the head and face, especially the eyes and jaw.46 There are many different triggers to the pain attacks that occur with this condition. This includes changes in temperature,44 emotional distress47 and spicy food.48 Paroxysmal extreme pain, as with the other SCN9A genetic mutation, leads to a peripheral neuropathy that affects the peripheral nervous system, affecting the pain sensation transmission to the brain.49 The data available associated with the SCN9A worldwide prevalence are not accurate. The medical literature using available medical data identified approximately 500 individuals with paroxysmal extreme pain disorder caused by SCN9A. This is a very rare disease.50,51 Due to the physiological nature of paroxysmal extreme pain disorder, it is usually misdiagnosed or underdiagnosed.17 It is an autosomal dominant disease where the mutated gene is inherited by both parents.16
The condition is associated with a missense mutation that occurs with the SCN9A gene associated with the NaV1.7 potential-dependent sodium channels potential dependent alpha subunits.16 The mutational change that occurs prevents the Nav1.7 sodium channels from completely turning off which allows for the flow of sodium into the nociceptor to be abnormal. The increased flow of the sodium ions allows for the pain signals to be exaggerated. The excitation rapid phase extension due to the interference of the inactivation process causing neuronal excitability.16
Small Fiber Neuropathy
Small fiber neuropathy is one of the other syndromes that is worth mentioning that is associated with a mutation of the SCN9A gene.52 The disease sequence consists of an attack of severe pain beginning at the feet and the hand.53 Even though the pain attack begins with the hand and feet as the person ages and in the more severe disease areas affected include the arm, ear, face, legs and legs.53 Patients with this condition may experience a whole body pain that is generalized. There are several different presentations of pain for this condition including stabbing and burning pain and itchiness and/or tingling.54 The pain, in some individuals, may be more severe with the patient resting or during the night.44 For this disease, the signs and symptoms during the teen years.55
In some individuals, the lack of being able to feel pain is concentrated in the extremely small area of the skin such as a pin prick.56 These individuals have an increased hyperalgesia (increased sensitivity to pain) and suffer from allodynia (pain to stimulation that does not typically cause pain).57 Individuals with this condition can have a reduced ability to differentiate between cold and hot. Even so, the pain attacks can be triggered by both cold and warm.58
There are several associated problems that are manifested outside the challenges with pain. They include bowel54 and urine problems,59 palpitation (rapid heartbeats),17,23 xerostomia (dry mouth),60 keratoconjunctivitis sicca (dry eyes)61, and (hyperhidrosis) abnormal sweating.54 Individuals with this condition suffer from orthostatic hypotension (when blood pressure drops suddenly upon standing) which can cause symptoms such as blurred vision, dizziness, and fainting.62,63 Peripheral neuropathy is one of the challenges associated with small fiber neuropathy as with the other conditions that are caused by mutations with SCN9A which prevents the proper transmission to the brain.17 The incidence of small fiber neuropathy is estimated to be 12 in every 100,000 individuals and the prevalence is 53 patients with every 100,000 individuals without the condition.64 Mutations in the genes SCN9A, SCN10A and SCN11A can be the cause of small fiber neuropathy (SFN).65 SCN9A codes for Nav1.7 which is presented as a type IX α subunit, voltage-gated sodium-channel1 SCN10A codes for Nav1.8 type IX α subunit, voltage-gated sodium-channel. SCN10A is encoded for the Nav1.9 subunits alpha voltage-gated sodium channel.66
Due to the SCN9A genetic mutation the malfunctioning Nav1.7 sodium channels are not completely turned off leading to increased sodium inflow.67 The same occurs with mutations of SCN10A which lead to the more easily opening of the Nav1.8 sodium channels.68 Both genes lead to altered channels that will allow for the abnormal flow of sodium ions in the nociceptors. The individuals with this condition have an increased insensitivity to stimuli that would not normally cause pain.69 There is an unknown cause of the degeneration that occurs in small fiber neuropathy. The neuronal degeneration that occurs could be a major factor for the temperature differentiation loss and the loss of pinprick discrimination.70 Thirty percent of the cases associated with idiopathic small fiber neuropathy is due to a mutation of SCN9A.65,71,72 SCN10A gene mutations are responsible for about 5% of the cases (drbonnie360.com, 2022).73 Small fiber neuropathy is found to be part of other diseases sequela but will not be highlighted within this review including diabetes mellitus, Fabry’s disease, celiac disease, Sjogren's syndrome, sarcoidosis, and human immunodeficiency virus.70 Mutations leading to alteration of the altered SCN9A gene or SCN10A gene are inherited in autosomal dominant manner.23
