")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 10
A review of the pharmacology and clinical efficacy of brivaracetam
Authors Klein P, Diaz A, Gasalla T, Whitesides J
Received 31 May 2017
Accepted for publication 23 August 2017
Published 19 January 2018 Volume 2018:10 Pages 1—22
DOI https://doi.org/10.2147/CPAA.S114072
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Pavel Klein,1 Anyzeila Diaz,2 Teresa Gasalla,3 John Whitesides4
1Mid-Atlantic Epilepsy and Sleep Center, Bethesda, MD, USA; 2Neurology Patient Value Unit, UCB Pharma, Smyrna, GA, USA; 3Neurology Patient Value Unit, UCB Pharma, Monheim am Rhein, Germany; 4Asset Development, UCB Pharma, Raleigh, NC, USA
Abstract: Brivaracetam (BRV; Briviact) is a new antiepileptic drug (AED) approved for adjunctive treatment of focal (partial-onset) seizures in adults. BRV is a selective, high-affinity ligand for synaptic vesicle 2A (SV2A) with 15- to 30-fold higher affinity than levetiracetam, the first AED acting on SV2A. It has high lipid solubility and rapid brain penetration, with engagement of the target molecule, SV2A, within minutes of administration. BRV has potent broad-spectrum antiepileptic activity in animal models. Phase I studies indicated BRV was well tolerated and showed a favorable pharmacokinetic profile over a wide dose range following single (10–1,000 mg) and multiple (200–800 mg/day) oral dosing. Three pivotal Phase III studies have demonstrated promising efficacy and a good safety and tolerability profile across doses of 50–200 mg/day in the adjunctive treatment of refractory focal seizures. Long-term data indicate that the response to BRV is sustained, with good tolerability and retention rate. BRV is highly effective in patients experiencing secondarily generalized tonic–clonic seizures. Safety data to date suggest a favorable psychiatric adverse effect profile in controlled studies, although limited postmarketing data are available. BRV is easy to use, with no titration and little drug–drug interaction. It can be initiated at target dose with no titration. Efficacy is seen on day 1 of oral use in a significant percentage of patients. Intravenous administration in a 2-minute bolus and 15-minute infusion is well tolerated. Here, we review the pharmacology, pharmacokinetics, and clinical data of BRV.
Keywords: brivaracetam, efficacy, epilepsy, focal seizures, pharmacokinetics, tolerability
Plain-language summary
Brivaracetam (BRV) is a new antiepileptic drug for add-on therapy for focal (also called partial-onset) seizures in adults with epilepsy. We report results from studies conducted throughout the development of BRV, including on how BRV is thought to work in the brain, how the drug is taken in and removed from the body, the effect of BRV on seizures, and side effects that have been reported in clinical trials of people with epilepsy. BRV is effective in many different animal models of seizures. BRV is easily absorbed by the body and quickly enters the brain. It has few interactions with other drugs. In three Phase III studies, patients with uncontrolled focal seizures taking BRV 50–200 mg/day had significantly fewer seizures than patients taking placebo, with good tolerability. BRV was good at treating a severe type of focal seizure: secondarily generalized tonic–clonic seizures. BRV is well tolerated, even when started at target doses, when taken as tablets, or when taken as an injection by infusion or bolus.
Introduction
Epilepsy affects more than 50 million people worldwide.1 Over 20 antiepileptic drugs (AEDs) are available for the treatment of epilepsy, and are approved for the treatment of several different types of seizure or syndrome. However, about a third of patients do not respond to AED treatment.2,3
In recent years, the choice of available AEDs has been expanded with the approval of lacosamide (Vimpat), rufinamide (Inovelon), ezogabine/retigabine (Potiga/Trobalt), perampanel (Fycompa), and eslicarbazepine acetate (Aptiom/Zebinix). However, ezogabine/retigabine will be discontinued during 2017 and will no longer be commercially available. These AEDs have been approved by the US Food and Drug Administration (FDA) for adjunctive treatment of focal (partial-onset) seizures,4–7 with perampanel also approved for treatment of primary generalized tonic–clonic seizures.5 Eslicarbazepine acetate is also approved by the European Medicines Agency for monotherapy treatment of focal seizures in adults with newly diagnosed epilepsy.8 The exception is rufinamide, which is approved by the FDA for adjunctive treatment of Lennox–Gastaut syndrome in adult and pediatric patients ≥1 year old.9 A brief overview of the main features of these five AEDs, including top-line pooled efficacy data from pivotal trials,10–14 is provided in Table 1.
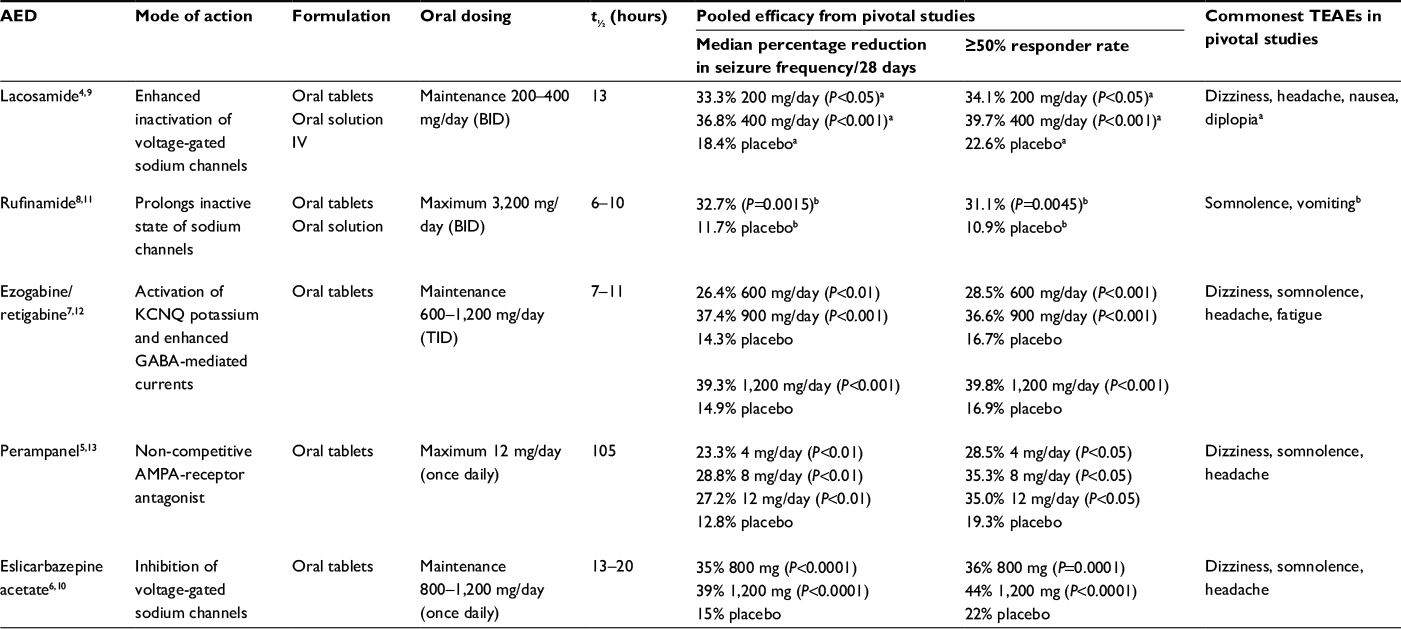 | Table 1 Overview of adjunctive AEDs: prescribing information and main findings from pooled analyses of pivotal studies Notes: aPooled analysis of Phase II and Phase III studies; bpooled analyses not available for rufinamide; data based on one pivotal study. Abbreviations: AED, antiepileptic drug; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BID, bis in die (twice daily); IV, intravenous; GABA, γ-aminobutyric acid; t½, half-life; TEAEs, treatment emergent adverse events; TID, ter in die (three times daily). |
Brivaracetam (BRV; Briviact; UCB Pharma SA, Brussels, Belgium) is a new AED, approved in 2016 by the FDA for adjunctive treatment of focal seizures in patients aged ≥16 years with epilepsy.15 BRV is a member of the racetam class of drugs, related to piracetam and the AED levetiracetam (LEV), and is the first selective ligand for SV2A.16 We review here the pharmacology of BRV, as well as its efficacy, safety, and tolerability in patients with epilepsy.
Mode of action
BRV’s mode of action was recently reviewed by Klitgaard et al and Löscher et al, who detailed current understanding of how BRV exerts its antiepileptogenic properties.17,18 BRV is a selective, high-affinity ligand for SV2A.16 SV2A is a transmembrane glycoprotein and galactose transporter.19 It is considered the master regulator molecule of neurotransmitter release.18 The human SV2A gene, located in the q arm of chromosome 1 at locus 21.2, is ~14,565 bp in size, and encodes a 4,353 bp mRNA with 13 exons, which is translated to an 82.6 kDa protein composed of 742 amino acids.20 SV2A is present ubiquitously throughout the central nervous system, except for the trigeminal and facial nuclei.18,20 It is present in both γ-aminobutyric acid (GABA)-ergic and glutamatergic neurons.21 Neurotransmitter release is a cyclical process that consists of several stages: uptake of the neurotransmitter into the vesicle in the cell interior, movement of the vesicle to the presynaptic wall, docking of the vesicle at the wall, priming of the vesicle for neurotransmitter release, calcium-dependent neurotransmitter release into the synapse, and movement of the vesicle back to the cell interior to join the pool of usable vesicles.18 It has been suggested that SV2A regulates neurotransmitter release at various steps of this synaptic vesicle cycle, including the rate of neurotransmitter uptake into the vesicle, vesicle priming at the presynaptic membrane, neurotransmitter release, and vesicle trafficking between the cytoplasm and the presynaptic membrane.18
SV2A absence or reduced function may facilitate seizures. SV2A–/–-knockout mice appear normal at birth, but develop seizures early postnatally, do not grow, and die at 3 weeks of age.22 Loss of SV2A in these animals is associated with a decrease in action potential-dependent (but not action potential-independent) GABA transmission in the CA3 region of the hippocampus. SV2A expression is reduced during kindling of epileptogenesis in spontaneously epileptic rats, and in pharmacoresistant amygdala-kindled rats compared with nonpharmacoresistant rats.23,24 SV2A is reduced in the hippocampus of patients with pharmacoresistant temporal lobe epilepsy and hippocampal sclerosis, observed in resected tissue specimens and in positron-emission tomography (PET) studies using the radioactive SV2A ligand [11C]UCB-J,25,26 as well as in epileptogenic cortical dysplasia and tubers.27 Recently, a case of intractable epilepsy due to a homozygous SV2A mutation was reported.28 The patient had an arginine–glutamine mutation in amino-acid position 383 (R383Q) in exon 5, and both parents were carriers for the R383Q variant, suggesting that R383Q is a recessive mutation.
However, increased expression of SV2A has been reported in the kindling model of temporal lobe epilepsy.29–32 Overexpression of SV2A in hippocampal neurons results in a neurotransmission phenotype that resembles that of neurons from SV2A-knockout mice, suggesting that too much SV2A is as detrimental to neuronal function as too little.33 LEV treatment during kindling reduces SV2A expression past the kindling phase.29 LEV antiseizure activity in the 6 Hz epilepsy model is reduced in heterozygous SV2A+/–-knockout mice with 50% SV2A-protein reduction.34
LEV and BRV are thought to exert their anticonvulsant action by binding to SV2A and modulating its effect on neurotransmitter release. However, details of how binding to SV2A results in an anticonvulsant effect are at present unknown.35 Of particular interest is the unanswered question of how modulation of a molecule that regulates the release of all neurotransmitters, excitatory and inhibitory alike, results in inhibition of seizures.
BRV was synthesized in a large medicinal chemistry-discovery effort by UCB Pharma to synthesize molecules with greater binding affinity than LEV to SV2A. Approximately 12,000 molecules were evaluated: ~1,200 were tested in the audiogenic seizure-susceptible mouse model, and ~30 of these were evaluated extensively in diverse animal epilepsy models. BRV and seletracetam were two molecules selected for clinical development, with BRV developed to the approval stage.17
BRV has 15- to 30-fold higher affinity for SV2A than LEV16 (Figure 1). Binding of LEV to SV2A results in reduced exocytosis of neurotransmitters17 and reduced neurotransmission in response to rapid-stimulus trains,36 possibly by blocking SV2A’s effect on vesicle priming at the presynaptic membrane. LEV reduces vesicular release in a stimulation-dependent manner, suggesting that repetitive stimulation is required to allow LEV entry into recycling synaptic vesicles and intravesicular binding to SV2A.37 LEV appears to target preferential hyperactive synapses,38 enter synaptic vesicles during recycling and endocytosis, and escape during synaptic fusion.20 LEV reverses the effects of overexpressed SV2A.33
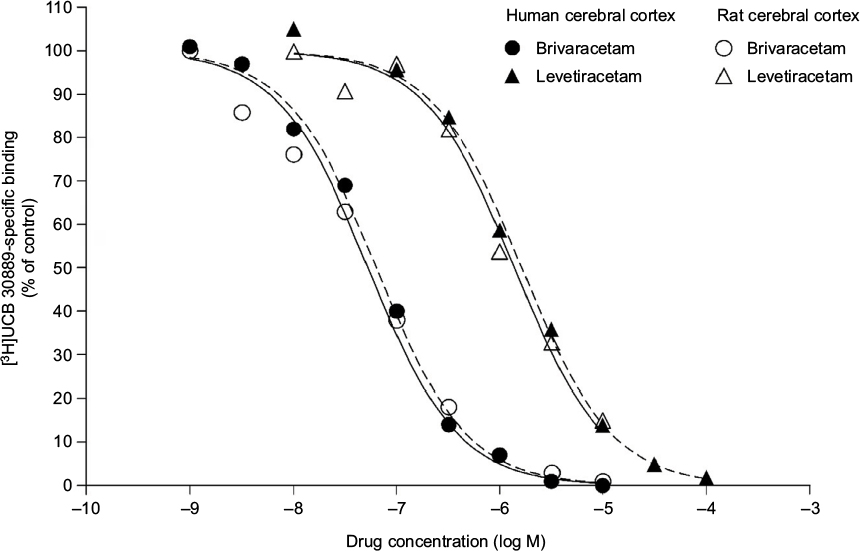 | Figure 1 Ex vivo binding of brivaracetam and levetiracetam in rat and human cerebral cortex. Notes: Republished with permission of Elsevier Science and Technology Journals, from Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: relationship to anti-convulsant properties, Gillard M, Fuks B, Leclercq K, Matagne A, Volume 664, Edition (1–3), 2011; permission conveyed through Copyright Clearance Center, Inc.16 |
While the anticonvulsant effect of BRV or LEV binding to SV2A is not fully understood, one hypothesis is that ligand binding to SV2A may stabilize a certain conformation of the protein, resulting in a potentiation or optimization of its function, thereby providing seizure protection.39 Rat hippocampal slice research comparing BRV and LEV using high-frequency neuronal stimulation have suggested that BRV augments synaptic depression and decreases synaptic transmission at 100-fold lower concentrations than LEV.40 In studies with dye-loaded vesicles, BRV slowed stimulation-induced vesicle release significantly more than LEV, indicating that BRV reduces vesicle mobilization more effectively than LEV.40
It is possible that different binding properties for LEV and BRV to the SV2A protein may contribute to their different pharmacological efficacies. In studies with the SV2A positive allosteric modulator UCB 1244283 in HEK293 cells, which express human recombinant SV2A, and in human cortex, [3H]BRV labeled twice as many sites as [3H]LEV.41 Moreover, coincubation of [3H]LEV with the allosteric modulator produced only a modest increase in affinity for SV2A but a twofold increase in maximum binding capacity. In contrast, coincubation of [3H]BRV with the modulator increased the affinity much more (around tenfold) while the increase in maximum binding capacity was more modest (1.3-fold)41 (Figure 2). This suggests differential interaction of BRV and LEV with SV2A, which may provide the molecular correlate to their distinct pharmacodynamic properties.
