")
Back to Journals » Drug Design, Development and Therapy » Volume 11
A review of the literature analyzing benefits and concerns of infliximab biosimilar CT-P13 for the treatment of rheumatologic diseases: focus on interchangeability
Authors Becciolini A , Raimondo MG, Crotti C , Agape E, Biggioggero M , Favalli EG
Received 31 March 2017
Accepted for publication 19 May 2017
Published 28 June 2017 Volume 2017:11 Pages 1969—1978
DOI https://doi.org/10.2147/DDDT.S138515
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Andrea Becciolini,1 Maria Gabriella Raimondo,2 Chiara Crotti,2 Elena Agape,2 Martina Biggioggero,2 Ennio Giulio Favalli1
1Department of Rheumatology, 2Department of Clinical Sciences and Health Community, University of Milan, Division of Rheumatology, Gaetano Pini Institute, Milan, Italy
Abstract: The introduction of biological agents drastically changed the treatment paradigm of inflammatory arthritides, ameliorating the natural history of the diseases but concomitantly increasing the drug costs due to the manufacturing process. On this concern, biosimilar drugs may represent a valid option for reducing this elevated cost and increasing the availability of these highly effective treatments. Recently, CT-P13, the first biosimilar of infliximab, has been approved with the same indications established for the reference product (RP), and its daily use is progressively increasing. However, the experience with biosimilar drugs in the field of rheumatology is still limited, raising potential doubts and concerns on their correct management in real-life settings. Comparability analysis between CT-P13 and its RP was evaluated in equivalence randomized controlled trials (RCTs) – PLANETRA and PLANETAS – performed on patients with rheumatoid arthritis and axial spondylitis, respectively. CT-P13 and RP showed similar profile in terms of quality, biological activity, safety, immunogenicity, and efficacy. However, the interchangeability between infliximab RP and its biosimilar still represents the most challenging issue because of a lack of a long-lasting experience. To date, reassuring preliminary data on this topic were reported in open-label extensions of PLANETRA and PLANETAS RCTs and in ongoing real-life observational studies. These findings, taken all together, significantly affect the landscape of biosimilar regulatory pathways and strongly support CT-P13 introduction as a great opportunity for expanding the accessibility to these very effective and high-cost therapies.
Keywords: biological therapy, biosimilars, interchangeability, TNF inhibitors, rheumatic diseases
Introduction
In the late 1990s, the introduction of tumor necrosis factor alpha inhibitors (TNFis) has dramatically revolutionized the management and the expected short- and long-term outcomes of inflammatory arthritides, such as rheumatoid arthritis (RA), psoriatic arthritis (PsA), and ankylosing spondylitis (AS).1,2 Infliximab (Janssen Biotech, Horsham, PA, USA), a human-murine chimeric monoclonal antibody (mAb) targeted on TNFα, was the first biological disease-modifying antirheumatic drug (bDMARD) licensed for the treatment of RA first and spondyloarthritis subsequently.3 Nowadays, the other four TNFis have become available for the treatment of RA, PsA, and AS: etanercept (Amgen Inc., Thousand Oaks, CA, USA) (a fusion protein of recombinant TNF receptor and Fc region of immunoglobulin), adalimumab (AbbVie Inc, North Chicago, Illinois, USA) and golimumab (Janssen Biotech, Horsham, PA, USA) (both human mAbs), and certolizumab pegol (UCB Inc, Smyrna, Georgia, USA) (a PEGylated Fab’ fragment from humanized mAb). The data obtained by several randomized controlled trials (RCTs) and by 15-year real-life experiences reported in observational registries have definitely demonstrated the favorable efficacy and safety profile of this drug class in all the above indications.4–6 According to this evidence and to what was suggested by international recommendations, TNFis represent the most used bDMARDs for the treatment of inflammatory arthritides.7–12 However, bDMARD introduction has significantly increased the amount of direct health-care costs intended for the management of inflammatory arthritides, leading in some countries to the implementation of budget restriction policies, potentially limiting the accessibility to bDMARDs for all those patients for whom the use of biological agents is clinically indicated according to international recommendations.13 In recent years, the expiration of data protection or patents for first-generation biopharmaceuticals, followed by patent expiration of the first-approved bDMARDs, has opened the possibility of developing biosimilar products.14,15 According to the World Health Organization (WHO), a biosimilar is defined as “a biotherapeutic product that is similar in term of quality, safety and efficacy to an already licensed reference biotherapeutic product”.16 Given the complexity of the molecular structure of biological agents and their manufacture, it is not possible to produce identical molecules or “generics” for biological drugs.17 For this reason, biosimilars may be approved only after a rigorous, although abbreviated, pathway that relies upon the extensive knowledge and experience gained by the reference product (RP).18,19 In fact, some of the principal regulatory authorities, such as the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA), stated that the development of biosimilars has to be accomplished by an extensive and comprehensive comparative program in order to compare quality requirements, biological activity, safety, and efficacy.20,21
The introduction of biosimilars could be beneficial to address unmet medical needs by widening the access to expensive biological therapy for rheumatologic disorders,22,23 as recommended by international guidelines.7–12 However, efficacy and safety concerns have been raised regarding the short- and long-term differences between biosimilars and RPs, as biological function, efficacy, and toxicity, due to the complexities of manufacturing “copies” of biological therapeutics.24,25
Being the first TNFi marketed for the treatment of rheumatic disorders, infliximab has been the first TNF blocker undergoing patent expiration, leading to the development of biosimilar agents already approved (CT-P13 [Celltrion, Yeonsu-gu, Incheon, South Korea]26,27 and SB228) or under evaluation (such as BOW15,29 PF-06438179,30 and ABP 71031). CT-P13, the first biosimilar of infliximab RP, has been recently approved in Europe by the EMA for all the indications of the RP. So far, a large number of biosimilars have been approved by the EMA since 2006, whereas CT-P13 has been the first biosimilar drug introduced in the rheumatology field. Approval was granted after a complete comparative program including two cardinal RCTs: the PLANETAS (Program evaluating the Autoimmune disease iNvEstigational drug cT-p13 in AS patients) trial is a Phase I, randomized, double-blind, multicenter, multinational, parallel-group study aiming to compare the pharmacokinetics (PK), safety, and efficacy of innovator infliximab (INX) and CT-P13, each administered as monotherapy at a dose of 5 mg/kg infused intravenously, in 250 patients with active AS26; and the PLANETRA (Program evaluating the Autoimmune disease iNvEstigational drug cT-p13 in RA patients) trial is a Phase III, randomized, double-blind, multicenter, multinational, parallel-group study aiming to compare the efficacy and safety of INX and CT-P13, each administered at 3 mg/kg intravenously in combination with methotrexate (MTX), in 606 active RA patients with inadequate response to MTX.27 On the basis of the results obtained from these two trials and EMA approval, quite all national agencies have initially licensed CT-P13 for the treatment of naive AS and RA patients, with the subsequent extrapolations to other indications granted to infliximab RP. Furthermore, the results of the 102-week open-label extensions of PLANETAS32 and PLANETRA33 trials have been recently published, providing crucial information about the safety and efficacy of switching from RP to CT-P13 in both AS and RA patients.