Therapeutic Interventions Targeting SCN9A and Nav1.7
Therapeutic interventions targeting SCN9A and Nav1.7 review focus on PubMed publications using the terms SCN9A and Nav1.7 and “clinical trial” to identify past and current trends of the human clinical therapeutic development. In addition, http://www.clinicaltrials.gov terms SCN9A and Nav1.7 were queried in order to identify current and past clinical trials. One of the first publications of a clinical trial involving a therapeutic intervention targeting SCN9A/Nav1.7 was by.74 The disease target was inherited erythromelalgia which is caused by a mutation of the SCN9A. The missense mutated gene leads to a non-functioning Nav1.7 sodium channel causing spontaneous pain in those individuals due to neuronal hyperexcitability within those peripheral nervous system neurons.74 This condition is opposite to the SCN9A mutation that occurs in congenital indifference to pain where there is under stimulation of the peripheral nervous system neuron of the non-functioning Nav1.7 sodium channel.74
The human clinical trial Phase I therapy given was XEN402 at a dose of 400mg twice daily topically.74 XEN402 is also known as Funapide. It has a molecular formula of C22H14F3NO5 with a molecular weight of 429.3.75 The name was changed TV-45070 from XEN-402 when it was in-licensed by Teva from Xenon Pharmaceuticals (https://tinyurl.com/2p8nh2bk 2022, https://tinyurl.com/38bu3z27). TV-45070 completed a Phase II trial (retrieved from clinicaltrials.gov 2022). However, the drug did not make it to Phase III for the treatment of inherited erythromelalgia (clinicaltrials.gov 2022).
The data associated with the results of efficacy and safety of the PF-05089771 therapeutic intervention for primary inherited erythromelalgia was published.76 This phase II studies consisted of only five individuals (http://www.clinicaltrials.gov, 2022). This is probably due to the small number of individuals that have this condition and the difficulty of obtaining patients for clinical trials. The small-molecule PF-05089771 selectively blocks the voltage-gated sodium channel Nav1.7 and Nav1.8. The therapy was developed by Pfizer. PF-05089771 completed a phase II clinical trial for the treatment of primary erythromelalgia and wisdom tooth removal.77–80 Even though PF-05089771 was used for other indications such as peripheral neuropathy, its efficacy was modest.81 There was another clinical trial with PF-05089771 looking at its ability to be analgesic to a battery of pain models. Those clinical trials did not show PF-05089771 to be clinically significant analgesic. The drug has not made it into clinical trial phase III for any indications.82
Jones et al (2016)83 published the results of the clinical trial that focused testing of PF-05089771, PF-05150122, PF-05186462 and PF-05241328. These compounds based on their pharmacokinetics were chosen due to their ability to selectively target the Nav1.7 sodium channels. This phase I study found compounds PF-05089771 based on the human clinical trial pharmacokinetic microdose and subsequent modeling, PF-05150122, PF-05186462 and PF-05241328.83 There were 13 different studies that were conducted using the molecules PF-05089771 to test their efficacy of different pain indications. None lead to the therapeutic reaching human clinical trial phase III (http://www.clinicaltrials.gov 2022).
Carbamazepine and vixotrigine are used as voltage-dependent Nav1.7 sodium channel blockers that underwent a human clinical phase I study. Both drugs were investigated for their ability to treat peripheral neuropathic pain conditions including the condition of trigeminal neuralgia.84 According to clinicaltrials.gov (2022), there are presently 21 clinical trials investigating Vixotrigine as a treatment intervention for different pain indications. Of the 21 trials mentioned on clinicaltrials.gov (2022), two are recruiting to use vixotrigine as a treatment for trigeminal neuralgia initiated by Convergence Pharmaceuticals. Carbamazepine has been approved by the FDA from treatment of acute manic disorder, bipolar I disorder, epilepsy, and trigeminal neuralgia, but none of the diseases that have been highlighted within this review specifically associated with SCN9A mutation.85 There have been other small molecules that have been used to target Nav1.7 expressions for the control of pain including lacosamide.