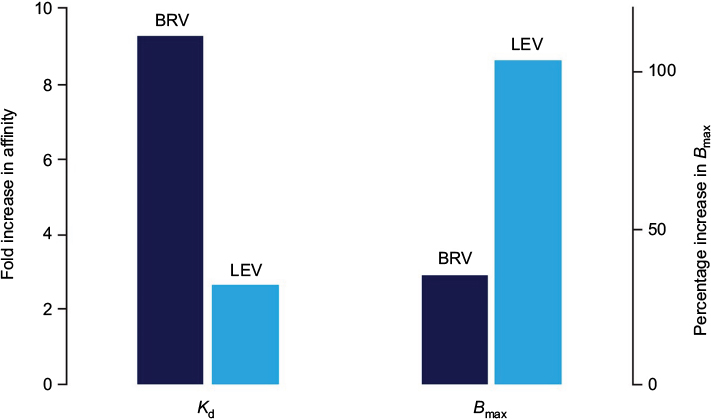 | Figure 2 Differential interaction of BRV and LEV with SV2A. Notes: Combination experiments assessing the impact of the allosteric SV2A modulator UCB 1244283 on [3H]BRV and [3H]LEV binding in recombinant cells expressing SV2A. A major increase in affinity (Kd) for SV2A was seen for [3H]BRV, and maximum binding capacity (Bmax) increased for [3H]LEV, suggesting that BRV and LEV have differential interactions with SV2A. Republished with permission of John Wiley and Sons Inc, from Brivaracetam: rationale for discovery and preclinical profile of a selective SV2A ligand for epilepsy treatment, Klitgaard H, Matagne A, Nicolas JM, et al, Volume 57, Edition 4, 2016; permission conveyed through Copyright Clearance Center, Inc.17 Abbreviations: BRV, brivaracetam; LEV, levetiracetam. |
Current evidence indicates that conventional modes of action of other AEDs do not contribute significantly to the antiepileptic properties of BRV. This contrasts with LEV, which inhibits α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors42 and high-voltage-gated calcium currents,43 in addition to SV2A binding. BRV exerts no direct effect on AMPA, GABA, glycine, or kainic acid-gated currents, and has only a minor inhibitory action on N-methyl-d-aspartate (NMDA)-receptor activity at supratherapeutic concentrations.17,44 It has no effect on voltage-gated potassium channels45 or high- and low-voltage-gated calcium channels at therapeutically relevant concentrations.46 Although an early patch-clamp study in rat neocortical neurons suggested that inhibition of voltage-gated sodium channels may contribute an additional mode of action for BRV,47 subsequent studies demonstrated that while BRV inhibited sodium currents to a lesser extent than in the initial study, this effect was not observed in CA1 pyramidal neurons from adult mice. Importantly, BRV had no effect against sustained repetitive firing in these neurons, suggesting that BRV’s antiepileptic mechanism is unrelated to effects on voltage-gated sodium channels.45,48,49
Preclinical profile
BRV is highly lipid-soluble (LogDpH7.4 1.04), comparable with benzodiazepines and phenytoin and more so than LEV.50 It enters the brain rapidly, crosses the blood–brain barrier via passive diffusion, and engages SV2A, the target molecule within minutes of intravenous (IV) administration.17,51,52 This speed of entry into the brain has been evaluated in a series of experiments. In audiogenic seizure-susceptible mice, a single oral administration of BRV resulted in simultaneous maximal peak plasma levels and maximal anticonvulsant effect.50 By contrast, LEV’s maximal anticonvulsant activity occurred 1 hour after maximum plasma peak level. In ex vivo binding studies in the same model, maximal SV2A occupancy and antiepileptic activity occurred 5–15 minutes after intraperitoneal injection with BRV versus 30–60 minutes with LEV.16 In rhesus monkeys, PET imaging using the SV2A radioligand PET tracer [11C]UCB-J after a single IV injection with BRV 5 mg/kg or LEV 30 mg/kg found that drug-entry half-time was 3 minutes for BRV and 23 minutes for LEV (Figure 3).50 In a related PET study in four healthy human volunteers, the drug entry half-time of [11C]UCB-J after a 5-minute IV infusion was 7 minutes with BRV 100 mg compared with 22 minutes for LEV 1,500 mg.51 New data from nine human volunteers found drug-entry half-time of [11C]UCB-J after a 5-minute IV infusion was 2, 10, and 22 minutes with BRV 200, 100, and 50 mg, respectively, compared with 20 minutes for LEV 1,500 mg.52 Both studies indicate a similarly fast speed of brain entry and SV2A binding by BRV in humans. There is no transporter-mediated efflux of BRV from the brain.53
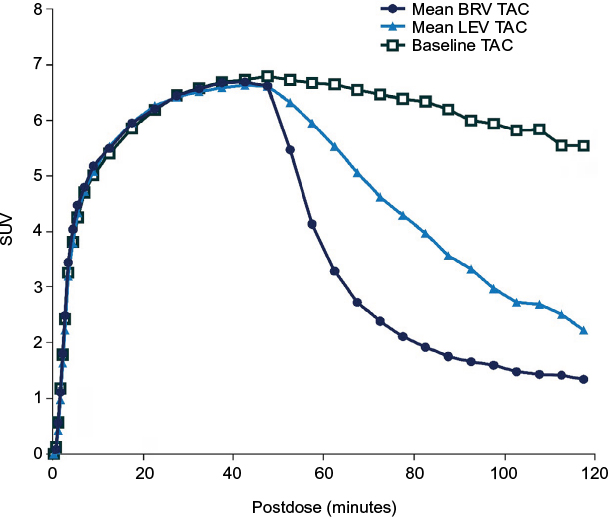 | Figure 3 Displacement of the SV2A PET tracer [11C]UCB-J after injection of 5 mg/kg BRV or 30 mg/kg LEV (administered intravenously 45 minutes after the PET tracer). Notes: The displacement graph shows the amount of radioactivity in the brain over time. The baseline curve (black squares) shows that the tracer by itself leaves the brain very slowly (gradual reduction in radioactivity over time). When BRV or LEV is administered, radioactivity in the brain is quickly reduced (dark blue circles and light blue triangles, respectively). This is because BRV and LEV displace the tracer. The BRV curve is steeper than the LEV curve, which means that BRV displaces the tracer more quickly than LEV does; therefore, it enters the brain more quickly than LEV. The steepness of these curves was used to estimate the speed of entry of LEV and BRV. Republished with permission of John Wiley and Sons Inc, from Brivaracetam, a selective high-affinity synaptic vesicle protein 2A (SV2A) ligand with preclinical evidence of high brain permeability and fast onset of action, Nicolas JM, Hannestad J, Holden D, et al, Volume 57, Edition 2, 2016; permission conveyed through Copyright Clearance Center, Inc.50 Abbreviations: BRV, brivaracetam; LEV, levetiracetam; PET, positron-emission tomography; SUV, standardized uptake value; TAC, time–activity curve. |
Preclinical seizure models
In line with its greater affinity for SV2A compared with LEV, BRV has demonstrated higher potency than LEV in a range of animal models of epilepsy (Table 2).17 Unlike LEV, BRV is effective in classical maximal electric seizure and pentylenetetrazole models, albeit at high concentrations. BRV provides more potent protection than LEV against secondary generalized seizures in models of focal epilepsy, such as cornea-kindled mice, hippocampus-kindled rats, and the 6 Hz seizure model in mice.54,55 In the 6 Hz cornea-kindling model in mice, BRV’s seizure-protection potency was several-fold higher than LEV against both partial and secondary generalized seizures.56,57 BRV appeared more effective than LEV for seizure suppression in the drug-resistant epilepsy model of amygdala-kindled mice: BRV dose-dependently (6.8–212.3 mg/kg) reduced seizure severity with near-complete suppression of seizures at the highest tested doses, while LEV (17–1,700 mg/kg) provided only limited protection.55
 | Table 2 Seizure suppression of BRV and LEV in animal models of seizures and epilepsy Note: Reproduced froim Klitgaard H, Matagne A, Nicolas JM, et al. Brivaracetam: rationale for discovery and preclinical profile of a selective SV2A ligand for epilepsy treatment. Epilepsia. 2016;57(4):538–548.17 Abbreviations: BRV, brivaracetam; ED50, effective dose, 50%; IP, intraperitoneal; IV, intravenous; LEV, levetiracetam; MAD, minimum active dose; SSSE, self-sustaining status epilepticus. |
BRV is also more effective than LEV in models of primary generalized epilepsy. It provides complete suppression of seizures in the genetic model of audiogenic seizure-susceptible mice and complete suppression of spontaneous spike-and-wave discharges in the genetic absence epilepsy rat from Strasbourg (an absence model), in contrast to LEV.17,55,58 BRV has tenfold-higher potency than LEV against both myoclonus and generalized seizures in rat posthypoxic myoclonus.59
Status epilepticus
BRV had a potent effect in a model of self-sustaining status epilepticus (SSSE).60 It shortened the cumulative seizure time in established SSSE from 467 minutes in control rats to 71 and 5 minutes at 20 and 300 mg/kg doses of BRV, respectively. Lower doses of BRV (0.3–10 mg/kg) were markedly more effective when administered in combination with diazepam 1 mg/kg, suggesting the possibility of a synergistic interaction. SSSE induction resulted in subsequent development of spontaneous recurrent seizures in control rats. However, acute treatment of SSSE with BRV 300 mg/kg alone or BRV 1–10 mg/kg in combination with diazepam prevented or reduced the frequency of spontaneous recurrent seizures.
Epileptogenesis
In addition to the reduction of spontaneous recurrent seizures after SSSE,60 BRV pretreatment with 6.8 mg/kg persistently reduces the incidence of seizures in cornea-kindled mice, another model of epileptogenesis, by approximately 50%, whereas LEV is ineffective.55 These studies suggest that BRV may have antiepileptogenic potential.