According to surveys conducted with European specialists, more than half claimed to have only a basic knowledge of biosimilars, and nearly a one-quarter is not able to define or previously had not heard of biosimilars,34,35 confirming the need for continued education aiming to a better understanding of the biosimilar landscape. To address this issue, the objective of this review is to critically analyze the clinical outcomes observed in CT-P13 RCT, double-blind, and open-label extension phases, as well as the first real-life data reported by observational studies.
PK and pharmacodynamic (PD) comparability
According to EMA and FDA guidelines on biosimilar drugs, PK evaluation is mandatory for demonstrating the comparability between a newly proposed biosimilar drug and the RP. As a part of the development program of CT-P13, the primary end point of the PLANETAS26 was the equivalence between INX and CT-P13 in terms of the observed maximum steady-state serum concentration (Cmax, ss) between weeks 22 and 30 and the area under the concentration-time curve at steady state. Moreover, PK and PD have been further evaluated as the secondary end points in the Phase III study PLANETRA.27 In both studies, steady-state trough concentration, total drug clearance, volume of distribution at steady state, half-life, geometric means (with a ratio near 100%), and peak serum concentrations (Cmax) were highly similar in the two treatment groups after each infusion of study treatment, as well as the overall range of geometric means across all doses. In conclusion, PLANETAS and PLANETRA studies confirmed the equivalence between CT-P13 and INX in terms of PK and PD end points.26,27
Efficacy and safety comparability in AS and RA patients
As requested by the EMA and the FDA, both PLANETAS and PLANETRA trials are designed as equivalence studies aiming to demonstrate the comparable efficacy and safety in a cohort of patients sensible enough for detecting differences in the measured end points. In an equivalence trial, two treatments are comparable if the difference observed between them lies within an established interval for the predefined clinical equivalence margin.36,37 The limits of this equivalence range are arbitrary and are usually established on the basis of a meta-analysis of previous RCTs conducted with the RP. If 95% CI of the difference between the two products is contained within this preestablished equivalence range, the two treatments may be considered equivalent.38 For CT-P13, this range was set at ±15%, meaning that clinical response to treatment with the biosimilar should stay within ±15% to that obtained with the innovator.
Efficacy end points
In the PLANETAS trial,26 the secondary efficacy end points included 20% and 40% improvement responses according to the criteria laid down by the Assessment of SpondyloArthritis International Society (ASAS). ASAS 20% response (ASAS20) has been similarly achieved in 62.6% and 64.8% of patients treated with CT-P13 and INX at 14 weeks (OR =0.91, 95% CI: 0.53–1.54) and in 70.5% and 72.4% of patients at 30 weeks (OR =0.91, 95% CI: 0.51–1.62), respectively. ASAS40 response has been observed in 41.7% and 45.9% patients at week 14 (OR =0.85, 95% CI: 0.51–1.42) and 51.8% and 47.4% patients at week 30 (OR =1.19, 95% CI: 0.70–2.00). The mean change from baseline in the Ankylosing Spondylitis Disease Activity Score (ASDAS)-C-reactive protein (CRP) score was comparable in both CT-P13 and INX groups at weeks 14 (−1.8, SD =1.1 vs −1.8, SD =1.1, respectively) and 30 (−1.8, SD =1.2 vs −1.7, SD =1.2, respectively). Similarly, no differences were found between CT-P13 and INX groups in the median change from baseline of Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score, Bath Ankylosing Spondylitis Functional Index (BASFI) score, and Bath Ankylosing Spondylitis Metrology Index (BASMI) score.26 In 210 AS patients (106 treated with CT-P13 and 104 treated with INX) who completed the 54-week extension of the PLANETAS study, no difference was observed between CT-P13 and INX groups in ASAS20 (67% vs 69.4%; OR =0.89, 95% CI: 0.50–1.59) and ASAS40 response rates (54.7% vs 49.1%; OR =1.26, 95% CI: 0.73–2.15). Similarly, no difference emerged in the mean change from the baseline of ASDAS-CRP (−1.7, SD =1.3 vs −1.7, SD =1.3), BASDAI (−3.1, SD =2.3 vs −2.8, SD =2.2), BASFI (−2.9, SD =2.3 vs −2.7, SD =2.1), and BASMI scores (−1.1, SD =1.5 vs −0.9, SD =1.6)32 (Figure 1).
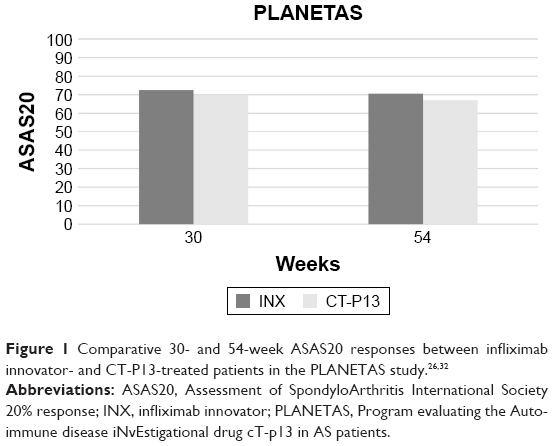 | Figure 1 Comparative 30- and 54-week ASAS20 responses between infliximab innovator- and CT-P13-treated patients in the PLANETAS study.26,32 |
The PLANETRA trial27 has been designed with the primary end point of evaluating the American College of Rheumatology 20% (ACR20) response at week 30, which has been achieved in CT-P13 and INX groups in 60.9% and 58.6% in intention-to-treat (ITT) (treatment difference =2%; 95% CI: −6% to 10%) and in 73.4% and 69.7% in per-protocol (PP) population (treatment difference =4%; 95% CI: −4% to 12%), respectively. Similarly, ACR50 and ACR70 response rates at week 30 for the PP population were 42.3% and 40.6% (treatment difference =2%; 95% CI: −7% to 10%) and 20.2% and 17.9% (treatment difference =2%; 95% CI: −5% to 9%) in the two groups of treatment, respectively. Moreover, additional efficacy end points (mean improvements from baseline of Clinical Disease Activity Index and Simplified Disease Activity Index, proportion of patients achieving good or moderate European League Against Rheumatism [EULAR] responses, and low disease activity or remission rates according to the Disease Activity Score on 28 joints) all confirmed the equivalence between the two products27 (Figure 2).