Lacosamide is an FDA-approved antiepileptic drug which has a mechanism of action of stabilizing the slow-inactivated state of Nav1.1 and Nav1.7 sodium channels found within neurons.86 Chronic pain can be associated with painful peripheral neuropathy and is presently a more health burden and a clinical need that is unmet. The Phase 3 study conclusion was mixed for the treatment of small fiber neuropathy due to a gain in function associated with Nav1.7 mutations. The possible determining factors of the variant outcomes are that in some patients, there was a hyperpolarizing shift associated with both of the fast and the slow voltage-dependent inactivation and thereby a use-dependent inhibition enhancement. In contrast, in some patient lacosamide enhanced the fast inactivation selectively.87
Conclusion
As one looks at the past and present human clinical trials targeting SCN9A and Nav1.7 a challenge picture emerges. Small molecules seem not to be very effectives in the management of pain associated with mutation of the SCN9A gene leading to the malfunction of the Nav1.7 sodium channel (clinicaltrials.gov). Gene therapy could be a more targeted way of engaging the SCN9A therapeutically.
For example, the CRISPR/Cas9 system has been used successfully in the area of genomic editing and genomic regulation.88 The CRISPR/Cas9 system was utilized to cause a down regulation of SCN9A leading to the repression of Nav1.7 within the dorsal root ganglia located within the lumbar region. The studied mouse models using this intervention did show decreased tactile allodynia when the mouse was in a neuropathic state. Also, there was the indication of thermal hyperalgesia that had been reduced, while there was no reduction in the motor function of the mice.89 Even with the preliminary results, there may be challenges associated with the use of adenovirus vector to deliver the CRISPR/Cas9 system therapeutics targeted intervention for the treatment of chronic pain via the down regulations of SCN9A.
There are several disadvantages for using an adenovirus vector in the area of gene therapy including immunogenicity,90 non-integration,91 and replication competence.92 The AAV vector packaging capacity is small. The adeno-associated virus (AAV) needs a genomic helper for replication. In addition, manufacturing to produce a stock can be very challenging.93 There is an alternative technology that could be utilized that could target SCN9A.
PepVax, Inc. has a drug delivery system called SMARTmid(tm) is a DNA plasmid vector drug delivery platform that has been configured to target and down regulated SCN9A gene to be used in the treatment of chronic pain. Preliminary data showed that there was a 50% reduction of the SCN9A gene expression which also coincided with the similar decrease in Nav1.7 protein expression in the cell line in vitro (data not shown). Using SMARTmid(tm) instead of an AAV delivery has many more advantages including increased insert packaging capability, replication competence, and lack of permanent integration. In addition, AAV cannot be used more than once due to its immunogenicity which can make it a limiting platform for gene therapy.94