Pharmacokinetic profile
Key pharmacokinetic (PK) parameters for BRV are summarized in Tables 3 and 4, and have been recently reviewed.17,61 Following single oral doses or multiple daily doses, BRV is rapidly absorbed with a median time to maximum concentration (tmax) of approximately 1 hour.62,63 High-fat food delays tmax (3 hours) and decreases maximum concentration (Cmax), but has no effect on area under the plasma concentration–time curve (AUC),63 and there is only weak plasma binding of <20%.64 Saliva and plasma levels of BRV are highly correlated,62 and the volume of distribution is slightly lower than total body water.62–64
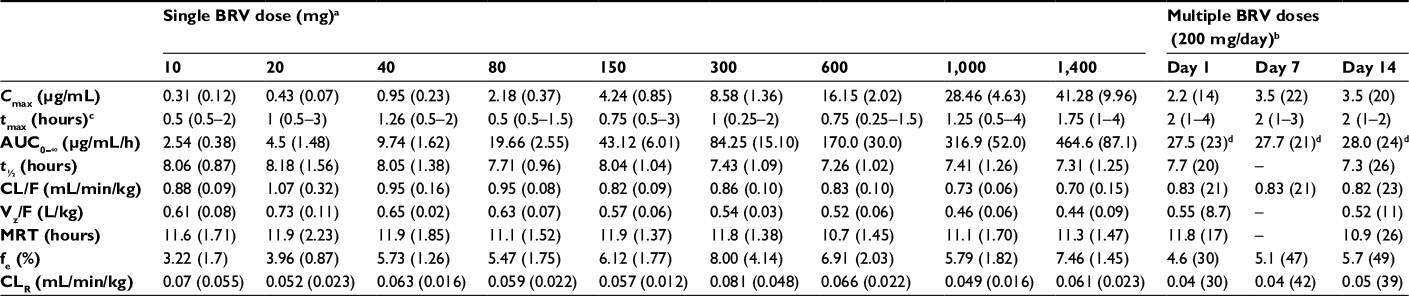 | Table 3 Pharmacokinetic parameters for BRV following single and multiple oral doses Notes: aArithmetic means (SD), n=6/dose; bgeometric means (CV%), n=9; cmedian (range); dAUC extrapolated to infinity on day 1, AUCt on days 7 and 14. Adapted with permission of British journal of clinical pharmacology, from The pharmacokinetics, CNS pharmacodynamics and adverse event profile of brivaracetam after multiple increasing oral doses in healthy men, Rolan P, Sargentini-Maier ML, Pigeolet E, Stockis A, Volume 66, Edition 1, 2008; permission conveyed through Copyright Clearance Center, Inc.62 Adapted with permission of British journal of clinical pharmacology, from The pharmacokinetics, CNS pharmacodynamics and adverse event profile of brivaracetam after single increasing oral doses in healthy males, Sargentini-Maier ML, Rolan P, Connell J, et al, Volume 63, Edition 6, 2007; permission conveyed through Copyright Clearance Center, Inc.63 Abbreviations: AUC, area under the (concentration–time) curve; BRV, brivaracetam; CL/F, apparent clearance; CLR, renal clearance; Cmax, maximum concentration; CV, coefficient of variation; fe, fraction of BRV dose excreted in urine over 24 hours (single dose and day 1) or over 12 hours (days 7 and 14); MRT, mean residence time; t½, half-life; tmax, time to Cmax; Vz/F, apparent volume of distribution. |
 | Table 4 Summary of key pharmacokinetic parameters Note: aFollowing a single 50 mg dose in healthy participants.66 Abbreviations: t½, half life; tmax, time to maximum plasma concentration; VD, volume of distribution. |
BRV is extensively metabolized, primarily by hydrolysis of the acetamide group to the carboxylic acid metabolite by an amidase,65,66 followed by hydroxylation by cytochrome P450 (CYP)2C9 to form a hydroxy-acid metabolite. β-Oxidation of the propyl side chain, mainly by CYP2C19, forms a secondary pathway. The three main metabolites (acid, hydroxy, and hydroxy acid) are not active. More than 95% of the BRV dose is eliminated in the urine within 72 hours, 8.6% is eliminated unchanged, and the rest is excreted as metabolites.64,67 Mean half-life (t½) was ~9 hours and plasma clearance was 3.4 L/hour following a single BRV 50 mg oral dose in healthy participants.68 In PK studies in healthy volunteers, the maximum tolerated single dose of BRV was 1,000 mg, and with 14-day repeated dosing persistent side effects (sedation) occurred at 800 mg/day.62,63
Factors affecting PK parameters
PK parameters in healthy participants are not significantly affected by age.69 In participants aged >75 years, t½ is slightly longer and clearance slightly lower compared with those aged 65–75 years; however, these changes are not considered significant enough to require a dose adjustment in elderly patients.70
PK parameters of BRV in adult epilepsy patients do not appear to be affected by age, sex, race, or creatinine clearance.71 Concomitant use of enzyme-inducing AEDs has been shown to increase BRV clearance.71 However, this PK effect is not considered to be clinically relevant: concomitant inducer AEDs did not influence the efficacy of BRV in a dose–response analysis of Phase II study data.72 In pediatric epilepsy patients, population PK analysis found that BRV exposure was reduced by 29% and 32% with concomitant administration of phenobarbital and carbamazepine, respectively, and increased by 11% with valproate.69
The influence of the CYP2C19 genotype on BRV PKs has been studied in Japanese patients,73 as the poor-metabolizer genotype is found in up to 20% of Asian populations compared with 2%–3% in Caucasian and black populations.74–76 In healthy Japanese men with poor-metabolizer genotype (mutations *2 and *3 of CYP2C19), formation of the hydroxy metabolite was reduced tenfold. However, there was only a 29% reduction in BRV clearance, which was not considered clinically significant. No dose adjustment was required in these patients.73
Renal clearance of the parent compound is low in healthy participants (~0.04 mL/min/kg).62 In patients with severe renal impairment, BRV exposure is increased by 21%, with a greater increase in levels of the acid, hydroxy, and hydroxy-acid metabolites.77 Toxicology research has identified no safety concerns arising from increased levels of these metabolites; therefore, it is unlikely that dose adjustment of BRV would be required in patients with renal dysfunction.77 It should be noted that this study did not include patients with end-stage renal disease undergoing dialysis. Metabolic clearance of BRV is reduced in patients with hepatic impairment, resulting in increased BRV exposure in mild, moderate, and severe liver disease of 50%–60%.78 This requires a BRV dose reduction of up to half in patients with liver impairment of any severity.15
Formulation-bioequivalence studies
In bioequivalence research on IV and oral tablet administration, an IV bolus of BRV 100 mg was bioequivalent to the oral formulation in terms of dose-normalized AUC, but not for dose-normalized Cmax, which was 28% higher versus the 50 mg oral tablet and 21% higher versus the 100 mg tablet.79 However, the plasma concentration–time profile was similar for the IV bolus and the two oral tablets after the first hour. Overall, the data indicated similar bioavailability, with no dose adjustment required when switching from oral to IV administration.79 An oral solution of BRV compared with the 50 mg oral tablet also demonstrated bioequivalence, with an earlier tmax but similar Cmax and AUC.68
Drug–drug interactions
The potential of BRV to reduce the activity of CYP450 enzymes is low. BRV has no significant effect on the activity of CYPs in human hepatocytes. When coadministered with midazolam, which is extensively metabolized by CYP3A isoenzymes, BRV doses of up to 150 mg/day have no significant effects on CYP3A activity in healthy participants.80
Gemfibrozil, a CYP2C9 inhibitor with the CYP2C8 inhibitor 1-O-β-glucuronide metabolite, has no effect on plasma- or urinary-excreted BRV, nor on hydroxy and carboxylic acid metabolites in healthy participants.81 The only observed change is a modest decrease in plasma and urinary levels of the hydroxy-acid metabolite. As gemfibrozil does not affect the PKs of unchanged BRV, BRV can be safely coadministered with CYP2C8 or CYP2C9 inhibitors.81 A study of the effects of the antibiotic rifampin (an inducer of CYP3A4, -2B6, -2C8, -2C9, and -2C19) on the PKs of BRV reported a 45% decrease in BRV AUC in the presence of rifampin, indicating that an increase in BRV dose may be required in patients receiving concomitant rifampin.66 Rifampin also increased the AUC of the BRV hydroxy metabolite by 109% and decreased the AUC of the carboxylic acid metabolite by 53%.66
A large study of adult epilepsy patients (n=1,771) enrolled across five BRV clinical studies analyzed the effects of BRV treatment on plasma concentrations of concomitant AEDs.82 BRV did not affect steady-state plasma concentrations of LEV, carbamazepine, lacosamide, lamotrigine, 10-hydroxyoxcarbazepine, phenobarbital, pregabalin, phenytoin, topiramate, valproate, or zonisamide. Levels of carbamazepine epoxide, the active metabolite of carbamazepine, were significantly increased due to BRV-mediated inhibition of epoxide hydrolase, the enzyme that metabolizes carbamazepine epoxide. Geometric mean plasma concentrations of 1.76 μg/mL without BRV increased to 2.53 μg/mL, 2.57 μg/mL, and 3.25 μg/mL with BRV doses of 50, 100, and 200 mg/day, respectively.82 This was not, however, associated with clinical adverse events (AEs). No dose adjustment is required when BRV up to 200 mg/day is added to other commonly prescribed AEDs.82 In a safety analysis of a pooled pivotal BRV Phase III population (n=1,558),83 BRV-treated patients had a similar incidence of treatment-emergent AEs (TEAEs) commonly associated with carbamazepine toxicity in the carbamazepine group (22.7%) and the non-carbamazepine group (22.6%). The non-carbamazepine group experienced more serious AEs (SAEs; 3.4% vs 1.1%) and more discontinuations due to TEAEs (8.6% vs 2.9%) than the carbamazepine group. There was no association between carbamazepine epoxide levels and toxicity symptoms.83 The effects of BRV administration on the PKs of carbamazepine in adults with epilepsy were further evaluated in a Phase I, open-label, dose-escalation study.84 In patients treated with carbamazepine (n=9) or carbamazepine and valproate (n=9), carbamazepine epoxide plasma concentrations were approximately doubled by BRV 100 mg or 200 mg twice daily. There was no change in carbamazepine, carbamazepine diol, or valproate levels, and no clinical AEs. Therefore, no adjustment in BRV dose is required when used concomitantly with carbamazepine.
In in vitro studies, BRV weakly inhibits phenytoin metabolism through CYP2C19 inhibition. A PK interaction study in patients with epilepsy demonstrated that a supratherapeutic BRV dose of 400 mg/day increased phenytoin exposure by approximately 20%.85 The recommended doses of BRV are not predicted to have an effect on phenytoin exposure.85
No interaction between BRV 100 mg/day and combination oral contraceptives (OCs; 30 µg ethinyl estradiol, 150 µg levonorgestrel) has been observed in healthy women (n=28), with no BRV effect on plasma concentrations of ethinyl estradiol or levonorgestrel and no OC effect on BRV trough levels.86 Supratherapeutic doses of BRV 400 mg/day and concomitant OC use in healthy women (n=24) resulted in a 27% reduction in plasma levels of ethinyl estradiol and a 23% reduction in levonorgestrel levels. However, there was no change in levels of endogenous hormones, and no ovulation occurred in any individual.87
Although no clinically relevant PK interactions have been observed between BRV and alcohol, administration of BRV has been reported to increase the effects of alcohol on psychomotor function, attention, and memory in healthy males.88 Unlike LEV, BRV is not a substrate of the transporters MDR1 (Pgp1), MRP1, and MRP2.53 MDR1 and MRP2 are efflux drug transporters thought to be overexpressed in patients with pharmacoresistant epilepsy.89
Efficacy, safety, and tolerability of adjunctive brivaracetam in patients with epilepsy
Phase IIA study
The clinical potential of BRV was first demonstrated in a subject-blind, placebo-controlled study in 18 patients with primary generalized epilepsy with photoparoxysmal electroencephalography response (NCT00401648).90 Intermittent photic stimulation was performed before and after a single dose of placebo or BRV. A single BRV dose of 10, 20, 40, or 80 mg abolished generalized photoparoxysmal electroencephalography response in 78% of patients compared with none after placebo. The overall effect was not dose-dependent, but time to maximal response was dose-related (shortest after BRV 80 mg [0.5 hours]), as was the duration of response, which was twice as long after BRV 80 mg (59.5 hours) compared with lower doses. BRV was well tolerated, with no reports of SAEs. The majority of TEAEs were mild or moderate in intensity. The most common was dizziness (n=5), which usually lasted <2 hours after BRV administration.
Phase IIB dose-ranging studies
Two double-blind, placebo-controlled, dose-ranging Phase IIB studies evaluated the efficacy and safety of adjunctive BRV doses of 5–150 mg/day in adults with uncontrolled focal epilepsy. NCT00175825 randomized BRV 5 mg/day, 20 mg/day, 50 mg/day, or placebo without uptitration (n=208).91 A significant difference in the primary efficacy outcome of percentage reduction over placebo in baseline-adjusted focal seizure frequency/week was observed for the 50 mg/day dose. In secondary efficacy outcomes, median percentage reduction from baseline in focal seizure frequency/week was significantly different from placebo for 50 mg/day and 20 mg/day, but not 5 mg/day; ≥50% responder rates were significantly higher than placebo with all BRV doses. BRV was well tolerated, with a low incidence of discontinuations due to TEAEs. The most common TEAEs were headache (placebo 7.4% vs BRV 5 mg/day 8%; 20 mg/day 3.8%; 50 mg/day 1.9%), somnolence (7.4% vs 2.0%, 5.8%, 5.8%), influenza (7.4% vs 8.0%, 0, 1.9%), dizziness (5.6% vs 2.0%, 0, 7.7%), neutropenia (1.9% vs 8.0%, 3.8%, 0), and fatigue (3.7% vs 0, 3.8%, 5.8%).
NCT00175929 randomized 157 patients to receive BRV 50 mg/day, BRV 150 mg/day, or placebo.92 A 3-week uptitration period was followed by a 7-week maintenance period. Percentage reduction over placebo in baseline-adjusted focal seizure frequency/week during the maintenance period (primary efficacy outcome) did not reach statistical significance for either dose of BRV, but significant differences were observed in several secondary efficacy outcomes. BRV was again well tolerated, with the most common TEAEs reported as headache, fatigue, nasopharyngitis, nausea, somnolence, dizziness, and urinary tract infection.
Phase III studies
Pivotal Phase III studies
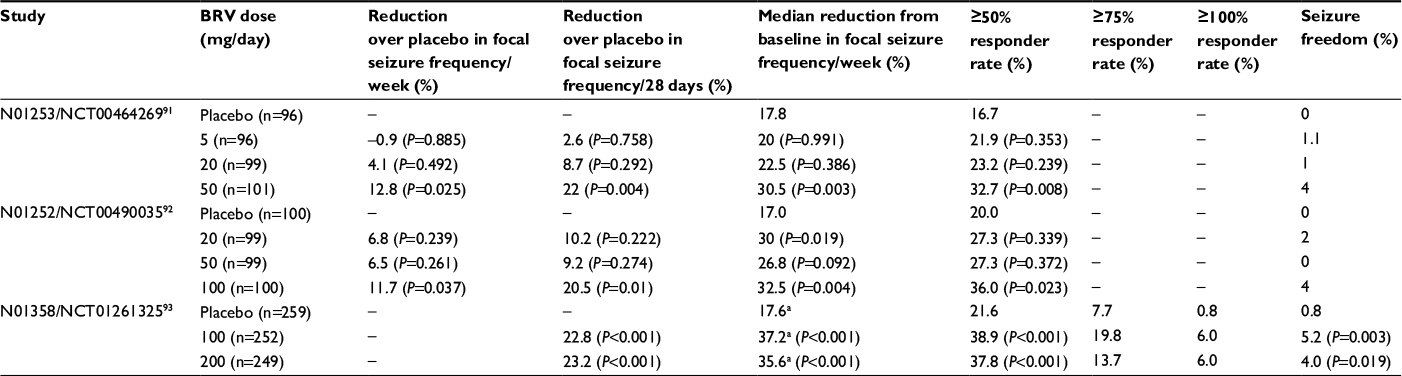
The pivotal Phase III BRV studies comprised three similarly designed, prospective, multicenter, randomized, double-blind, placebo-controlled, parallel-group trials that evaluated fixed dosing of adjunctive BRV (5–200 mg/day) with no uptitration over a 12-week treatment period following an 8-week baseline period, in adult patients with uncontrolled focal seizures treated with one or two AEDs. Efficacy results for the three studies are summarized in Table 5.
 | Table 5 Efficacy results from Phase III pivotal studies of adjunctive BRV in patients with uncontrolled focal seizures Note: aPer 28 days. Abbreviation: BRV, brivaracetam |
In NCT00464269 (N01253), patients were randomized to receive BRV 5 mg/day, 20 mg/day, or 50 mg/day.93 Percentage reduction over placebo in baseline-adjusted partial-onset seizure frequency/week during the 12-week treatment period (the primary efficacy end point) was significant for the 50 mg/day dose only (5 mg/day –0.9, P=0.885; 20 mg/day 4.1, P=0.492; 50 mg/day 12.8, P=0.025) (Table 5). Secondary end points, including ≥50% responder rate and median percentage reduction from baseline in seizure frequency/week, were also significantly different from placebo for the 50 mg/day dose only (Table 5).