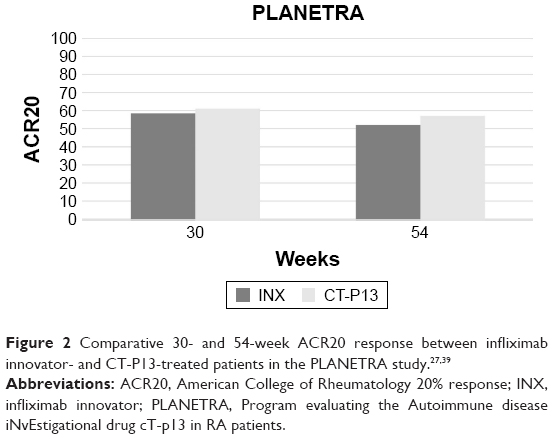 | Figure 2 Comparative 30- and 54-week ACR20 response between infliximab innovator- and CT-P13-treated patients in the PLANETRA study.27,39 |
In the first extension of the PLANETRA study,39 455 patients (233 in the CT-P13 and 222 in the INX subgroup) were treated up to 54 weeks. The ACR20, ACR50, and ACR70 response rates at 54 weeks in the PP population were 74.7% vs 71.3% (treatment difference =3%; 95% CI: −5% to 12%), 43.6% vs 43.1% (treatment difference =0%; 95% CI: −9% to 10%), and 21.3% vs 19.9% (treatment difference =1%; 95% CI: −6% to 9%), respectively, for CT-P13- and INX-treated patients. Regarding radiographic damage progression, the mean total Sharp score change from baseline was similar between CT-P13 and INX treatment groups (1.0±6.3 vs 0.6±5.6, P=0.54), and the proportion of patients classified as nonprogressor was 51.7% vs 51.4% (P=1.00), respectively.39
A recent 54-week Japanese RCT including 101 RA patients treated with either INX or CT-P13 confirmed the results of the PLANETRA study.40 The proportion of patients achieving ACR20 response in the two subgroups was 70.6% and 74% (P=0.82) at week 14, 64.7% and 78% (P=0.18) at week 30, and 49% and 64% (P=0.16) at week 54, respectively. Similarly, the proportion of patients achieving a EULAR good–moderate response in the same subgroups was 82.4% vs 84% (P=1.00) at week 14, 80.4% vs 82% (P=1.00) at week 30, and 66.7% vs 72% (P=0.66) at week 54, respectively.40
In conclusion, the overall comparative experience of CT-P13 clearly demonstrated the short- and long-term clinical and radiographic progression equivalence between biosimilar and infliximab RP.
Safety end points
The main concerns about the extensive use of biosimilars are related to safety. Developing a biosimilar with a safety profile comparable with the RP may be challenging because of the complex molecular structure and complicated manufacturing process involved.41 However, safety data coming from comparative studies of CT-P13 seem to be very reassuring. The proportion of patients who experienced at least one treatment-emergent adverse event (TEAE) in the first 54 weeks of the PLANETAS study in CT-P13 and INX groups was 74.2% and 67.2% (50% and 51.6% related to treatment), respectively. The most frequently reported drug-related TEAEs were abnormal liver function test (12.5% vs 12.3%), upper respiratory tract infection (9.4% vs 6.6%), infusion-related reaction (8.6% vs 12.3%), and latent tuberculosis (7% vs 4.9%).26,27,32
Similarly, in the PLANETRA trial, 70.5% of CT-P13-treated patients and 70.3% of INX-treated patients experienced at least one TEAE in the first 54 weeks (43.7% and 45% related to treatment, respectively). The most frequently reported drug-related TEAEs in both groups were as follows: infusion-related reaction (9.9% vs 14.3%), latent tuberculosis (7.3% vs 6.7%), upper respiratory tract infection (7.6% vs 4.7%), abnormal liver function test (7.3% vs 4.7%), urinary tract infection (3% vs 3.7%), lower respiratory tract infection (3.3% vs 3%), RA flare (2.3% vs 1.7%), Herpes virus infection (1% vs 2.3%), anemia (1.3% vs 1.7%), and headache (1.3% vs 1.7%).27,32–39 Moreover, according to what was reported in the PLANETRA study, the previously mentioned Japanese RCT showed no difference between the two treatment groups (CT-P13 vs INX) among the patients who experienced at least one TEAE (88.2% and 86.8%, respectively).40
To summarize, in the double-blind, randomized phase of equivalence studies conducted with CT-P13, the safety profile has been shown as comparable with INX, even if it should be notable that neither PLANETAS nor PLANETRA was statistically powered to detect significant differences in adverse events between the two treatment groups. In this scenario, the postapproval pharmacovigilance plan will be crucial to assess the risk of less common adverse events that may emerge as related to the biosimilar, similar to the postmarketing identification of risks including congestive heart failure, opportunistic infections, tuberculosis, and demyelinating events associated with infliximab RP.