Based on the review of the information (Tables 1–3), a conclusion can be reached that small molecules may not be effective in the treatment of consideration of mutations associated with SCN9A. The best treatment could be the use of DNA plasmid vectors which can be used over and over again without having to be reconfigured. Further studies on the development of treatments targeting the SCN9A gene as a substitute for opioid for the treatment of chronic pain should continue.
 |
Table 1 List of Publications Found in PubMed That are Associated with Human Clinical Trial Targeting the Pain Gene SCN9A and Its Protein Expression Nav1.7 as of December 2022 |
 |
Table 2 The SCN9A is a Gene Which Belongs to a Gene Family That is Involved in the Development of Sodium Channels |
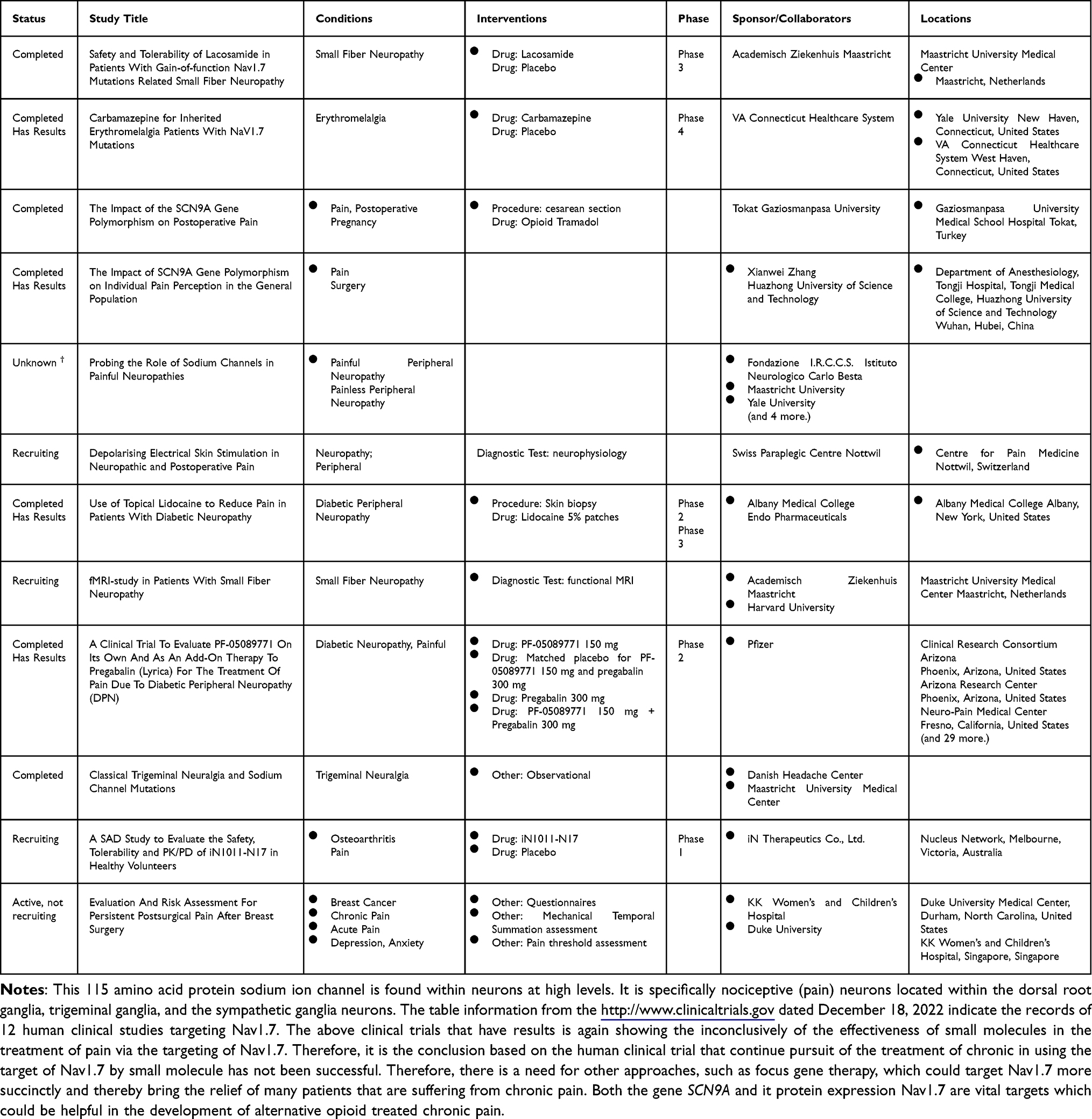 |
Table 3 Nav1.7 is the Gene Expression of the SCN9A in Humans |
Funding
There is no funding to report.
Disclosure
Dr Gary Jay reports personal fees from Creative Biopeptides, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Drenth JP, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117(12):3603–3609. doi:10.1172/JCI33297
2. Wang Y, Mi J, Lu K, Lu Y, Wang K. Comparison of gating properties and use-dependent block of Nav1.5 and Nav1.7 channels by anti-arrhythmics mexiletine and lidocaine. PLoS One. 2015;10(6):e0128653. doi:10.1371/journal.pone.0128653
3. Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57(4):397–409. doi:10.1124/pr.57.4.4
4. Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14(6):1084–1090. doi:10.1002/j.1460-2075.1995.tb07091.x
5. Plummer NW, Meisler MH. Evolution and diversity of mammalian sodium channel genes. Genomics. 1999;57(2):323–331. doi:10.1006/geno.1998.5735
6. Marchi M, D’Amato I, Andelic M, et al. Congenital insensitivity to pain: a novel mutation affecting a U12-type intron causes multiple aberrant splicing of SCN9A. Pain. 2022;163(7):e882–e887. doi:10.1097/j.pain.0000000000002535
7. Chen L, Effraim PR, Carrara J, et al. Pharmacological characterization of a rat Nav1.7 loss-of-function model with insensitivity to pain. Pain. 2020;161(6):1350–1360. doi:10.1097/j.pain.0000000000001807
8. Hameed S. Nav1.7 and Nav1.8: role in the pathophysiology of pain. Mol Pain. 2019;15:1744806919858801. doi:10.1177/1744806919858801
9. Thau L, Reddy V, Singh P. Anatomy, Central Nervous System. Available from. Treasure Island (FL): StatPearls Publishing;2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK542179/.
10. Gadhvi M, Waseem M. Physiology, Sensory System. Treasure Island (FL): StatPearls Publishing; 2022. Available from https://www.ncbi.nlm.nih.gov/books/NBK547656/.