BRV doses of 20 mg/day, 50 mg/day, and 100 mg/day were evaluated in NCT00490035 (N01252).94 To control for multiplicity, comparison of BRV with placebo was sequential (50 mg/day, 100 mg/day, 20 mg/day). BRV 50 mg/day was required to demonstrate superiority over placebo to meet the primary efficacy end point: percentage reduction over placebo in baseline-adjusted seizure frequency/week during the 12-week treatment period. Based on this criterion, the primary efficacy end point was not met (20 mg/day 6.8, P=0.239; 50 mg/day 6.5, P=0.261; 100 mg/day 11.7, P=0.037), although percentage reduction over placebo in seizure frequency, median percentage reduction from baseline in seizure frequency, and ≥50% responder rate were significant for BRV 100 mg/day (Table 5).
Due to the negative outcome of the primary efficacy end point of NCT00490035, a confirmatory study (NCT01261325; N01358) assessed higher BRV doses of 100 mg/day and 200 mg/day (n=768).95 Based on the results of the two previous studies, patients exposed to LEV within 90 days prior to enrollment were excluded from this study. The coprimary efficacy outcomes reached statistical significance for both doses: percentage reduction over placebo in baseline-adjusted seizure frequency/28 days (100 mg/day 22.8, P<0.001; 200 mg/day 23.2, P<0.001) and ≥50% responder rate (placebo, 21.6%; 100 mg/day 38.9%, P<0.001; 200 mg/day 37.8%, P<0.001) (Table 5). Secondary outcomes of median percentage reduction in seizure frequency from baseline and seizure freedom were also significantly different from placebo for both BRV doses. In this study, there did not appear to be a dose–response relationship between the 100 and 200 mg/day dosages, though previously one Phase III93 and one Phase II study92 had both observed dose–response effects. The study was carried out on four continents. Subgroup analysis showed regional variation in responses to the 200 mg/day dose. A notably higher response was seen with the 200 mg/day dose than with the 100 mg/day dose in North America and Latin America, but not in Europe or Asia.95 Placebo response for ≥50% responder rates also varied: it was highest in non-EU countries in Europe and lowest in Asia-Pacific/other countries. There was no clear explanation for these differences.
Efficacy analyses of LEV treatment in the first two studies (NCT00464269, NCT00490035) showed that patients never exposed to LEV had the highest response, while patients previously treated with LEV, but who had discontinued it, had lesser but still generally substantial responses. Patients treated concomitantly with LEV and BRV during the studies had a placebo-like response. In NCT00464269, ≥50% responder rates for placebo and BRV 50 mg/day were 16.4% and 40.3% in LEV-naïve patients, 18.2% and 35.0% in those with prior (failed) LEV use, and 15.8% and 5.3% in those with concomitant LEV use, respectively.93 In NCT00490035, ≥50% responder rates for placebo, BRV 50 mg/day, and 100 mg/day were 18.2%, 33.3%, and 43.1% for LEV-naïve, 22.2%, 21.4%, and 36.4% for prior LEV use, and 22.2%, 20.0%, and 15.0% for the concomitant LEV group, respectively.94 Because of this, the third pivotal Phase III study, NCT01261325, disallowed concomitant LEV use.95 The design of the study included predetermined stratified subgroup analysis by prior LEV exposure. Response was higher in LEV-naïve patients than in patients previously treated with LEV, although BRV response was higher than for placebo in both subgroups. Percentage reduction over placebo in baseline-adjusted seizure frequency/28 days was 29.5% (P<0.001) and 27.1% (P<0.001) for BRV 100 mg/day and 200 mg/day, respectively, in LEV-naïve patients, and 15.8% (P=0.024) and 19.4% (P=0.005) in patients with previous LEV exposure. The ≥50% responder rates in LEV-naïve patients were 27.6%, 50.9% (P<0.001), and 45.2% (P=0.008) for placebo, BRV 100 mg/day, and 200 mg/day, respectively, and 16.8%, 28.7% (P=0.016), and 31.3% (P=0.007) in patients with previous LEV exposure. Response appeared to be greater in patients who had failed LEV due to TEAEs rather than for lack of efficacy, although the subgroup sizes were insufficient for robust statistical comparison.
BRV was well tolerated in all the pivotal trials, with the majority of TEAEs being mild to moderate in intensity. The most commonly reported TEAEs seen more in BRV- than placebo-treated patients were somnolence, dizziness, and fatigue.
Pooled Phase III efficacy and tolerability
A pooled analysis of all three pivotal Phase III studies (NCT00490035, NCT00464269, and NCT01261325) was performed that included only those patients exposed to the effective doses, namely BRV 50, 100, and 200 mg/day.96 Patients treated concomitantly with LEV were excluded from the pooled efficacy analysis (n=1,160) but were included in the safety analysis (n=1,262). Median percentage reduction over placebo in baseline-adjusted focal seizure frequency/28 days was 19.5% for BRV 50 mg/day (P=0.0015), 24.4% for 100 mg/day (P<0.00001), and 24.0% for 200 mg/day (P<0.00001). Pooled ≥50% responder rates were 20.3% for placebo, 34.2% for 50 mg/day (P=0.0015), 39.5% for 100 mg/day (P<0.00001), and 37.8% for 200 mg/day (P<0.00003). Efficacy was greater for BRV 100 and 200 mg/day than BRV 50 mg/day, and there was no clear dose response from 100 to 200 mg/day. Secondary efficacy end points showed seizure-freedom rates of 2.5% (P=0.053), 5.1% (P<0.001), and 4.0% (P=0.001) for BRV 50, 100, and 200 mg/day, respectively, compared with 0.5% for placebo. Categorized percentage reduction in focal seizure frequency is shown in Figure 4. The ≥75% responder rates were 6.9% for placebo and 13.0%, 21.4%, and 19.7% for BRV 50, 100, and 200 mg/day, respectively. Patients previously treated with five or more AEDs tended to have smaller reductions in seizure frequency than patients with less prior AED exposure (Figure 5).
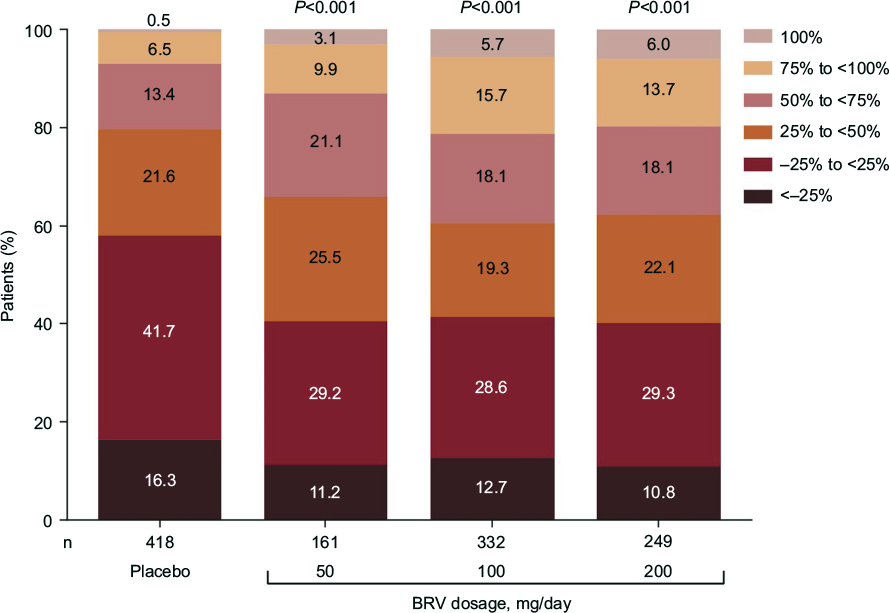 | Figure 4 Categorized percentage reduction in focal seizure frequency per 28 days in the pooled Phase III population. Adapted from Ben-Menachem E, Mameniškienė R, Quarato PP, et al. Efficacy and safety of brivaracetam for partial-onset seizures in three pooled clinical studies. Neurology. 2016;87(3):314–323.96 Promotional and commercial use of the material in print, digital or mobile device format is prohibited without the permission from the publisher Wolters Kluwer. Please contact [email protected] for further information. Abbreviation: BRV, brivaracetam. |
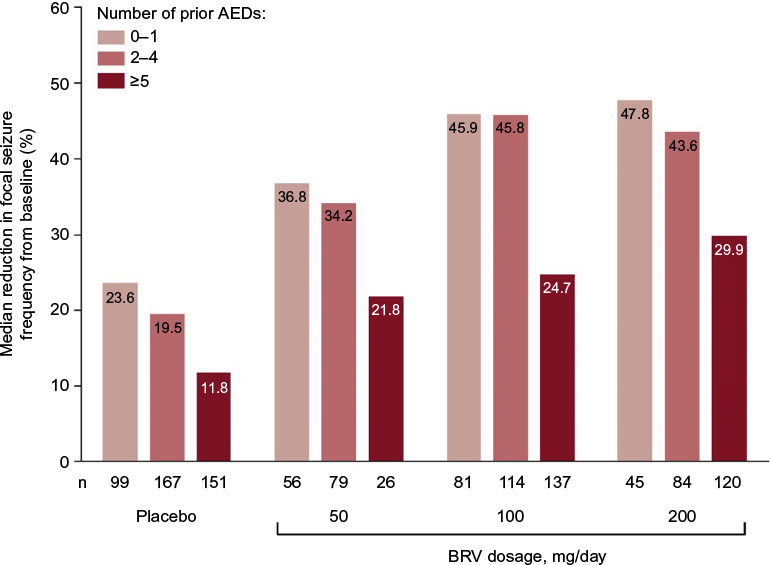 | Figure 5 Percentage reduction from baseline in focal seizure frequency by number of prior AEDs in the pooled Phase III population. Reproduced from Ben-Menachem E, Mameniškienė R, Quarato PP, et al. Efficacy and safety of brivaracetam for partial-onset seizures in three pooled clinical studies. Neurology. 2016;87(3):314–323.96 Promotional and commercial use of the material in print, digital or mobile device format is prohibited without the permission from the publisher Wolters Kluwer. Please contact [email protected] for further information. Abbreviations: AED, antiepileptic drug; BRV, brivaracetam. |
The pooled Phase III safety population data showed that 90.0%–93.9% of patients completed the studies.96 TEAEs were reported in 68.0% of patients receiving BRV and 62.1% of patients receiving placebo (Table 6). Most of the TEAEs were mild to moderate. Discontinuation rates because of TEAEs were 5.0%, 7.6%, and 6.8% for patients treated with 50, 100, and 200 mg/day, compared with 3.9% of placebo-treated patients. The most common TEAEs in BRV-treated patients were somnolence (15.2% vs 8.5% for placebo), dizziness (11.2% vs 7.2%), headache (9.6% vs 10.2%), and fatigue (8.7% vs 3.7%) (Table 6). There were no clinically relevant differences in TEAE incidence for the different starting doses, except for a dose–response relationship for somnolence and fatigue. SAEs occurred in 3.0% of BRV-treated patients and 2.8% of placebo-treated patients. There were four deaths: one sepsis (placebo) and three sudden unexpected deaths in epilepsy (SUDEPs; one with BRV 50 mg/day 2 weeks after the last confirmed intake of BRV, and two with BRV 200 mg/day 1 and 9 days after the last confirmed dose of BRV). Two patients (0.2%) taking BRV and three (0.7%) taking placebo reported suicidal ideation. There were no reported suicide attempts. There were no significant changes in clinical chemistry, urine analysis, vital signs, or body weight, and no clinically meaningful changes for hematology parameters, apart from a small reduction in neutrophil counts with BRV (four patients, 0.5%) compared with placebo (no patients), but no associated infections were reported.