Immunogenicity
All bDMARDs, even when totally humanized, are immunogenic molecules that can be recognized as nonself antigens able to trigger an immune response with the production of detectable antidrug antibodies (ADAs).42–44 Moreover, the parenteral (often subcutaneous) route of administration may further increase their immunogenicity, resembling the mechanism of an active immunization (vaccination).45 ADA formation may be associated with lower drug levels and clinical nonresponse through two possible mechanisms. First, the presence of ADAs may lead to the formation of immune complexes, which might result in accelerating the clearance of the drug.46 Second, neutralizing ADAs may affect the binding of the drug to its target.47 Furthermore, infusion-related reactions have been indicated as the most frequent potential safety consequence of biological drug immunogenicity.48,49
Thus, efficacy and safety considerations about biosimilar comparability should certainly include immunogenicity and hypersensitivity reactions. In fact, the EMA requires preapproval immunogenicity testing, including the head-to-head demonstration of a not increased immune response to a biosimilar product compared with RP.20 However, immunogenicity is not fully evaluable by the comparability exercise’s analytical assays and may depend on other product- and patient-related factors (such as patient variability, comorbidities, and concomitant medications), leading to the need for strict postapproval monitoring by a postmarket pharmacovigilance program.20
In the PLANETAS study, ADAs were detected at weeks 14, 30, and 54 in 9.1%, 27.4%, and 19.5% in the CT-P13 group, and 11%, 22.5%, and 23% in the RP group, respectively.26,32 As expected, the 14-, 30-, and 54-week incidences of ADAs in the PLANETRA study were higher compared with AS population, but once again similar between CT-P13 and INX (25.4%, 48.4%, and 36% vs 25.8%, 48.2%, and 41.1%, respectively).27,39 These findings have been recently confirmed by an observational real-life Japanese 54-week study conducted in a RA cohort, showing detectable ADA levels in 19.6% (vs 15.1%) at week 14, 25.5% (vs 26.4%) at week 30, and 25.5% (vs 32.1%) at week 54 CT-P13-treated patients compared with INX.40
Beyond the evaluation of ADA incidence in naive patients receiving CT-P13, the key point of immunogenicity is the potential development of ADAs in patients switching between RP and a biosimilar drug. In fact, a sudden change in epitopes presented to the immune system can elicit an immune response,50 and such antigenic discontinuity may arise when switching or alternating between originator and biosimilar therapies, potentially increasing the likelihood of developing ADAs. Moreover, the reiterated presentation of different molecular motifs (as could be the case for an originator and its biosimilar) may induce a synergistic boost of immune response primed by the initial therapy switch51 and potentially dependent on the timing between these switches.52
In the open-label extension of both AS and RA studies, ADA detections at 78 and 102 weeks revealed similar rates between patients maintaining CT-P13 and switching from INX, 23.3% vs 29.8% (P=0.39) and 23.3% vs 27.4% (P=0.60) in the PLANETAS study53 and 44.7% vs 46.2% (P=0.82) and 40.3% vs 44.8% (P=0.48) in the PLANETRA study, respectively.33
Recently, it has been shown that anti-infliximab antibodies in INX-treated RA patients may cross-react with CT-P13, thus suggesting that epitopes influencing immune response to RP are shared with the biosimilar molecular structure.54
In both PLANETAS and PLANETRA double-blind phases, administration reactions seemed to be numerically less frequent in CT-P13-treated patients compared with INX ones. Efficacy analyses of both the studies showed that ADA-positive patients had reduced therapeutic responses when compared with patients who did not develop ADAs, confirming the potential role of immunogenicity in influencing both clinical response and efficacy. However, no substantial differences were observed either in efficacy or in ADA production between patients treated with INX and those treated with CT-P13, thus partially reassuring about the immunogenicity of this biosimilar product.26,27,32,39
Interchangeability and substitutability: data on switching from infliximab RP to CT-P13
Interchangeability and substitution remain key concerns regarding the use of biosimilars. According to the European Generic and Biosimilar Medicines Association (EGA), interchangeability refers to the medical prescription of a biosimilar in place of the RP, while substitution means that pharmacists are allowed to dispense a biosimilar without the prescriber’s knowledge or their explicit request.55 However, the FDA very recently defined interchangeability as the option to introduce a biosimilar agent for the RP without the intervention of the health-care provider who prescribed the RP, whereas a definition of substitutability has not been provided.56 Moreover, in the same draft guidance, the FDA provided the indications about the study design requirements of confirmatory trials evaluating and supporting multiple interchangeability. In particular, the switching arm of the study is expected to incorporate at least two separate exposure periods to each of the two products (ie, at least three switches with each switch crossing over to the alternate product).56
Both PLANETAS and PLANETRA trials have been designed by including an open-label extension phase with the aim of exploring the efficacy and safety of switching from infliximab RP to CT-P13 (Table 1). Among 210 patients enrolled in the PLANETAS trial, 174 (90 treated with CT-P13 and 84 with INX) completed the double-blind phase of the study and entered the open-label extension phase, in which all the patients were treated with the biosimilar drug. At week 102 (ie, 48 weeks after the end of the PLANETAS study), ASAS20 and ASAS40 response rates were 76.9% and 61.5% in the switch (from INX to CT-P13) group and 80.7% and 63.9% in the CT-P13 maintenance group, respectively. The response rates were maintained from weeks 54 to 102 in both groups.32 The proportion of patients who experienced at least one TEAE was apparently higher in the switch group (71.4%) when compared with the maintenance (48.9%) one, but it matched the range reported in historical studies conducted with infliximab RP.57 Moreover, the majority of TEAEs in the switch group had mild-to-moderate severity, while the incidence of infusion-related reactions did not increase in patients who switched from INX to CT-P13 (7.1% vs 7.8% in the maintenance group). The frequency of serious AEs, serious infections including active TB, and TEAEs leading to discontinuation (3/90 and 4/84, respectively) was similar in the two groups.53