11. Osterweis M, Kleinman A, Mechanic D, editors,; Institute of Medicine (US) Committee on Pain, Disability, and Chronic Illness Behavior. Pain and Disability: Clinical, Behavioral, and Public Policy Perspectives. Washington (DC): National Academies Press (US); 1987. 7. Available from https://www.ncbi.nlm.nih.gov/books/NBK219252/.
12. Yam MF, Loh YC, Tan CS, Khadijah Adam S, Abdul Manan N, Basir R. General Pathways of Pain Sensation and the Major Neurotransmitters Involved in Pain Regulation. Int J Mol Sci. 2018;19(8):2164.
13. Cummins TR, Sheets PL, Waxman SG. The roles of sodium channels in nociception: implications for mechanisms of pain. Pain. 2007;131(3):243–257. doi:10.1016/j.pain.2007.07.026
14. Chen Z, Zhang H, Dai SM. Charcot ankle, congenital insensitivity to pain & a mutation in the SCN9A gene. QJM. 2022;5:hcac117.
15. Świtała WW, Szymańska-Adamcewicz O, Jurga S, Pilchowska-Ujma E, Krakowiak J. Genetic aspects of, pain and its variability in the human population. Ann Agric Environ Med. 2021;8(4):569–574. doi:10.26444/aaem/134151
16. Stępień A, Sałacińska D, Staszewski J, Durka-Kęsy M, Dobrogowski J. Paroxysmal extreme pain disorder in family with c.3892G > T (p.Val1298Phe) in the SCN9A gene mutation - case report. BMC Neurol. 2020;20(1):182. doi:10.1186/s12883-020-01770-9
17. Hisama FM, Dib-Hajj SD, Waxman SG. SCN9A Neuropathic Pain Syndromes. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 2006:1993–2022.
18. Rajasekharan S, Martens L, Domingues L, Cauwels R. SCN9A channelopathy associated autosomal recessive congenital indifference to pain. A case report. Eur J Paediatr Dent. 2017;18(1):66–68. doi:10.23804/ejpd.2017.18.01.14
19. Schon KR, Parker APJ, Woods CG. Congenital Insensitivity to Pain Overview. In: Adam MP, Mirzaa GM, Pagon RA, et al.editors.GeneReviews® [Internet].Seattle: University of Washington, Seattle; 2018:1993–2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK481553/.
20. Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898.
21. Marchi M, Provitera V, Nolano M, et al. A novel SCN9A splicing mutation in a compound heterozygous girl with congenital insensitivity to pain, hyposmia and hypogeusia. J Peripher Nerv Syst. 2018;23(3):202–206. doi:10.1111/jns.12280
22. Ravichandra KS, Kandregula CR, Koya S, Lakhotia D Congenital Insensitivity to Pain and Anhydrosis: Diagnostic and Therapeutic Dilemmas revisited. Int J Clin Pediatr Dent. 2015;8(1):75–81.
23. Kelley MA, Oaklander AL. (2020) Association of small-fiber polyneuropathy with three previously unassociated rare missense SCN9A variants. Can J Pain. 2020;4(1):19–29. doi:10.1080/24740527.2020.1712652
24. Finnerup NB, Kuner R, Jensen TS. Neuropathic pain: from mechanisms to treatment. Physiol Rev. 2021;101(1):259–301. doi:10.1152/physrev.00045.2019
25. Kurban M, Wajid M, Shimomura Y, Christiano AM. (2010) A nonsense mutation in the SCN9A gene in congenital insensitivity to pain. Dermatology. 2010;221(2):179–183. doi:10.1159/000314692
26. Jha SK, Karna B, Goodman MB. Erythromelalgia. Available from. Treasure Island (FL): StatPearls Publishing; 2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557787/.