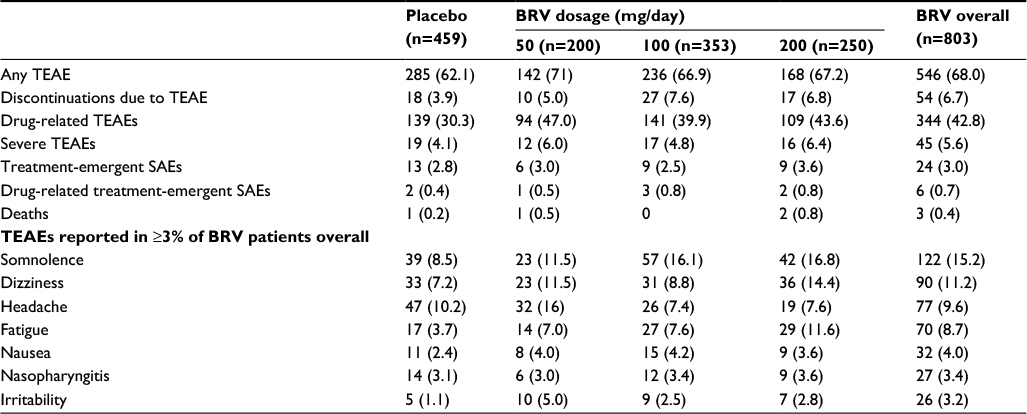 | Table 6 Summary and incidence of TEAEs reported in ≥3% of patients taking BRV in the pooled Phase III safety population Notes: Data represent number of (%) patients reporting a TEAE at any point during the entire study. Reproduced from Ben-Menachem E, Mameniškienė R, Quarato PP, et al. Efficacy and safety of brivaracetam for partial-onset seizures in three pooled clinical studies. Neurology. 2016;87(3):314–323.96 Promotional and commercial use of the material in print, digital or mobile device format is prohibited without the permission from the publisher Wolters Kluwer. Please contact [email protected] for further information. Abbreviations: BRV, brivaracetam; SAE, serious adverse event; TEAE, treatment-emergent AE. |
Phase III flexible-dose safety study
The fourth oral Phase III study was a flexible-dose study of BRV 20–150 mg/day (NCT00504881).97 This was the only Phase III study that included patients with primary generalized epilepsy (n=49 of 480 patients). BRV was initiated at 20 mg/day and increased to 50, 100, or 150 mg/day, at 2-week intervals during an 8-week dose-finding period, followed by an 8-week maintenance period. TEAEs were similar to the three pivotal studies. The ≥50% responder rate for focal seizures was significantly higher with BRV (30.3%, P=0.006) compared with placebo (16.7%). In an exploratory analysis of patients with generalized seizures, the median percentage reduction from baseline in primary generalized seizure days/week was 42.6% with BRV compared with 20.7% with placebo; ≥50% responder rate was 44.4% with BRV versus 15.4% in placebo-treated patients.
Secondary Phase III analyses and other studies
A number of secondary analyses of pooled data from patients receiving BRV 50, 100, and 200 mg/day in the three pivotal Phase III studies have been published.
Efficacy in secondarily generalized seizures
In the pooled Phase III population, 409 patients had secondarily generalized tonic–clonic seizure (SGTCS) at baseline.98 Median baseline SGTCS frequency was 3.0 per 28 days. The median percentage reduction from baseline in SGTCS frequency/28 days was 33.3% for placebo, 66.6% for BRV 50 mg/day (P<0.001), 61.2% for BRV 100 mg/day (P=0.002), and 82.1% for BRV 200 mg/day (P<0.001). The ≥50% responder rate for SGTCS was 33.0% for placebo, 61.3% for BRV 50 mg/day (P=0.003), 55.0% for BRV 100 mg/day (P<0.001), and 64.0% for BRV 200 mg/day (P<0.001) (Figure 6). SGTCS freedom occurred in 14.8%, 22.6%, 31.0%, and 36.0% of patients treated with placebo, BRV 50, 100, and 200 mg/day, respectively (Figure 6). Overall, 30.4% of patients became free from SGTCS during the 12-week treatment period when taking BRV ≥50 mg/day. Reported TEAEs were similar in the SGTCS safety population compared with the overall patient population.
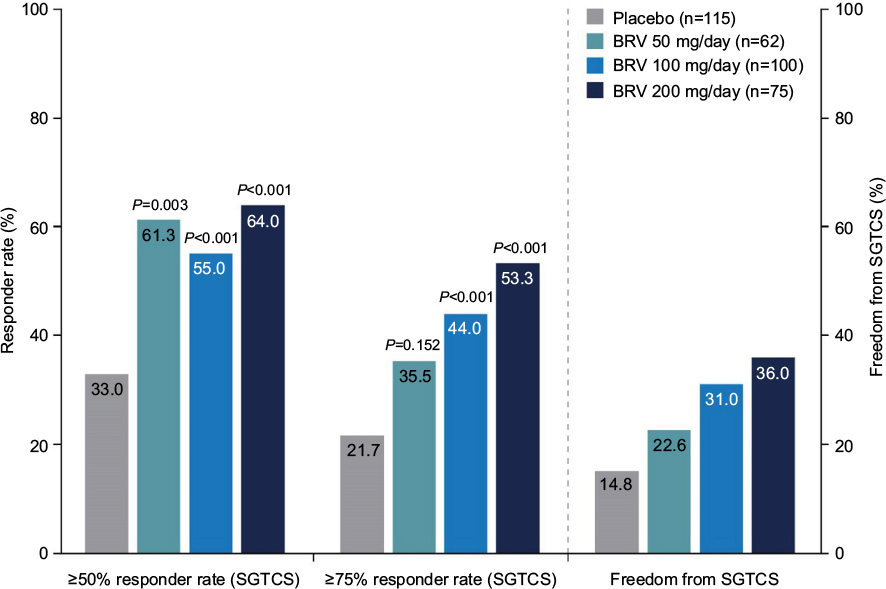 | Figure 6 Responder rates (≥50% and ≥75%) and complete freedom from SGTCS in the pooled Phase III population. Adapted from Elsevier Science and Technology Journals, Efficacy, safety, and tolerability of adjunctive brivaracetam for secondarily generalized tonic-clonic seizures: pooled results from three phase III studies, Moseley BD, Sperling MR, Asadi-Pooya AA, et al, Volume 127, 2016; permission conveyed through Copyright Clearance Center, Inc.98 Abbreviations: BRV, brivaracetam; SGTCS, secondarily generalized tonic–clonic seizure. |
Speed of onset
As noted, BRV crosses the blood–brain barrier and engages the target SV2A molecule quickly. A drug-entry half-time of 7 minutes after IV injection was reported in initial data in human PET studies.51 Because BRV is well tolerated, it was initiated at target dose without titration in the three pivotal Phase III studies. The question whether these attributes, rapid brain entry and no titration, translate into clinical benefit was addressed in a post hoc analysis that evaluated speed of onset of efficacy of BRV across the pooled Phase III studies. Time to onset of sustained ≥50% responder status was assessed for patients receiving placebo or BRV 50 mg/day, 100 mg/day, or 200 mg/day (n=1,160).99 Sustained responders were defined as patients who completed the entire treatment period, and were ≥50% responders from the first day of 50% reduction in seizure frequency until the end of the treatment period. The proportion of patients achieving sustained ≥50% responder status on day 1 was 6.7% for placebo, 15.5% for BRV 50 mg/day, 18.1% for BRV 100 mg/day, and 19.4% for BRV 200 mg/day (all P<0.001 vs placebo; Figure 7), indicating that BRV has early, sustained efficacy in a significant proportion of patients. The incidence of TEAEs during the first week was similar to that in the overall treatment period.
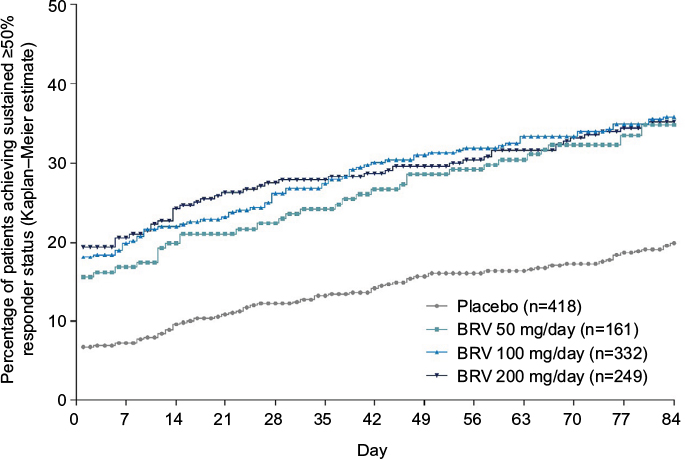 | Figure 7 Kaplan–Meier plot of time to onset of sustained ≥50% responder status in the pooled Phase III population. Notes: For each day, P versus placebo was ≤0.003 for BRV 50 mg/day and <0.001 for BRV 100 mg/day and 200 mg/day. Reproduced from John Wiley and Sons Inc, Time to onset of sustained ≥50% responder status in patients with focal (partial-onset) seizures in three phase III studies of adjunctive brivaracetam treatment, Klein P, Johnson ME, Schiemann J, Whitesides J, Volume 58, 2017; permission conveyed through Copyright Clearance Center, Inc.99 Abbreviation: BRV, brivaracetam. |
Prior exposure to AEDs
The observation that response to BRV is lower in patients with prior LEV-treatment failure raises the question whether this may be due to BRV and LEV acting on the same molecule – SV2A. An analysis using pooled data from the three pivotal Phase III studies suggested a different possible reason. In this analysis, evaluation of patients receiving BRV 50–200 mg/day (n=1,160) by prior exposure to LEV and three other commonly used AEDs – carbamazepine, topiramate, and lamotrigine – showed that patients previously treated by any of these four AEDs all had reduced response to BRV compared with patients not previously treated by the same AEDs.100 A total of 578 patients had prior exposure to LEV, 424 to carbamazepine, 428 to topiramate, and 349 to lamotrigine. The proportion of patients who had previously had more than five AEDs fail was higher among previously LEV-, carbamazepine-, topiramate-, and lamotrigine-exposed subgroups (61%–69%) than among subgroups never exposed to those AEDs (13%–17%). Similarly, baseline seizure frequency among patients previously exposed to these four AEDs was higher (9.6–11.3 seizures/28 days) than among the unexposed patients (7.9–8.5 seizures/28 days) (response in the “prior fail” groups was higher than placebo for all of the AEDs studied). This suggests that the reduced response to BRV in patients previously treated by these AEDs compared with naïve patients may have been associated with greater disease severity in the exposed patients. Table 7 summarizes the ≥50% responder rate by prior AED and number of prior AEDs.
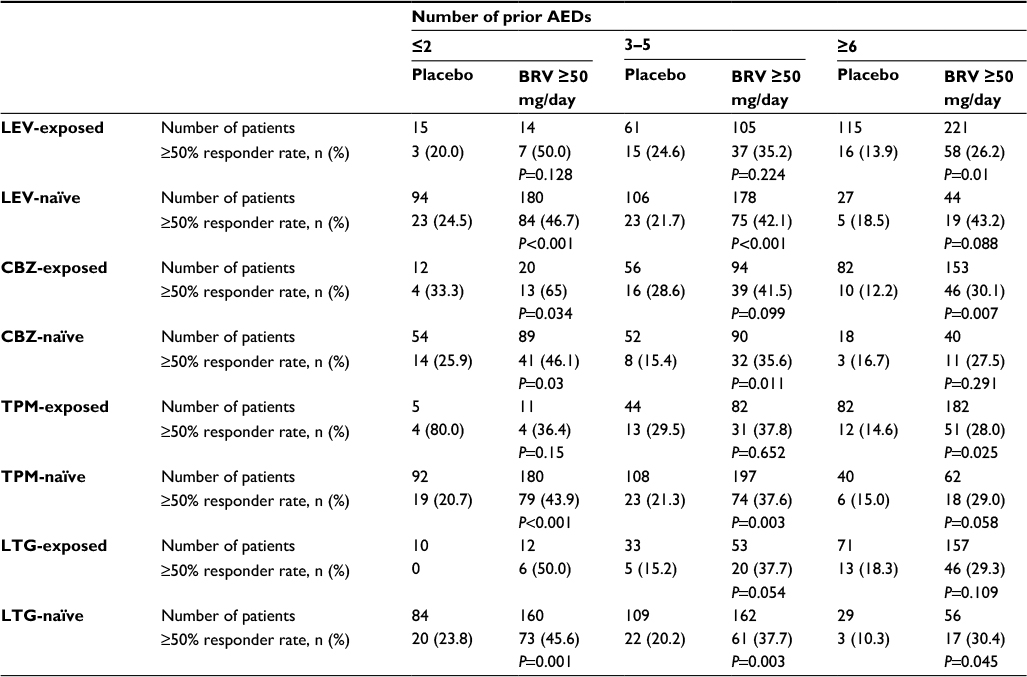 | Table 7 ≥50% Responder rate by prior AED exposure and number of prior AEDs in post hoc analysis using pooled data from Phase III studies Notes: All P-values were exploratory. The modified efficacy population comprised randomized patients who received at least one dose of study drug and had at least one postbaseline seizure diary entry; patients taking concomitant LEV were included. Republished with permission of Elsevier Science and Technology Journals, from Efficacy and tolerability of adjunctive brivaracetam in patients with prior antiepileptic drug exposure: a post-hoc study, Asadi-Pooya AA, Sperling MR, Chung S, et al, Volume 131, 2017; permission conveyed through Copyright Clearance Center, Inc.100 Abbreviations: AED, antiepileptic drug; BRV, brivaracetam; CBZ, carbamazepine; LEV, levetiracetam; LTG, lamotrigine; TPM, topiramate. |
Concomitant AED use
There has been much interest in the potential of combination therapy to improve treatment outcomes in refractory epilepsy. A separate post hoc analysis of the pooled pivotal Phase III trials studied the efficacy of BRV 50–200 mg/day with concomitant use of lamotrigine or topiramate in patients treated with only one of these two medications with or without a second concomitant AED (UCB Pharma, data on file, 28 October 2016). Percentage reduction over placebo in baseline-adjusted focal seizure frequency/28 days for BRV 50, 100, and 200 mg/day was 8.7%, 5.3%, and 8.9% in the lamotrigine subgroup (n=220) and 8.4%, 21.3%, and –4.2% in the topiramate subgroup (n=122). The ≥50% responder rate with concomitant lamotrigine or topiramate with BRV 50, 100, and 200 mg/day or placebo was 28.1%, 36.1%, 34.1%, and 29.1% for lamotrigine and 14.3%, 44.4%, 25.0%, and 17.5% for topiramate. This would suggest that there is minimal synergistic effect between BRV and lamotrigine or topiramate. The efficacy data are limited by the small number of patients in the groups, particularly in the topiramate group, and by the fact that the majority of patients were also on another AED. TEAEs were similar to the overall patient population, as were discontinuations due to TEAEs (lamotrigine 7.3% vs 6.3% with placebo, topiramate 8.2% vs 4.7% with placebo).