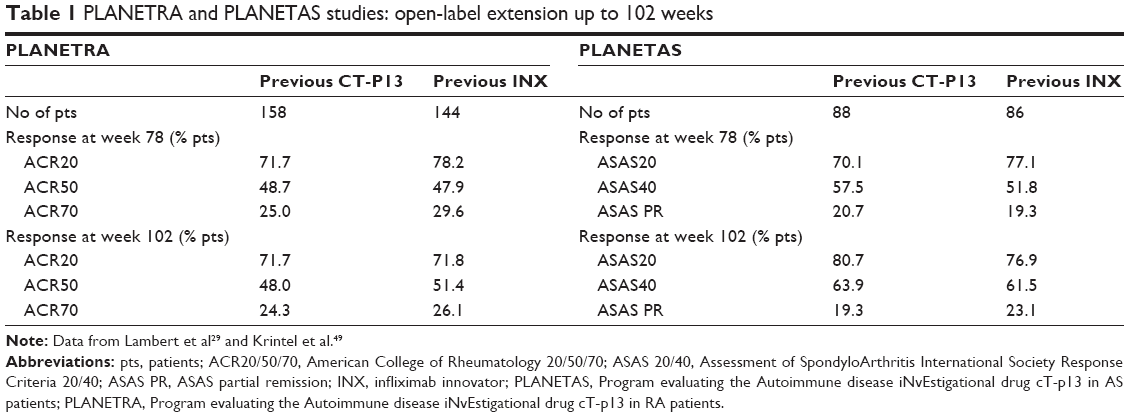 | Table 1 PLANETRA and PLANETAS studies: open-label extension up to 102 weeks |
Similarly, 302 of 455 RA patients who completed the PLANETRA study (158 previously treated with CT-P13 and 144 with RP) were enrolled in an open-label extension, all receiving the biosimilar drug. At week 102, ACR20, ACR50, and ACR70 response rates were 71.8%, 51.4%, and 26.1% in the switch (from INX to CT-P13) group and 71.7%, 48%, and 24.3% in the maintenance group, respectively. The response rates were maintained from weeks 54 to 102 in both groups.33 In this case, the proportion of patients reporting at least one TEAE was more balanced between the maintenance and the switch group (63.5% vs 62.2%), as well as the rate of TEAEs classified as related to treatment (22% vs 18.9%), and the incidence of infusion-related reaction (6.9% vs 2.8%), latent tuberculosis (5.7% vs 2.8%), upper respiratory tract infection (3.8% vs 2.1%), lower respiratory tract infection (2.5% vs 2.8%), abnormal liver function test (0.6% vs 2.8%), and urinary tract infection (1.3% vs 1.4%).33
Besides RCTs, several observational postmarketing studies are ongoing, with the aim of evaluating the efficacy and safety profile of switching from infliximab RP to CT-P13 in a real-life setting (Table 2), and some preliminary data are now available. A Finnish group observed that in 39 patients (15 with RA, 14 with AS, 7 with PsA, 2 with juvenile idiopathic arthritis, and 1 with chronic reactive arthritis) the clinical effectiveness of switching to CT-P13 was comparable to infliximab RP during the first year, without safety concerns.58 Conversely, an Italian group reported that 7 of 23 patients (11 with PsA, 8 with SA, 2 with RA, 2 with Crohn’s disease associated with axial spondyloarthritis, and 1 with Behçet’s disease associated with axial spondyloarthritis) switching from infliximab RP to CT-P13 experienced a disease relapse within 1.71 months.59 These findings have not been confirmed by another Italian study demonstrating the persistence of a 6-month efficacy in switcher patients in an SpA cohort, without any significant change in circulating infliximab or anti-infliximab antibody levels.60
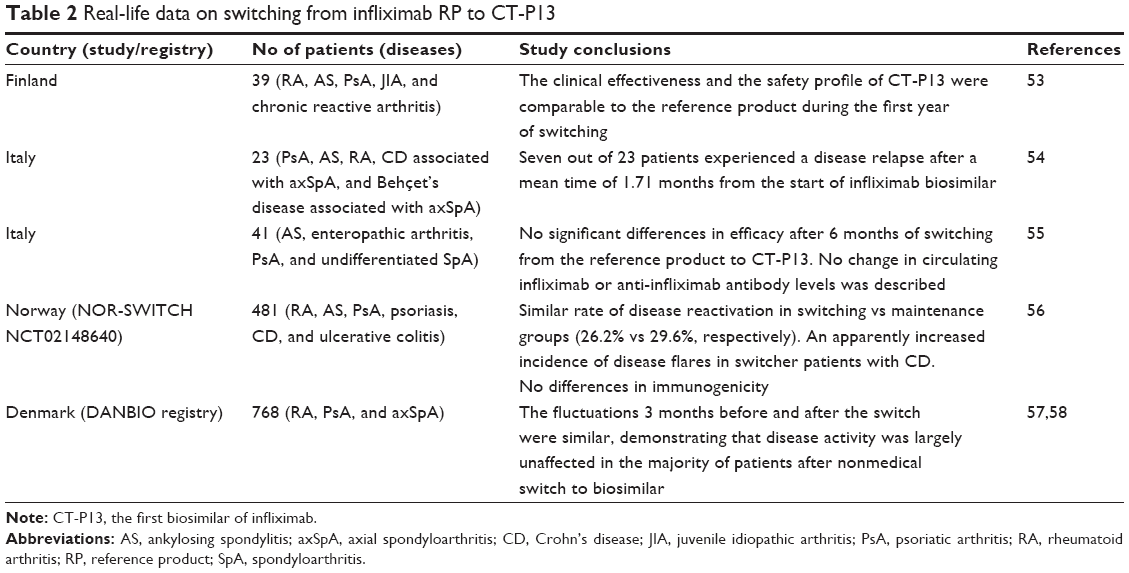 | Table 2 Real-life data on switching from infliximab RP to CT-P13 |
The NOR-SWITCH study is a 52-week randomized, double-blind trial comparing switching to CT-P13 with infliximab RP maintenance therapy in 481 patients with RA (n=77), AS (n=91), PsA (n=30), psoriasis (n=35), and inflammatory bowel disease (Crohn’s disease [CD], n=155; ulcerative colitis =93).61 Enrolled population was selected among subjects receiving a stable infliximab originator treatment during the previous 6 months, and the primary end point was noninferiority (margin set to 15%) in disease worsening (defined by disease-specific efficacy assessment scores) of switching compared with maintenance strategy. Disease reactivation across indication has been reported in 26.2% vs 29.6% in switching vs maintenance groups, respectively (adjusted rate difference [95% CI]: −4.4 [-12.7% to −3.9%]). An apparently increased incidence of disease flares in switcher patients was observed only in CD subgroup (36.5% vs 21.2%), but the trial was not powered to analyze noninferiority in disease worsening by individual indication; thus, this was classified as an exploratory analysis. Finally, the frequency of ADA development (7.1% vs 7.9% in switching vs maintenance patients, respectively) and trough drug levels were similar in the two groups.61
In the Danish DANBIO registry including all RA, PsA, and axSpA patients treated with bDMARDs, 768 subjects switched from RP to CT-P13 between May 2015 and February 2016.62 According to the study design, disease activity was calculated for each patient at the time of switch as well as 3 months before and 3 months after the starting treatment with biosimilar, and changes in disease activity over time were also evaluated. The fluctuations 3 months after the switch were comparable to the fluctuations observed 3 months prior to the switch, demonstrating that disease activity was largely unaffected in the majority of patients after nonmedical switch to biosimilar. Overall, 117 patients (15%) stopped CT-P13 treatment between switch and end of follow-up (AEs 35, inefficacy 51, remission 1, death 2, pregnancy 1, and other 27).62 Moreover, in a subgroup from the same population of 231 patients treated with infliximab RP for more than 5 years, blood samples were drawn at baseline (immediately before switching) and during follow-up (after 3, 6, and 12 months), in order to measure serum infliximab trough levels (sIFX) and ADA. The analysis showed that trough sIFX levels increased from 2.5 mg/L (at the moment of the switch) to 2.9 and 3.1 mg/L (3 and 6 months after the switch, respectively). The presence of medium-high ADA was unchanged over time, and 94% of patients had similar ADA levels at baseline and at 3 months.63
In conclusion, long-term open-label extensions of comparative trials and the first real-life switching experiences did not show unexpected differences in the efficacy, safety profile, and immunogenicity of patients who switched from infliximab RP to CT-P13, when compared with the group treated only with CT-P13. Although all these studies are not adequate to completely satisfy the new study design requirements proposed by the FDA about interchangeability, the favorable findings may be reassuring about single switching between infliximab RP and CT-P13.