27. Kang BC, Nam DJ, Ahn EK, Yoon DM, Cho JG. Secondary erythromelalgia - a case report. Int J Med. 2013:26(3);299–302.
28. Waxman SG. Alabama to Beijing and Back: the Search for a Pain Gene. Cerebrum. 2018;2018:cer-02–18.
29. Latessa V. Erythromelalgia: a rare microvascular disease. J Vasc Nurs. 2010;28(2):67–71. doi:10.1016/j.jvn.2009.11.002
30. Kundu A, Rafiq M, Warren PS, Tobias JD. Erythromelalgia in the pediatric patient: role of computed-tomography-guided lumbar sympathetic blockade. J Pain Res. 2016;9:837–845. doi:10.2147/JPR.S110688
31. Tang Z, Chen Z, Tang B, Jiang H. Primary erythromelalgia: a review. Orphanet J Rare Dis. 2015;30(10):127. doi:10.1186/s13023-015-0347-1
32. Tham SW, Giles M. Current pain management strategies for patients with erythromelalgia: a critical review. J Pain Res. 2018;11:1689–1698. doi:10.2147/JPR.S154462
33. Arthur L, Keen K, Verriotis M, et al. Pediatric Erythromelalgia and SCN9A Mutations: systematic Review and Single-Center Case Series. J Pediatr. 2019;206(217–224.e9):217–224.e9. doi:10.1016/j.jpeds.2018.10.024
34. Reed KB, Davis MD. Incidence of erythromelalgia: a population-based study in Olmsted County, Minnesota. J Eur Acad Dermatol Venereol. 2009;23(1):13–15. doi:10.1111/j.1468-3083.2008.02938.x
35. Choi JS, Dib-Hajj SD, Waxman SG. Inherited erythermalgia. Limb pain from an S4 charge-neutral Na channelopathy. Neurology. 2006;67:1563–1567. doi:10.1212/01.wnl.0000231514.33603.1e
36. Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci. 2004;24::8232–8236. doi:10.1523/JNEUROSCI.2695-04.2004
37. Dib-Hajj SD, Rush AM, Cummins TR, et al. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain. 2005;128(Pt8):1847–1854. doi:10.1093/brain/awh514
38. Han C, Rush AM, Dib-Hajj SD, et al. Sporadic onset of erythermalgia: a gain-of-function mutation in Nav1.7. Ann Neurol. 2006;59(3):553–558. doi:10.1002/ana.20776
39. Harty TP, Dib-Hajj SD, Tyrrell L, et al. Na(V)1.7 mutant A863P in erythromelalgia: effects of altered activation and steady-state inactivation on excitability of nociceptive dorsal root ganglion neurons. J Neurosci. 2006;26(48):12566–12575. doi:10.1523/JNEUROSCI.3424-06.2006
40. Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci U S A. 2006;103(21):8245–8250. doi:10.1073/pnas.0602813103
41. Toro CP, Lipscombe D. Erythromelalgia and Paroxysmal Extreme Pain Disorder (PEPD) Biophysics of Voltage-Gated Ion Channels. From Mol Net. 2014:377. doi:10.1016/B978-0-12-397179-1.00013-0
42. Bjeloševič M, Kušíková K, Tomko J. Paroxysmal extreme pain disorder: a very rare genetic aetiology of syncope with bizarre flushing in an infant. J Paediatr Child Health. 2021;57(6):938–940.
43. Choi JS, Boralevi F, Brissaud O, et al. Paroxysmal extreme pain disorder: a molecular lesion of peripheral neurons. Nat Rev Neurol. 2011;7(1):51–55. doi:10.1038/nrneurol.2010.162
44. Hisama FM, Dib-Hajj SD, Waxman SG SCN9A Neuropathic Pain Syndromes. [Updated 2020 Jan 23]. In: Adam M, Mirzaa G Pagon R, et al.., editors [Internet]. Seattle (WA): University of Washington, Seattle (WA).
45. Stutchfield CJ, Loh NR. Focal epilepsy presenting as a bath-induced paroxysmal event/breath-holding attack. Epilepsy Behav Case Rep. 2014;2:102–104.
46. Estacion M, Dib-Hajj SD, Benke PJ, et al. NaV1.7 gain-of-function mutations as a continuum: A1632E displays physiological changes associated with erythromelalgia and paroxysmal extreme pain disorder mutations and produces symptoms of both disorders. J Neurosci. 2008;28(43):11079–11088. doi:10.1523/JNEUROSCI.3443-08.2008
47. Martinez-Lavin M. Fibromyalgia: when distress becomes (un)sympathetic pain. Pain Res Treat. 2012;2012:981565. doi:10.1155/2012/981565
48. de Lera Ruiz M, Kraus RL. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J Med Chem. 2015;58(18):7093–7118.
49. Zorina-Lichtenwalter K, Meloto CB, Khoury S, Diatchenko L. Genetic predictors of human chronic pain conditions. Neuroscience. 2016;338:36–62.
50. Wordliczek J, Banach M, Garlicki J, Wordliczek J, Dobrogowski J. Influence of pre- or intraoperational use of tramadol (preemptive or preventive analgesia) on tramadol requirement in the early postoperative period. Pol J Pharmacol. 2002;54(6):693–697.