Behavioral adverse events
The rate of psychiatric TEAEs in the pooled Phase III studies was relatively low. Overall, 13% of BRV-treated patients across the 50–200 mg/day doses experienced a psychiatric adverse reaction compared with 8% of placebo-treated patients, and 1.7% of BRV-treated patients discontinued study medication due to a psychiatric adverse reaction compared with 1.3% of placebo-treated patients.15 The most common psychiatric TEAEs were irritability (3.2% of BRV-treated patients vs 1.1% of placebo-treated patients), insomnia (2.9% BRV vs 1.5% placebo), anxiety (2.0% BRV vs 1.3% placebo), and depression (2.0% BRV vs 1.1% placebo). There were three cases of psychosis in the overall Phase III pooled patient population: one each in the placebo, BRV 50 mg/day, and BRV 100 mg/day groups (UCB Pharma, data on file, 18 September 2015).
LEV has been associated with nonpsychotic behavioral AEs. An exploratory, prospective, Phase IIIB open-label study was conducted in 29 adults with epilepsy who had experienced seizure control on LEV but had disabling nonpsychotic behavioral AEs (NCT01653262).101 Patients were switched abruptly from their last full dose of LEV (1–3 g/day) to BRV target dose 200 mg/day without titration, and received BRV 200 mg for 12 weeks. Overall, 26 of 29 patients (89.7%) completed the study and 27 of 29 (93.1%) had clinically meaningful reductions in behavioral AEs, of which 18 of 29 (62.1%) achieved complete resolution of behavioral AEs. One patient experienced slight worsening and one marked worsening of behavioral AEs. Median time to behavioral AE resolution was 15 days after BRV initiation. One patient stopped BRV because of lack of efficacy. The results suggest that patients who experience behavioral AEs with LEV may benefit from switching to BRV treatment, but the data are limited by the open-label nature of the study and the small sample size.
Patients with Unverricht–Lundborg disease
Two Phase III studies (NCT00357669 and NCT00368251) were conducted in patients with Unverricht–Lundborg disease (also called epilepsy with progressive myoclonus type I [EPM1]).102 In two randomized, double-blind, placebo-controlled, multicenter studies, patients aged ≥16 years with moderate–severe myoclonus and genetically confirmed EPM1 received placebo, BRV 50 mg/day, or BRV 150 mg/day (n=50) in one study, or placebo, BRV 5 mg/day, or BRV 150 mg/day (n=56) in the other study for a 2-week uptitration period and a 12-week maintenance period. BRV did not significantly improve median percentage reduction from baseline in action myoclonus score on the Unified Myoclonus Rating Scale, the primary outcome measure, versus placebo in either study. Action myoclonus scores showed wide intrapatient variability, and thus may not have been the best possible instrument for measuring myoclonus severity in this population. The tolerability profile of BRV across both studies was similar to Phase III studies in patients with focal seizures.
Pooled long-term follow-up
Data from the BRV Phase IIB and Phase III long-term follow-up studies were pooled for an analysis of long-term, open-label safety/tolerability (n=2,186) and seizure-outcome data (n=1,836; NCT00175929, NCT00175825, NCT00490035, NCT00464269, NCT00504881, NCT01261325, NCT00175916, NCT00150800, NCT01339559, NCT01405508, NCT01653262, and NCT01728077).103 Total BRV exposure was 5,339 patient-years, with 41 patients treated for ≥8 years. Among patients with a modal dose of 50–200 mg/day, 6-month, 12-month, 24-month, and 60-month retention rates were 91.0%, 79.8%, 68.1%, and 54.4%, respectively. The most frequent TEAEs were headache (20.9%), dizziness (17.5%), somnolence (15.2%), nasopharyngitis (13.2%), fatigue (11.3%), and convulsion (10.6%). A total of 264 patients (12.1%) had a TEAE that resulted in discontinuation of BRV. The most frequently reported TEAEs leading to discontinuation were convulsion (1.4%), pregnancy (0.9%), somnolence (0.7%), depression (0.6%), dizziness (0.6%), fatigue (0.5%), suicidal ideation (0.5%), and suicide attempt (0.5%). SAEs were reported in 18.3% of patients, including 4.3% considered to be treatment-related by the investigator. The most common SAEs were convulsion (2.6%), status epilepticus (0.9%), epilepsy (0.6%), pneumonia (0.5%), suicidal ideation (0.5%), suicide attempt (0.5%), and fall (0.5%). There were 28 deaths (1.3%), including cancer (n=6), drowning (n=4), sudden unexpected death in epilepsy (SUDEP) (n=4), myocardial infarction (n=3), accident (n=2), and suicide (n=2). Four of the 28 deaths were considered to be possibly related to treatment with BRV by the investigator: two cases of SUDEP, one possible SUDEP event, and one suicide.
The most frequently reported psychiatric/behavioral side effects were depression (7.1%), insomnia (6.2%), irritability (5.2%), anxiety (4.9%), suicidal ideation (2.0%), depressed mood (1.8%), aggression (1.7%), nervousness (1.6%), and sleep disorder (1.6%). Thirteen patients (0.6%) attempted suicide. There was no evidence that these TEAEs increased with dose.
The median percentage reduction from baseline in focal seizure frequency/28 days was 48.8%, and the ≥50% responder rate was 48.7%. The median percentage reduction from baseline in focal seizure frequency/28 days increased over time from 43.1% for the 1,834 patients treated for 3 months to 77.0% for the 540 patients treated for 58–60 months (Figure 8A). The ≥50% responder rate increased from 43.5% for patients treated for 3 months to 71.0% for patients who were treated for 58–60 months (Figure 8B). Complete seizure freedom (all seizure types) was achieved in 4.9% patients for the first 6 months of BRV treatment, 4.2% for 12 months, 3% for 24 months, and 3.3% for 60 months.
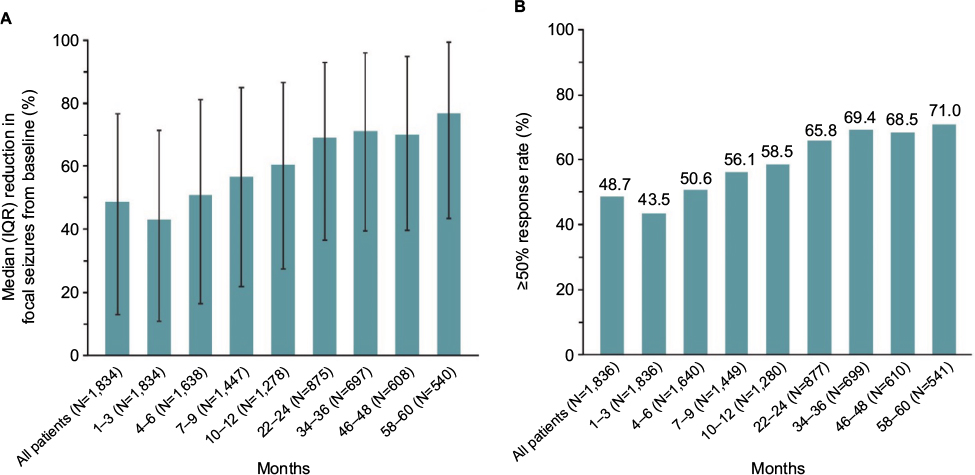 | Figure 8 Seizure reduction and responder rates. (A) Median (IQR) percentage reduction in focal seizures from baseline; (B) ≥50% responder rates over time for BRV modal doses 50–200 mg/day in pooled long-term follow-up analysis. Notes: Adapted with permission of John Wiley and Sons Inc, from Safety, tolerability, and seizure control during long-term treatment with adjunctive brivaracetam for partial-onset seizures, Toledo M, Whitesides J, Schiemann J, et al, Volume 57, Edition 7, 2016; permission conveyed through Copyright Clearance Center, Inc. © 2016 The Authors. Epilepsia published by Wiley Periodicals Inc. on behalf of International League Against Epilepsy.103 Abbreviations: BRV, brivaracetam; IQR, interquartile range. |
Special populations
Elderly patients
Data from patients aged ≥65 years in the pivotal Phase III studies were pooled to assess the safety and efficacy of BRV 50–200 mg/day in elderly patients (n=32).104 Findings were consistent with those of the overall patient population. The most common TEAEs were headache (25.0% placebo, 12.5% BRV), paresthesia (0 placebo, 12.5% BRV), and somnolence (50.0% placebo, 12.5% BRV). SAEs were reported in no patients in the placebo group and 4.2% of patients in the BRV group. There were no deaths or treatment-related SAEs. Median percentage reduction from baseline in focal seizure frequency/28 days was 14.0% for placebo, 25.5% for BRV 50 mg/day, 49.6% for BRV 100 mg/day, and 74.9% for BRV 200 mg/day; and ≥50% responder rates were 14.3% for placebo, 25 .0% for BRV 50 mg/day, 50.0% for BRV 100 mg/day, and 66.7% for BRV 200 mg/day.
Pediatric studies
An oral solution of BRV was well tolerated in pediatric epilepsy patients aged 1 month to 16 years (n=99) in a Phase IIA, open-label, single-arm, multicenter study (NCT00422422; N01263).105 During a 3-week evaluation period, BRV was titrated based on patient age. TEAEs were reported in 66.7% of patients and treatment-related TEAEs were reported in 32.3% of patients. The most common TEAEs were convulsion (10.1%), irritability (8.1%), pyrexia (8.1%), and somnolence (8.1%). The most common treatment-related TEAEs were somnolence (7.1%) and decreased appetite (6.1%). The incidence of TEAEs was greater in patients aged between 1 month and 2 years compared with patients aged ≥2 years; however, drug-related TEAEs were less frequent in patients aged between 1 month and 2 years. SAEs were reported in 8.1% of patients; no deaths occurred.
Using data from this study, a population PK model of oral BRV in pediatric patients has been developed to provide guidance on dosing in the pediatric patient population.69 Simulations demonstrated that with an age-independent dosing regimen of 2 mg/kg twice daily up to a maximum of 100 mg twice daily for body weight >50 kg, the majority of children were predicted to have an exposure similar to that in adult patients receiving 100 mg twice daily (the highest recommended dose).
IV formulation
Several IV BRV-bioequivalence, PK, and safety/tolerability studies have been performed in healthy volunteers and in patients with refractory epilepsy (see the “Pharmacokinetic profile” section for bioequivalence studies). In an open-label Phase III randomized study evaluating the safety, tolerability, and PKs of IV BRV (NCT01405508), 105 patients with uncontrolled partial or generalized epilepsy treated with 1–2 AEDs received placebo or oral BRV 200 mg/day for 7 days, followed by a 2-minute bolus or 15-minute infusion of IV BRV 200 mg/day for 4.5 days.106 The study was completed by 98% of patients, and only two discontinued: one during the run-in period prior to BRV exposure because of a rash, and one during BRV infusion because of anxiety. IV BRV was well tolerated. The incidence of TEAEs was similar with the 2-minute bolus (71.2%) and 15-minute infusion (65.4%), and there was little difference in incidence of individual TEAEs between bolus- and infusion-treated patients. The two main AEs were somnolence and dizziness, which occurred in 19% bolus versus 25% infusion groups, and dizziness in 10% versus 6%. Other TEAEs included headache in 8% versus 6% in the bolus versus infusion groups, dysgeusia in 8% versus 0, infusion-site pain in 4% versus 6%, fatigue in 6% versus 2%, and postural orthostatic tachycardia syndrome in 6% versus 0. Dizziness tended to be more common in patients who received IV BRV after oral placebo (12%) than in patients who received IV BRV after oral BRV (4%), rash was also more common in the placebo/BRV group (6%) than the BRV/BRV group (0), and other TEAEs had similar frequencies in both groups. Severe TEAEs were reported by one patient (vertigo and nausea 12 days after the first BRV dose). There were no SAEs. Injection-related TEAEs were reported by 9.6% of patients who received bolus and 11.5% who received infusion. There were no clinically relevant abnormal findings in vital signs, laboratory tests, or electrocardiography.
Postmarketing studies
Status epilepticus (SE)
A retrospective chart-review study evaluated adjunctive BRV treatment in eleven patients with refractory SE at two German centers.107 The median number of previous anticonvulsants used since the onset of SE was four. Initial BRV doses were 50–400 mg (median 100 mg), titrated to 100–400 mg/day (median 200 mg/day). SE was stopped in the first 24 hours after BRV initiation in three patients (27%) and no further anticonvulsants given, in seven patients one to three further anticonvulsants were given before resolution, and one patient received one further anticonvulsant before death. No serious side effects were reported.
Clinical practice experience
Two retrospective-review studies of BRV treatment have been published since BRV approval, both in Germany. In one, 101 patients with uncontrolled epilepsy were treated with adjunctive BRV for 6 months at a German tertiary epilepsy center.108 The median number of AEDs used prior to BRV was ten (range two to 18). BRV was started at ≥50 mg/day, and the mean maintenance dose was 168.6 mg/day (median 200 mg/day, range 50–400 mg/day). The responder rate was 27.8%, and 7% of patients achieved seizure freedom. TEAEs occurred in 37% of patients. The most common TEAEs were dizziness (16%) and somnolence (11%). Psychiatric TEAEs included irritability, aggression, depression, and psychosis (all n=1). The retention rate after 6 months was 51.5%. The main reason for discontinuation was lack of efficacy. LEV was switched to BRV in 43 patients abruptly and without complications. In 26 of these patients, BRV was reswitched to LEV within weeks, mainly due to a lack of better efficacy. Five of the 43 patients who switched from LEV to BRV experienced seizure exacerbation.