The prescription of CT-P13: rationale and current regulatory landscape
The EMA pioneered the development of regulatory requirements for biosimilars in 2005 through the publication of a general framework guidelines, establishing the principle of biosimilarity and providing the authorization for the first-generation biosimilar products in the market.64 Regarding biosimilar positioning, EMA regulatory guidelines consider this drug class as a preferable and valid therapeutic option with highly economic advantage, especially in naive patients. The definition of “naive patient” refers to two specific categories: 1) patients never previously exposed to RP (primary naive), and 2) patients with previous exposure to the RP as suitable candidates for the biosimilar after an adequate washout period (secondary naive).45 However, the latter definition, and specifically the washout period, are still challenging due to the persistence for an undefined timeframe of antireference product antibodies that potentially might cross-react with the biosimilar drug. Thus, the identification of an adequate washout period for introducing the biosimilar agent in secondary naive subjects is still left to the judgment of the physician.45 However, interchangeability and substitutability still remain crucial issues regarding biosimilars, particularly in a scenario of progressively increasing number of available biosimilars of the same RP, leading to the need for specific guidance on multiple switching. Data about this condition are still lacking, and newly designed clinical trials according to requirements proposed by the FDA56 should be advocated in order to improve our knowledge about this potential issue. Moreover, since postapproval pharmacovigilance may be important for a comprehensive assessment of safety risks related to the single biosimilar agent or to multiple interchangeability between RP and all its biosimilars, the traceability of each biological product through a proprietary brand name along with the same international nonproprietary name will be crucial.
The EMA has not included specific recommendations on interchangeability so far; thus, the decision-making processes for these issues should be regulated at a national level.20
Recently published results of open-label extension phase of PLANETAS and PLANETRA trials along with observational data from the NOR-SWITCH study and the DANBIO registry represent the most important and comprehensive source of data on biosimilar today and may be crucial for improving the knowledge on the efficacy and safety of switching strategy. Nevertheless, as suggested for all bDMARDs, most regulatory agencies, including the EMA, require further postmarketing surveillance and encourage the use of registries for biosimilars, in order to obtain detailed information on risks and safety concerns, including immunogenicity and detection of any new safety signals. Till date, several postmarketing evaluations are ongoing, with the aim of analyzing the efficacy and safety profile of switching from infliximab RP to CT-P13 in a real-life setting, especially with regard to interchangeability and substitutability. Most countries of the EU oppose automatic substitution of biosimilars, and many, such as Italy, Spain, and the UK, have enacted laws prohibiting the practice.65,66 However, positions of agencies about this issue are also evolving, as more information on biosimilars becomes available. As an example, the Dutch Medicines Evaluation Board now accepts interchangeability of biosimilars, but only if adequate clinical monitoring is performed, the patient is properly informed, and if detailed product and batch information are recorded in the patient notes in order to guarantee the traceability of the product.67
Conclusion
In summary, available data from comparative RCTs and observational studies strongly confirmed the equivalence between CT-P13 and infliximab RP from a pharmacodynamic and a clinical point of view. Moreover, open-label extension of PLANETRA and PLANETAS trials provided reassuring data about the switching approach from originator to biosimilar drug, more recently supported by preliminary results from prospective pragmatic trials (such as the NOR-SWITCH study) and from retrospective analyses of national registries (such as the Danish DANBIO registry). These findings suggested interchangeability between infliximab RP and CT-P13 as a feasible and safe strategy to be applied in real-life clinical practice. Additional data from future clinical trials designed with the aim of specifically evaluating multiple interchangeability between RP and all its biosimilars should be advocated in order to improve our approach in the management of this condition.
Disclosure
The authors report no conflicts of interest in this work.