51. Cannon A, Kurklinsky S, Guthrie KJ, Riegert-Johnson DL. Advanced genetic testing comes to the pain clinic to make a diagnosis of paroxysmal extreme pain disorder. Case Rep Neurol Med. 2016;2016:9212369. doi:10.1155/2016/9212369
52. Xue Y, Kremer M, Muniz Moreno MDM, et al. The human SCN9AR185H point mutation induces pain hypersensitivity and spontaneous pain in mice. Front Mol Neurosci. 2022;15::913990. doi:10.3389/fnmol.2022.913990
53. Devigili G, Rinaldo S, Lombardi R, et al. Diagnostic criteria for small fibre neuropathy in clinical practice and research. Brain. 2019;142(12):3728–3736. doi:10.1093/brain/awz333
54. Levine TD. Small fiber neuropathy: disease classification beyond pain and burning. J Cent Nerv Syst Dis. 2018;10:117957351877170. doi:10.1177/1179573518771703
55. Oaklander AL, Klein MM. (2013) Evidence of small-fiber polyneuropathy in unexplained, juvenile-onset, widespread pain syndromes. Pediatrics. 2013;131(4):e1091–e1100. doi:10.1542/peds.2012-2597
56. McDermott LA, Weir GA, Themistocleous AC, et al. Defining the Functional Role of NaV1.7 in Human Nociception. Neuron. 2019;101(5):905–919.e8.
57. He Y, Kim PY Allodynia. [Updated 2022 Sep 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan
58. Fealey RD. Thermoregulation in neuropathies. Handb Clin Neurol. 2018;157:777–787.
59. Mozafarpour S, Chen A, Paredes Mogica JA, et al. Urodynamic autonomic bladder dysfunction in women with complex chronic pelvic pain is associated with small fiber polyneuropathy. Neurourol Urodyn. 2022;41(1):482–489. doi:10.1002/nau.24858
60. Shinu P, Morsy MA, Nair AB, et al. Novel Therapies for the Treatment of Neuropathic Pain: Potential and Pitfalls. J Clin Med. 2022;11(11):3002
61. Elsana B, Imtirat A, Yagev R, et al. Ocular manifestations among patients with congenital insensitivity to pain due to variants in PRDM12 and SCN9A genes. Am J Med Genet A. 2022;188(12):3463–3468
62. Le Cann K, Meents JE, Bhagavath Eswaran V S, et al. Assessing the impact of pain-linked Nav1.7 variants: An example of two variants with no biophysical effect. Channels (Austin). 2021;15(1):208–228.
63. Birnbaum J, DuncanT, Owoyemi, K, Wang KC, Carrino J, Chhabra A. Use of a novel high-resolution magnetic resonance neurography protocol to detect abnormal dorsal root Ganglia in Sjögren patients with neuropathic pain: case series of 10 patients and review of the literature. Medicine (Baltimore). 2014;93(3):121–134. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4632907/.
64. Raasing LRM, Vogels OJM, Veltkamp M, van Swol CFP, Grutters JC. Current view of diagnosing small fiber neuropathy. J Neuromuscul Dis. 2021;8(2):185–207. doi:10.3233/JND-200490
65. Faber CG, Hoeijmakers JG, Ahn HS, et al. Gain of function Naν1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol. 2012;71(1):26–39. doi:10.1002/ana.22485
66. Blasius AL, Dubin AE, Petrus MJ, et al. Hypermorphic mutation of the voltage-gated sodium channel encoding gene Scn10a causes a dramatic stimulus-dependent neurobehavioral phenotype. Proc Natl Acad Sci USA. 2011;108(48):19413–19418
67. Vasylyev DV, Han C, Zhao P, Dib-Hajj S, Waxman SG. Dynamic-clamp analysis of wild-type human Nav1.7 and erythromelalgia mutant channel L858H. J Neurophysiol. 2014;111(7):1429–1443.
68. Nettuwakul C, Praditsap O, Sawasdee N, et al. Loss-of-function mutations of SCN10A encoding NaV1.8 α subunit of voltage-gated sodium channel in patients with human kidney stone disease. Sci Rep. 2018;8(1):10453. doi:10.1038/s41598-018-28623-3
69. Brouwer BA, Merkies IS, Gerrits MM, Waxman SG, Hoeijmakers JG, Faber CG Painful neuropathies: the emerging role of sodium channelopathies. J Peripher Nerv Syst. 2014;19(2):53–65.
70. Themistocleous AC, Ramirez JD, Serra J, Bennett DL The clinical approach to small fibre neuropathy and painful channelopathy. Pract Neurol. 2014;14(6):368–379.