In the other retrospective-review study, a cohort of 262 epilepsy patients were treated with adjunctive BRV for at least 3 months and up to 12 months at multiple centers in Germany.109 Most patients (87%) had a diagnosis of focal epilepsy. The median number of previously used AEDs was four (range 0–17). Half of the patients (50.4%) in the cohort were receiving LEV before initiation of BRV and switched to BRV. Dose conversion from LEV to BRV used a median ratio of 10:1 for LEV doses ≤2,000 mg/day and 15:1 for LEV doses >2,000 mg/day. In patients on LEV, BRV was started at 25–400 mg/day and reached a mean target dose of 175.7 mg/day (median 200 mg/day, range 50–400 mg/day). In 105 patients, LEV was switched to BRV from one dose to the next (median 1 day); in 28 patients LEV and BRV overlapped and the switch took a median of 12 days. In patients not on LEV, BRV was started at an initial dose of 10–200 mg/day and reached a mean target dose of 128.1 mg/day (median 100 mg/day, range 50–200 mg/day) in a median 7 days. The ≥50% responder rate at 6 months was 40.5% (77 of 192). Seizure freedom was achieved by 15.3% (29 of 192) of patients. TEAEs were reported in 37.8% of patients, most frequently somnolence/sedation (16.0%), dizziness (11.8%), and behavioral AEs, of which depressive mood change (9.1%) was the most common, followed by irritability (4.2%) and aggression (3.8%). Retention rate at 6 months was 75.8%. Of 51 patients previously on LEV who were switched to BRV due to TEAEs, 30 reported improvement in TEAEs, 17 had no change/undetermined, and four reported worsened TEAEs.
Conclusion
BRV is the first selective and high-affinity SV2A ligand with high lipid-solubility, rapid brain penetration, and rapid onset of action. It has broad-spectrum antiepileptic activity in animal models of epilepsy, and Phase I studies indicated that single oral doses of 10–1,000 mg and repeated oral doses of up to 800 mg/day were well tolerated and showed a favorable PK profile. Three pivotal Phase III studies have demonstrated efficacy and a favorable safety and tolerability profile across doses of 50–200 mg/day of adjunctive treatment of refractory focal seizures. Long-term data indicate that the response to BRV is sustained, with good tolerability and retention rate. BRV is highly effective in patients experiencing SGTCS. Safety data to date suggest a favorable psychiatric AE profile, although limited postmarketing data are available to confirm the controlled-study data.
BRV penetrates the blood–brain barrier rapidly and engages the target molecule, SV2A, within minutes, faster than LEV. Its good tolerability allows dosing initiation at target doses without uptitration. Efficacy is seen on day 1 of oral use in a significant proportion of patients. IV administration as a 2-minute bolus or as a 15-minute infusion is well tolerated. This, together with its rapid occupancy of SV2A, suggests possible use in the treatment of acute seizures, to be explored in future studies.
Acknowledgments
The authors wish to thank the patients, their caregivers, and the investigators and their teams who contributed to the original studies. The authors acknowledge Cédric Laloyaux, Strategic Publication Lead Neurology, UCB Pharma, for critical review of the manuscript and coordination of manuscript development. The authors acknowledge Sally Cotterill, PhD of QXV Comms, an Ashfield company, part of UDG Healthcare plc, and Sian-Marie Lucas, PhD of Lucas Medical for medical writing support that was funded by UCB Pharma.
Disclosure
PK has served as a consultant for Eisai, Lundbeck, Sunovion, and UCB Pharma, is on the speakers bureaux for Eisai, Sunovion, and UCB Pharma, and has received grant support from Eisai and Lundbeck. AD, TG, and JW are employees of UCB Pharma. The authors report no other conflicts of interest in this work.
References
World Health Organization. Infographics on epilepsy: 2016–2017. 2017. Available from: http://www.who.int/mediacentre/infographic/mental-health/epilepsy/en. Accessed July 4, 2017. | ||
Brodie MJ, Barry SJ, Bamagous GA, Norrie JD, Kwan P. Patterns of treatment response in newly diagnosed epilepsy. Neurology. 2012; 78(20):1548−1554. | ||
Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342(5):314–319. | ||
US Food and Drug Administration. Lacosamide [prescribing information]. 2009. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=022253. Accessed July 4, 2017. | ||
US Food and Drug Administration. Perampanel [prescribing information]. 2015. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202834lbl.pdf. Accessed May 5, 2017. | ||
US Food and Drug Administration. Eslicarbazepine [prescribing information]. 2013. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&applno=022416. Accessed July 4, 2017. | ||
US Food and Drug Administration. Ezogabine [prescribing information]. 2015. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&applno=022345. Accessed July 4, 2017. | ||
European Medicines Agency. Zebinix: eslicarbazepine acetate. 2017. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000988/human_med_001172.jsp&mid=WC0b01ac058001d124. Accessed January 8, 2017. | ||
US Food and Drug Administration. Rufinamide [prescribing information]. 2015. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=021911. Accessed July 4, 2017. | ||
Chung S, Sperling MR, Biton V, et al. Lacosamide as adjunctive therapy for partial-onset seizures: a randomized controlled trial. Epilepsia. 2010;51(6):958–967. | ||
Gil-Nagel A, Elger C, Ben-Menachem E, et al. Efficacy and safety of eslicarbazepine acetate as add-on treatment in patients with focal-onset seizures: integrated analysis of pooled data from double-blind phase III clinical studies. Epilepsia. 2013;54(1):98–107. | ||
Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70(21):1950–1958. | ||
Porter RJ, Burdette DE, Gil-Nagel A, et al. Retigabine as adjunctive therapy in adults with partial-onset seizures: integrated analysis of three pivotal controlled trials. Epilepsy Res. 2012;101(1–2):103–112. | ||
Steinhoff BJ, Ben-Menachem E, Ryvlin P, et al. Efficacy and safety of adjunctive perampanel for the treatment of refractory partial seizures: a pooled analysis of three phase III studies. Epilepsia. 2013;54(8):1481–1489. | ||
US Food and Drug Administration. Brivaracetam [prescribing information]. 2016. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=205836. Accessed July 4, 2017. | ||
Gillard M, Fuks B, Leclercq K, Matagne A. Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: relationship to anti-convulsant properties. Eur J Pharmacol. 2011;664(1–3):36−44. | ||
Klitgaard H, Matagne A, Nicolas JM, et al. Brivaracetam: rationale for discovery and preclinical profile of a selective SV2A ligand for epilepsy treatment. Epilepsia. 2016;57(4):538–548. | ||
Löscher W, Gillard M, Sands ZA, Kaminski RM, Klitgaard H. Synaptic vesicle glycoprotein 2A ligands in the treatment of epilepsy and beyond. CNS Drugs. 2016;30(11):1055–1077. | ||
Madeo M, Kovacs AD, Pearce DA. The human synaptic vesicle protein, SV2A, functions as a galactose transporter in Saccharomyces cerevisiae. J Biol Chem. 2014;289(48):33066–33071. | ||
Mendoza-Torreblanca JG, Vanoye-Carlo A, Phillips-Farfan BV, Carmona-Aparicio L, Gomez-Lira G. Synaptic vesicle protein 2A: basic facts and role in synaptic function. Eur J Neurosci. 2013;38(11):3529–3539. | ||
Grønborg M, Pavlos NJ, Brunk I, et al. Quantitative comparison of glutamatergic and GABAergic synaptic vesicles unveils selectivity for few proteins including MAL2, a novel synaptic vesicle protein. J Neurosci. 2010;30(1):2–12. | ||
Crowder KM, Gunther JM, Jones TA, et al. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc Natl Acad Sci U S A. 1999;96(26):15268–15273. | ||
van Vliet EA, Aronica E, Redeker S, Boer K, Gorter JA. Decreased expression of synaptic vesicle protein 2A, the binding site for levetiracetam, during epileptogenesis and chronic epilepsy. Epilepsia. 2009;50(3):422–433. | ||
Hanaya R, Hosoyama H, Sugata S, et al. Low distribution of synaptic vesicle protein 2A and synaptotagimin-1 in the cerebral cortex and hippocampus of spontaneously epileptic rats exhibiting both tonic convulsion and absence seizure. Neuroscience. 2012;221:12–20. | ||
Feng G, Xiao F, Lu Y, et al. Down-regulation synaptic vesicle protein 2A in the anterior temporal neocortex of patients with intractable epilepsy. J Mol Neurosci. 2009;39(3):354–359. | ||
Finnema SJ, Nabulsi NB, Eid T, et al. Imaging synaptic density in the living human brain. Sci Transl Med. 2016;8(348):348ra96. | ||
Toering ST, Boer K, de Groot M, et al. Expression patterns of synaptic vesicle protein 2A in focal cortical dysplasia and TSC-cortical tubers. Epilepsia. 2009;50(6):1409–1418. | ||
Serajee FJ, Huq AM. Homozygous mutation in synaptic vesicle glycoprotein 2A gene results in intractable epilepsy, involuntary movements, microcephaly, and developmental and growth retardation. Pediatr Neurol. 2015;52(6):642–646.e1. | ||
Matveeva EA, Vanaman TC, Whiteheart SW, Slevin JT. Levetiracetam prevents kindling-induced asymmetric accumulation of hippocampal 7S SNARE complexes. Epilepsia. 2008;49(10):1749–1758. | ||
Matveeva EA, Vanaman TC, Whiteheart SW, Slevin JT. Asymmetric accumulation of hippocampal 7S SNARE complexes occurs regardless of kindling paradigm. Epilepsy Res. 2007;73(3):266–274. | ||
Ohno Y, Okumura T, Terada R, Ishihara S, Serikawa T, Sasa M. Kindling-associated SV2A expression in hilar GABAergic interneurons of the mouse dentate gyrus. Neurosci Lett. 2012;510(2):93–98. | ||
Ohno Y, Ishihara S, Terada R, et al. Preferential increase in the hippocampal synaptic vesicle protein 2A (SV2A) by pentylenetetrazole kindling. Biochem Biophys Res Commun. 2009;390(3):415–420. | ||
Nowack A, Malarkey EB, Yao J, Bleckert A, Hill J, Bajjalieh SM. Levetiracetam reverses synaptic deficits produced by overexpression of SV2A. PLoS One. 2011;6(12):e29560. | ||
Kaminski RM, Gillard M, Leclercq K, et al. Pro-epileptic phenotype of SV2A deficient mice is associated with reduced anticonvulsant efficacy of levetiracetam. Epilepsia. 2009;50(7):1729–1740. | ||
Wood MD, Gillard M. Evidence for a differential interaction of brivaracetam and levetiracetam with the synaptic vesicle 2A protein. Epilepsia. 2017;58(2):255–262. | ||
Yang XF, Weisenfeld A, Rothman SM. Prolonged exposure to levetiracetam reveals a presynaptic effect on neurotransmission. Epilepsia. 2007;48(10):1861–1869. | ||
Yang XF, Rothman SM. Levetiracetam has a time- and stimulation-dependent effect on synaptic transmission. Seizure. 2009;18(9):615–619. | ||
Meehan AL, Yang X, McAdams BD, Yuan L, Rothman SM. A new mechanism for antiepileptic drug action: vesicular entry may mediate the effects of levetiracetam. J Neurophysiol. 2011;106(3):1227–1239. | ||
Daniels V, Wood M, Leclercq K, Kaminski RM, Gillard M. Modulation of the conformational state of the SV2A protein by an allosteric mechanism as evidenced by ligand binding assays. Br J Pharmacol. 2013;169(5):1091–1101. | ||
Yang X, Bognar J Jr, He T, et al. Brivaracetam augments short-term depression and slows vesicle recycling. Epilepsia. 2015;56:1899–1909. | ||
Wood MD, Gillard M. Evidence for a differential interaction of brivaracetam and levetiracetam with the synaptic vesicle 2A protein. Epilepsia. 2017;58(2):255–262. | ||
Margineanu D, Klitgaard H. Levetiracetam mechanisms of action. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, editors. Antiepileptic Drugs. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2002:419–427. | ||
Pisani A, Bonsi P, Martella G, et al. Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine. Epilepsia. 2004;45(7):719–728. | ||
Rigo JM, Nguyen L, Hans G, et al. UCB 34714: effect on inhibitory and excitatory neurotransmission. Epilepsia. 2004;45(s3):56. | ||
Niespodziany I, Lukyanetz EA, Matagne A, Klitgaard H, Wolff C. Brivaracetam does not modulate the major ionic conductances in neurons. Epilepsia. 2015;56 Suppl 1:192–193. | ||
Kostyuk PG, Lukyanetz EA, Klitgaard H, Margineanu DG. UCB 34714, a new pyrrolidone derivative without impact on high and low voltage activated calcium currents in rat isolated neurons. Epilepsia. 2004;45 Suppl 7:141. | ||
Zona C, Pieri M, Carunchio I, Curcio L, Klitgaard H, Margineanu D. Brivaracetam (UCB 34714) inhibits Na+ current in rat cortical neurons in culture. Epilepsy Res. 2010;88(1):46–54. | ||
Niespodziany I, André V, Leclère N, Ghisdal P, Wolff C. Anticonvulsant properties of brivaracetam are not mediated by its effects on voltage-gated sodium channels. Epilepsy Curr. 2014;14 Suppl 1:97. | ||