References
Caporali R, Pallavicini FB, Filippini M, et al. Treatment of rheumatoid arthritis with anti-TNF-alpha agents: a reappraisal. Autoimmun Rev. 2009;8(3):274–280. | ||
Sieper J, Poddubnyy D. New evidence on the management of spondyloarthritis. Nat Rev Rheumatol. 2016;12(5):282–295. | ||
Klotz U, Teml A, Schwab M. Clinical pharmacokinetics and use of infliximab. Clin Pharmacokinet. 2007;46(8):645–660. | ||
Nam JL, Ramiro S, Gaujoux-Viala C, et al. Efficacy of biological disease-modifying antirheumatic drugs: a systematic literature review informing the 2013 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. 2014;73(3):516–528. | ||
Maxwell LJ, Zochling J, Boonen A, Singh JA, Veras MM, Tanjong Ghogomu E, et al. TNF-alpha inhibitors for ankylosing spondylitis. Cochrane Database Syst Rev. 2015;CD005468. | ||
Ash Z, Gaujoux-Viala C, Gossec L, et al. A systematic literature review of drug therapies for the treatment of psoriatic arthritis: current evidence and meta-analysis informing the EULAR recommendations for the management of psoriatic arthritis. Ann Rheum Dis. 2012;71(3):319–326. | ||
Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73(3):492–509. | ||
Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2016;68(1):1–26. | ||
Braun J, van den Berg R, Baraliakos X, et al. 2010 Update of the ASAS/EULAR recommendations for the management of ankylosing spondylitis. Ann Rheum Dis. 2011;70(6):896–904. | ||
Ward MM, Deodhar A, Akl EA, et al. American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network 2015 Recommendations for the Treatment of Ankylosing Spondylitis and Nonradiographic Axial Spondyloarthritis. Arthritis Rheumatol. 2016;68(2):282–298. | ||
Coates LC, Kavanaugh A, Mease PJ, et al. Group for research and assessment of psoriasis and psoriatic arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis Rheumatol. 2016;68(5):1060–1071. | ||
Gossec L, Smolen JS, Ramiro S, et al. European League Against Rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Ann Rheum Dis. 2016;75(3):499–510. | ||
Lapadula G, Ferraccioli GF. Biosimilars in rheumatology: pharmacological and pharmacoeconomic issues. Clin Exp Rheumatol. 2012;30(4 Suppl 73):S102–S106. | ||
Dranitsaris G, Amir E, Dorward K. Biosimilars of biological drug therapies: regulatory, clinical and commercial considerations. Drugs. 2011;71(12):1527–1536. | ||
Goel N, Chance K. Biosimilars in rheumatology: understanding the rigor of their development. Rheumatology (Oxford). 2017;56(2):187–197. | ||
World Health Organization. WHO Expert Committee on Biological Standardization. Sixty-fifth report. World Health Organ Tech Rep Ser. 2015;(993):1–262. | ||
Crommelin DJA, Shah VP, Klebovich I, et al. The similarity question for biologicals and non-biological complex drugs. Eur J Pharm Sci. 2015;76:10–17. | ||
Dörner T, Kay J. Biosimilars in rheumatology: current perspectives and lessons learnt. Nat Rev Rheumatol. 2015;11(12):713–724. | ||
Alten R, Cronstein BN. Clinical trial development for biosimilars. Semin Arthritis Rheum. 2015;44(Suppl 6):S2–S8. | ||
European Medicines Agency [Internet]. Guideline on similar biological medicinal products; 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768. Accessed June 14, 2017. | ||
Food and Drug Administration. Biosimilars: Additional Questions and Answers Regarding Implementation of the Biologics Price Competition and Innovation Act of 2009 Guidance for Industry; 2009. Available from: http://www.fda.gov/downloads/Drugs/.../Guidances/UCM273001.pdf. Accessed June 14, 2017. | ||
Gulacsi L, Brodszky V, Baji P, et al. Biosimilars for the management of rheumatoid arthritis: economic considerations. Expert Rev Clin Immunol. 2015;11(Suppl 1):S43–S52. | ||
Jha A, Upton A, Dunlop WCN, Akehurst R. The budget impact of biosimilar infliximab (Remsima®) for the treatment of autoimmune diseases in five European Countries. Adv Ther. 2015;32(8):742–756. | ||
Scheinberg MA, Kay J. The advent of biosimilar therapies in rheumatology – “O brave new world”. Nat Rev Rheumatol. 2012;8(7):430–436. | ||
Roger SD, Mikhail A. Biosimilars: opportunity or cause for concern? J Pharm Pharm Sci. 2007;10(3):405–410. | ||
Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605–1612. | ||
Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72(10):1613–1620. | ||
Choe J-Y, Prodanovic N, Niebrzydowski J, et al. A randomised, double-blind, phase III study comparing SB2, an infliximab biosimilar, to the infliximab reference product Remicade in patients with moderate to severe rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2017;76(1):58–64. | ||
Lambert J, Wyand M, Lassen C, et al. Bioavailability, safety and immunogenicity of biosimilar infliximab (BOW015) compared to reference infliximab. Int J Clin Pharmacol Ther. 2016;54(4):315–322. | ||
Derzi M, Johnson TR, Shoieb AM, et al. Nonclinical Evaluation of PF-0,64,38,179: A Potential Biosimilar to Remicade(®) (Infliximab). Adv Ther. 2016;33(11):1964–1982. | ||
Kaur P, Chow V, Zhang N, Krishnan E. Pharmacokinetic similarity of ABP 710 relative to infliximab: results from a randomized, single-blind, single-dose, parallel group study in healthy subjects. Arthritis Rheumatol. 2016;68(Suppl 10). Available from: http://acrabstracts.org/abstract/pharmacokinetic-similarity-of-abp-710-relative-to-infliximab-results-from-a-randomized-single-blind-single-dose-parallel-group-study-in-healthy-subjects/. Accessed May 31, 2017. | ||
Park W, Yoo DH, Jaworski J, et al. Comparable long-term efficacy, as assessed by patient-reported outcomes, safety and pharmacokinetics, of CT-P13 and reference infliximab in patients with ankylosing spondylitis: 54-week results from the randomized, parallel-group PLANETAS study. Arthritis Res Ther. 2016;18(1):25. | ||
Yoo DH, Suh CH, Shim SC, et al. A multicentre randomised controlled trial to compare the pharmacokinetics, efficacy and safety of CT-P10 and innovator rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76:566–570. | ||
Dolinar RO, Reilly MS. Biosimilars naming, label transparency and authority of choice – survey findings among European physicians. G B I J. 2014;3(2):58–62. | ||
Danese S, Fiorino G, Michetti P. Viewpoint: Knowledge and viewpoints on biosimilar monoclonal antibodies among members of the European Crohn’s and Colitis Organization. Journal Crohn’s Colitis. 2014;8(11):1548–1550. | ||
Castañeda-Hernández G, González-Ramírez R, Kay J, Scheinberg MA. Biosimilars in rheumatology: what the clinician should know. RMD Open. 2015;1(1):e000010. | ||
Njue C. Statistical considerations for confirmatory clinical trials for similar biotherapeutic products. Biologicals. 2011;39(5):266–269. | ||
Kay J, Smolen JS. Biosimilars to treat inflammatory arthritis: the challenge of proving identity. Ann Rheum Dis. 2013;72(10):1589–1593. | ||