71. Fertleman CR, Baker MD, Parker KA, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–774. doi:10.1016/j.neuron.2006.10.006
72. Yang Y, Wang Y, Li S, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41(3):171–174. doi:10.1136/jmg.2003.012153
73. Cojocaru M, Cojocaru IM, Silosi I. Multiple autoimmune syndrome. Mædica. 2010;5(2):132–134.
74. Goldberg YP, Price N, Namdari R, et al. Treatment of Na(v)1.7-mediated pain in inherited erythromelalgia using a novel sodium channel blocker. Pain. 2012;153(1):80–85.
75. National Center for Biotechnology Information (2022). PubChem Compound Summary for CID 49836093, Funapide. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Funapide.
76. Cao L, McDonnell A, Nitzsche A, et al. Pharmacological reversal of a pain phenotype in Ipsc-derived sensory neurons and patients with inherited erythromelalgia. Sci Transl Med. 2016;8(335):335ra56.
77. Alexandrou AJ, Brown AR, Chapman ML, et al. Subtype-selective small molecule inhibitors reveal a fundamental role for Nav1.7 in nociceptor electrogenesis, axonal conduction and presynaptic release. PLoS One. 2016;11(4):e0152405. doi:10.1371/journal.pone.0152405
78. Bagal SK, Chapman ML, Marron BE, Prime R, Storer RI, Swain NA. Recent progress in sodium channel modulators for pain. Bioorg Med Chem Lett. 2014;24(16):3690–3699. doi:10.1016/j.bmcl.2014.06.038
79. Martz L. Nav-i-gating antibodies for pain. Sci Business Exchange. 2014;7(23):662.
80. McMahon SB, Koltzenburg M, Tracey I, Turk D. Wall & Melzack’s Textbook of Pain. Elsevier Health Sciences; 2013:508.
81. McDonnell A, Collins S, Ali Z, et al. Efficacy of the Nav1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain. 2018;159(8):1465–1476. doi:10.1097/j.pain.0000000000001227
82. Siebenga P, van Amerongen G, Hay JL, et al. Lack of detection of the analgesic properties of PF-05089771, a selective Nav 1.7 inhibitor, using a battery of pain models in healthy subjects. Clin Transl Sci. 2020;13(2):318–324. doi:10.1111/cts.12712
83. Jones HM, Butt RP, Webster RW, et al. Clinical Micro-Dose Studies to Explore the Human Pharmacokinetics of Four Selective Inhibitors of Human Nav1.7 Voltage-Dependent Sodium Channels. Clin Pharmacokinet. 2016;55(7):875–887.
84. Dunbar J, Versavel M, Zhao Y, et al. Evaluation of the Pharmacokinetic Interaction Between the Voltage- and Use-Dependent Nav1.7 Channel Blocker Vixotrigine and Carbamazepine in Healthy Volunteers. Clin Pharmacol Drug Dev. 2020;9(1):62–73.
85. Mann N, King T, Murphy R Review of primary and secondary erythromelalgia. Clin Exp Dermatol. 2019;44(5):477–482.
86. Abdelsayed M, Sokolov S Voltage-gated sodium channels: pharmaceutical targets via anticonvulsants to treat epileptic syndromes. Channels (Austin). 2013;7(3):146–152.
87. Labau JIR, Estacion M, Tanaka BS, et al. Differential effect of lacosamide on Nav1.7 variants from responsive and non-responsive patients with small fibre neuropathy. Brain. 2020;143(3):771–782. doi:10.1093/brain/awaa016
88. Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482.
89. Moreno AM, Alemán F, Catroli GF, et al. Long-lasting analgesia via targeted in situ repression of NaV1.7 in mice. Sci Transl Med. 2021;3(584):eaay9056. doi:10.1126/scitranslmed.aay9056
90. Thaci B, Ulasov IV, Wainwright DA, Lesniak MS. The challenge for gene therapy: innate immune response to adenoviruses. Oncotarget. 2011;2(3):113–121. doi:10.18632/oncotarget.231
91. Athanasopoulos T, Munye MM, Yáñez-Muñoz RJ. Nonintegrating gene therapy vectors. Hematol Oncol Clin North Am. 2017;31(5):753–770. doi:10.1016/j.hoc.2017.06.007
92. Vannucci L, Lai M, Chiuppesi F, Ceccherini-Nelli L, Pistello M. Viral vectors: a look back and ahead on gene transfer technology. New Microbiol. 2013;36(1):1–22.
93. Lee CS, Bishop ES, Zhang R, et al. Adenovirus-mediated gene delivery: potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017;4(2):43–63. doi:10.1016/j.gendis.2017.04.001
94. Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122(1):23–36. doi:10.1182/blood-2013-01-306647
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.