Niespodziany I, André VM, Leclère N, Hanon E, Ghisdal P, Wolff C. Brivaracetam differentially affects voltage-gated sodium currents without impairing sustained repetitive firing in neurons. CNS Neurosci Ther. 2015;21(3):241–251. | ||
Nicolas JM, Hannestad J, Holden D, et al. Brivaracetam, a selective high-affinity synaptic vesicle protein 2A (SV2A) ligand with preclinical evidence of high brain permeability and fast onset of action. Epilepsia. 2016;57(2):201−209. | ||
Hannestad J, Finnema SJ, Mercier J, et al. Brivaracetam enters the human brain faster than levetiracetam. Epilepsia. 2016;57 Suppl 2:128. | ||
Finnema SJ, Mercier J, Naganawa M, et al. Brivaracetam enters the brain faster than levetiracetam: a PET study in healthy volunteers. Neurology. 2017;88(16 Suppl):P6.233. | ||
Chanteux H, Kervyn S, Gerin B, et al. In vitro pharmacokinetic profile of brivaracetam reveals low risk of drug-drug interaction and unrestricted brain permeability. Epilepsy Curr. 2015;15 Suppl 1:333. | ||
Detrait E, Leclercq K, Matagne A, Klitgaard H. Protective activity of brivaracetam in the 6 Hz model of partial epilepsy: comparison with levetiracetam and older antiepileptic drugs. Epilepsia. 2008;49 Suppl 7:1252. | ||
Matagne A, Margineanu DG, Kenda B, Michel P, Klitgaard H. Anti-convulsive and anti-epileptic properties of brivaracetam (UCB 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A. Br J Pharmacol. 2008;154(8):1662–1671. | ||
Leclercq K, Kaminski RM. Anticonvulsant effects of brivaracetam in the 6 Hz fully-kindled mice. Epilepsia. 2015;56(1):53. | ||
Leclercq K, Matagne A, Kaminski RM. Low potency and limited efficacy of antiepileptic drugs in the mouse 6 Hz corneal kindling model. Epilepsy Res. 2014;108(4):675–683. | ||
Kenda BM, Matagne AC, Talaga PE, et al. Discovery of 4-substituted pyrrolidone butanamides as new agents with significant antiepileptic activity. J Med Chem. 2004;47(3):530–549. | ||
Tai KK, Truong DD. Brivaracetam is superior to levetiracetam in a rat model of post-hypoxic myoclonus. J Neural Transm. 2007;114(12):1547–1551. | ||
Niquet N, Suchomelova L, Thompson K, Klitgaard H, Matagne A, Wasterlain C. Acute and long-term effects of brivaracetam and brivaracetam-diazepam combinations in an experimental model of status epilepticus. Epilepsia. 2017;58(7):1199–1207. | ||
Klein P, Tyrlikova I, Brazdil M, Rektor I. Brivaracetam for the treatment of epilepsy. Expert Opin Pharmacother. 2016;17(2):283–295. | ||
Rolan P, Sargentini-Maier ML, Pigeolet E, Stockis A. The pharmacokinetics, CNS pharmacodynamics and adverse event profile of brivaracetam after multiple increasing oral doses in healthy men. Br J Clin Pharmacol. 2008;66(1):71–75. | ||
Sargentini-Maier ML, Rolan P, Connell J, et al. The pharmacokinetics, CNS pharmacodynamics and adverse event profile of brivaracetam after single increasing oral doses in healthy males. Br J Clin Pharmacol. 2007;63(6):680–688. | ||
Sargentini-Maier ML, Espie P, Coquette A, Stockis A. Pharmacokinetics and metabolism of 14C-brivaracetam, a novel SV2A ligand, in healthy subjects. Drug Metab Dispos. 2008;36(1):36–45. | ||
Stockis A, Watanabe S, Rouits E, Matsuguma K, Irie S. Brivaracetam single and multiple rising oral dose study in healthy Japanese participants: influence of CYP2C19 genotype. Drug Metab Pharmacokinet. 2014;29(5):394–399. | ||
Stockis A, Watanabe S, Scheen AJ, et al. Effect of rifampin on the disposition of brivaracetam in human subjects: further insights into brivaracetam hydrolysis. Drug Metab Dispos. 2016;44(6):792–799. | ||
Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS. Progress report on new antiepileptic drugs: a summary of the Tenth Eilat Conference (EILAT X). Epilepsy Res. 2010;92(2–3):89–124. | ||
Otoul C, Watanabe S, McCabe S, Stockis A. Relative bioavailability and bioequivalence of brivaracetam 10 mg/mL oral solution and 50-mg film-coated tablet. Clin Pharmacol Drug Dev. 2017;6(3):313–317. | ||
Schoemaker R, Wade JR, Stockis A. Brivaracetam population pharmacokinetics in children with epilepsy aged 1 month to 16 years. Eur J Clin Pharmacol. 2017;73(6):727–733. | ||
Sargentini-Maier M-L, Homery MC, Stockis A. Pharmacokinetics, safety and tolerability of brivaracetam in healthy elderly subjects. Epilepsia. 2008;49 Suppl 7:452. | ||
Lacroix B, von Rosenstiel P, Sargentini-Maier ML. Population pharmacokinetics of brivaracetam in patients with partial epilepsy. Epilepsia. 2007;48 Suppl 6:333. | ||
Laveille C, Lacroix B, Snoeck E, Sargentini-Maier ML, von Rosenstiel P, Stockis A. Dose- and exposure-response modeling of brivaracetam add-on treatment in patients with partial epilepsy. Epilepsia. 2007;48 Suppl 6:328. | ||
Stockis A, Watanabe S, Rouits E, Matsuguma K, Irie S. Brivaracetam single and multiple rising oral dose study in healthy Japanese participants: influence of CYP2C19 genotype. Drug Metab Pharmacokinet. 2014;29(5):394–399. | ||
Goldstein JA, Ishizaki T, Chiba K, et al. Frequencies of the defective CYP2C19 alleles responsible for the mephenytoin poor metabolizer phenotype in various Oriental, Caucasian, Saudi Arabian and American black populations. Pharmacogenetics. 1997;7(1):59–64. | ||
Inomata S, Nagashima A, Itagaki F, et al. CYP2C19 genotype affects diazepam pharmacokinetics and emergence from general anesthesia. Clin Pharmacol Ther. 2005;78(6):647–655. | ||
Kimura M, Ieiri I, Mamiya K, Urae A, Higuchi S. Genetic polymorphism of cytochrome P450s, CYP2C19, and CYP2C9 in a Japanese population. Ther Drug Monit. 1998;20(3):243–247. | ||
Sargentini-Maier ML, Sokalski A, Boulanger P, Jacobs T, Stockis A. Brivaracetam disposition in renal impairment. J Clin Pharmacol. 2012;52(12):1927–1933. | ||
Stockis A, Sargentini-Maier ML, Horsmans Y. Brivaracetam disposition in mild to severe hepatic impairment. J Clin Pharmacol. 2013;53(6):633–641. | ||
Stockis A, Hartstra J, Mollet M, Hadi S. Bioavailability and bioequivalence comparison of brivaracetam 10, 50, 75, and 100 mg tablets and 100 mg intravenous bolus. Epilepsia. 2016;57(8):1288–1293. | ||
Stockis A, Watanabe S, Scheen AJ. Effect of brivaracetam on CYP3A activity, measured by oral midazolam. J Clin Pharmacol. 2015;55(5):543–548. | ||
Nicolas JM, Chanteux H, Rosa M, Watanabe S, Stockis A. Effect of gemfibrozil on the metabolism of brivaracetam in vitro and in human subjects. Drug Metab Dispos. 2012;40(8):1466–1472. | ||
Otoul C, Stockis A. Evaluation of the pharmacokinetic interaction of brivaracetam on other antiepileptic drugs in adults with partial-onset seizures. Poster presented at: American Epilepsy Society 69th Annual Meeting; December 4–8, 2015; Philadelphia, PA. | ||
McDonough B, Colson A, Stockis A, Johnson M, Whitesides J. Brivaracetam-induced elevation of carbamazepine-epoxide levels: a safety analysis. Poster presented at: American Epilepsy Society 69th Annual Meeting; December 4–8, 2015; Philadelphia, PA. | ||
Stockis A, Sargentini-Maier ML, Brodie MJ. Pharmacokinetic interaction of brivaracetam on carbamazepine in adult patients with epilepsy, with and without valproate co-administration. Epilepsy Res. 2016;128:163–168. | ||
Moseley BD, Nicolas JM, Yeo KR, et al. The effect of brivaracetam on the pharmacokinetics of phenytoin: in vivo, in vitro, and modeling studies. Poster presented at: American Epilepsy Society 70th Annual Meeting; December 2–6, 2016; Houston, TX. | ||
Stockis A, Watanabe S, Fauchoux N. Interaction between brivaracetam (100 mg/day) and a combination oral contraceptive: a randomized, double-blind, placebo-controlled study. Epilepsia. 2014;55(3):e27–e31. | ||
Stockis A, Rolan P. Effect of brivaracetam (400 mg/day) on the pharmacokinetics and pharmacodynamics of a combination oral contraceptive in healthy women. J Clin Pharmacol. 2013;53(12):1313–1321. | ||
Kruithof AC, Watanabe S, Peeters PA, et al. Pharmacological interactions between brivaracetam and ethanol in healthy males. J Psychopharmacol. 2016;31(7):915–926. | ||
Löscher W, Luna-Tortós C, Römermann K, Fedrowitz M. Do ATP-binding cassette transporters cause pharmacoresistance in epilepsy? Problems and approaches in determining which antiepileptic drugs are affected. Curr Pharm Des. 2011;17(26):2808–2828. | ||
Kasteleijn-Nolst Trenité DG, Genton P, Parain D, et al. Evaluation of brivaracetam, a novel SV2A ligand, in the photosensitivity model. Neurology. 2007;69(10):1027–1034. | ||
French JA, Costantini C, Brodsky A, von Rosenstiel P. Adjunctive brivaracetam for refractory partial-onset seizures: a randomized, controlled trial. Neurology. 2010;75(6):519–525. | ||
van Paesschen W, Hirsch E, Johnson M, Falter U, von Rosenstiel P. Efficacy and tolerability of adjunctive brivaracetam in adults with uncontrolled partial-onset seizures: a phase IIB, randomized, controlled trial. Epilepsia. 2013;54(1):89–97. | ||
Biton V, Berkovic SF, Abou-Khalil B, Sperling MR, Johnson ME, Lu S. Brivaracetam as adjunctive treatment for uncontrolled partial epilepsy in adults: a phase III randomized, double-blind, placebo-controlled trial. Epilepsia. 2014;55(1):57−66. | ||
Ryvlin P, Werhahn KJ, Blaszczyk B, Johnson ME, Lu S. Adjunctive brivaracetam in adults with uncontrolled focal epilepsy: results from a double-blind, randomized, placebo-controlled trial. Epilepsia. 2014;55(1):47−56. | ||
Klein P, Schiemann J, Sperling MR, et al. A randomized, double-blind, placebo-controlled, multicenter, parallel-group study to evaluate the efficacy and safety of adjunctive brivaracetam in adult patients with uncontrolled partial-onset seizures. Epilepsia. 2015;56(12):1890−1898. | ||
Ben-Menachem E, Mameniškienė R, Quarato PP, et al. Efficacy and safety of brivaracetam for partial-onset seizures in three pooled clinical studies. Neurology. 2016;87(3):314–323. | ||
Kwan P, Trinka E, van Paesschen W, Rektor I, Johnson ME, Lu S. Adjunctive brivaracetam for uncontrolled focal and generalized epilepsies: results of a phase III, double-blind, randomized, placebo-controlled, flexible-dose trial. Epilepsia. 2014;55(1):38–46. | ||
Moseley BD, Sperling MR, Asadi-Pooya AA, et al. Efficacy, safety, and tolerability of adjunctive brivaracetam for secondarily generalized tonic-clonic seizures: pooled results from three phase III studies. Epilepsy Res. 2016;127:179–185. | ||
Klein P, Johnson ME, Schiemann J, Whitesides J. Time to onset of sustained ≥50% responder status in patients with focal (partial-onset) seizures in three phase III studies of adjunctive brivaracetam treatment. Epilepsia. 2017;58(2):e21–e25. | ||
Asadi-Pooya AA, Sperling MR, Chung S, et al. Efficacy and tolerability of adjunctive brivaracetam in patients with prior antiepileptic drug exposure: a post-hoc study. Epilepsy Res. 2017;131:70–75. | ||
Yates SL, Fakhoury T, Liang W, Eckhardt K, Borghs S, D’Souza J. An open-label, prospective, exploratory study of patients with epilepsy switching from levetiracetam to brivaracetam. Epilepsy Behav. 2015;52(Pt A):165–168. | ||
Kälviäinen R, Genton P, Andermann E, et al. Brivaracetam in Unverricht-Lundborg disease (EPM1): results from two randomized, double-blind, placebo-controlled studies. Epilepsia. 2016;57(2):210–221. | ||
Toledo M, Whitesides J, Schiemann J, et al. Safety, tolerability, and seizure control during long-term treatment with adjunctive brivaracetam for partial-onset seizures. Epilepsia. 2016;57(7):1139–1151. | ||
Brodie MJ, Whitesides J, Schiemann J, D’Souza J, Johnson ME. Tolerability, safety, and efficacy of adjunctive brivaracetam for focal seizures in older patients: a pooled analysis from three phase III studies. Epilepsy Res. 2016;127:114–118. | ||
Liu E, Hepner A, Dilley D, Stockis A, Daniels A. Safety and tolerability of adjunctive brivaracetam administration as oral solution in pediatric patients aged at least 1 month to 16 years with epilepsy. Epilepsy Curr. 2013;14 Suppl 1:390. | ||
Klein P, Biton V, Dilley D, Barnes M, Schiemann J, Lu S. Safety and tolerability of adjunctive brivaracetam as intravenous infusion or bolus in patients with epilepsy. Epilepsia. 2016;57(7):1130–1138. | ||
Strzelczyk A, Steinig I, Willems LM, et al. Treatment of refractory and super-refractory status epilepticus with brivaracetam: a cohort study from two German university hospitals. Epilepsy Behav. 2017;70(Pt A):177–181. | ||
Steinhoff BJ, Bacher M, Bucurenciu I, et al. Real-life experience with brivaracetam in 101 patients with difficult-to-treat epilepsy - A monocenter survey. Seizure. 2017;48:11–14. | ||
Steinig I, von Podewils F, Moddel G, et al. Postmarketing experience with brivaracetam in the treatment of epilepsies: a multicenter cohort study from Germany. Epilepsia. 2017;58(7):1208–1216. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.