Yoo DH, Racewicz A, Brzezicki J, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2016;18(1):82. | ||
Takeuchi T, Yamanaka H, Tanaka Y, et al. Evaluation of the pharmacokinetic equivalence and 54-week efficacy and safety of CT-P13 and innovator infliximab in Japanese patients with rheumatoid arthritis. Mod Rheumatol. 2015;25(6):817–824. | ||
Declerck PJ. Biosimilar monoclonal antibodies: a science-based regulatory challenge. Expert Opin Biol Ther. 2013;13(2):153–156. | ||
Vincent FB, Morand EF, Murphy K, Mackay F, Mariette X, Marcelli C. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)-specific neutralising agents in chronic inflammatory diseases: a real issue, a clinical perspective. Ann Rheum Dis. 2013;72(2):165–178. | ||
Schaeverbeke T, Truchetet ME, Kostine M, Barnetche T, Bannwarth B, Richez C. Immunogenicity of biologic agents in rheumatoid arthritis patients: lessons for clinical practice. Rheumatology (Oxford). 2016;55(2):210–220. | ||
Thomas SS, Borazan N, Barroso N, et al. Comparative immunogenicity of TNF inhibitors: impact on clinical efficacy and tolerability in the management of autoimmune diseases. A systematic review and meta-analysis. BioDrugs. 2015;29(4):241–258. | ||
Biggioggero M, Danova M, Genovese U, et al. The challenging definition of naïve patient for biological drug use. Autoimmun Rev. 2015;14(6):543–546. | ||
van Schouwenburg PA, van de Stadt LA, de Jong RN, et al. Adalimumab elicits a restricted anti-idiotypic antibody response in autoimmune patients resulting in functional neutralisation. Ann Rheum Dis. 2013;72(1):104–109. | ||
Aarden LA, Ruuls SR, Wolbink G. Immunogenicity of anti-tumor necrosis factor antibodies-toward improved methods of anti-antibody measurement. Curr Opin Immunol. 2008;20(4):431–435. | ||
Emi Aikawa N, de Carvalho JF, Artur Almeida Silva C, Bonfá E. Immunogenicity of anti-TNF-alpha agents in autoimmune diseases. Clin Rev Allergy Immunol. 2010;38(2–3):82–89. | ||
Krintel SB, Grunert VP, Hetland ML, et al. The frequency of anti-infliximab antibodies in patients with rheumatoid arthritis treated in routine care and the associations with adverse drug reactions and treatment failure. Rheumatology (Oxford). 2013;52(7):1245–1253. | ||
Pradeu T, Jaeger S, Vivier E. The speed of change: towards a discontinuity theory of immunity? Nature Publishing Group. 2013;13(10):764–769. | ||
Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol. 2004;25(2):98–104. | ||
Castiglione F, Mantile F, De Berardinis P, Prisco A. How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput Math Methods Med. 2012;2012:842329. | ||
Park W, Yoo DH, Miranda P, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76(2):346–354. | ||
Ruiz-Argüello MB, Maguregui A, Ruiz Del Agua A, et al. Antibodies to infliximab in Remicade-treated rheumatic patients show identical reactivity towards biosimilars. Ann Rheum Dis. 2016;75(9):1693–1696. | ||
Olech E. Biosimilars: rationale and current regulatory landscape. Semin Arthritis Rheum. 2016;45(Suppl 5):S1–S10. | ||
Food and Drug Administration. Considerations in Demonstrating Interchangeability with a Reference Product Guidance for Industry; 2017. Available from: https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm537135.pdf. Accessed June 14, 2017. | ||
Braun J, Deodhar A, Dijkmans B, et al. Efficacy and safety of infliximab in patients with ankylosing spondylitis over a two-year period. Arthritis Rheum. 2008;59(9):1270–1278. | ||
Nikiphorou E, Kautiainen H, Hannonen P, et al. Clinical effectiveness of CT-P13 (Infliximab biosimilar) used as a switch from Remicade (infliximab) in patients with established rheumatic disease. Report of clinical experience based on prospective observational data. Expert Opin Biol Ther. 2015;15(12):1677–1683. | ||
Gentileschi S, Barreca C, Bellisai F, et al. Switch from infliximab to infliximab biosimilar: efficacy and safety in a cohort of patients with different rheumatic diseases. Expert Opin Biol Ther. 2016;16(10):1311–1312. | ||
Benucci M, Gobbi FL, Bandinelli F, et al. Safety, efficacy and immunogenicity of switching from innovator to biosimilar infliximab in patients with spondyloarthritis: a 6-month real-life observational study. Immunol Res. 2017;65(1):419–422. | ||
Løvik Goll G, Olsen IC, Jorgensen KK, et al. Biosimilar Infliximab (CT-P13) Is Not Inferior to Originator Infliximab: Results from a 52-Week Randomized Switch Trial in Norway [abstract]. Arthritis Rheumatol. 2016;68(Suppl 10). Available from: http://acrabstracts.org/abstract/biosimilar-infliximab-ct-p13-is-not-inferior-to-originator-infliximab-results-from-a-52-week-randomized-switch-trial-in-norway/. Accessed May 31, 2017. | ||
Glintborg B, Sørensen IJ, Jensen DV, et al. A Nationwide Non-Medical Switch from Originator to Biosimilar Infliximab in Patients with Inflammatory Arthritis. Eleven Months’ Clinical Outcomes from the Danbio Registry [abstract]. Arthritis Rheumatol. 2016;68(Suppl 10). Available from: http://acrabstracts.org/abstract/a-nationwide-non-medical-switch-from-originator-to-biosimilar-infliximab-in-patients-with-inflammatory-arthritis-eleven-months-clinical-outcomes-from-the-danbio-registry/. Accessed May 31, 2017. | ||
Glintborg B, Kringelbach TM, Høgdall E, et al. Non-medical switch from originator to biosimilar infliximab in patients with inflammatory arthritis – impact on s-infliximab and antidrug-antibodies. Results from the Danish Rheumatologic Biobank and the Danbio Registry [abstract]. Arthritis Rheumatol. 2016;68(Suppl 10). Available from: http://acrabstracts.org/abstract/non-medical-switch-from-originator-to-biosimilar-infliximab-in-patients-with-inflammatory-arthritis-impact-on-s-infliximab-and-antidrug-antibodies-results-from-the-danish-rheumatologic-biobank-and/. Accessed May 31, 2017. | ||
European Medicines Agency. Guideline on Similar Biological Medicinal Products; 2005. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf. Accessed June 14, 2017. | ||
Rovira J, Espin J, Garcia L, Olry A. The impact of biosimilars’ entry in the EU market [Internet]; 2011 [cited January 10, 2017]. Available from: http://ec.europa.eu/DocsRoom/documents/7651/attachments/1/translations/en/renditions/pdf. Accessed June 14, 2017. | ||
Italian Society of Rheumatology. Italian Society of Rheumatology position paper on biosimilar [Internet]; 2015. [cited January 10, 2017]. Available from: http://www.reumatologia.it/obj/files/Biosimiliari/BIOSIMILARI2015.pdf | ||
Netherlands Medicines Evaluation Board. Clarification of stance on biological and biosimilar medicines [Internet]; 2015 [cited January 10, 2017]. Available from: http://english.cbg-meb.nl/latest/news/2015/08/17/clarification-of-stance-on-biological-and-biosimilar-medicines. Accessed June 14, 2017. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.