")
Back to Journals » Clinical Ophthalmology » Volume 11
A review and update on orphan drugs for the treatment of noninfectious uveitis
Authors You C, Sahawneh HF, Ma L, Kubaisi B , Schmidt A, Foster CS
Received 7 September 2016
Accepted for publication 16 November 2016
Published 31 January 2017 Volume 2017:11 Pages 257—265
DOI https://doi.org/10.2147/OPTH.S121734
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Caiyun You,1–3 Haitham F Sahawneh,1,2 Lina Ma,1,2 Buraa Kubaisi,1,2 Alexander Schmidt,1,2 C Stephen Foster1,2,4
1Massachusetts Eye Research and Surgery Institution (MERSI), Waltham, 2Ocular Immunology and Uveitis Foundation, Weston, MA, USA; 3Department of Ophthalmology, Tianjin Medical University General Hospital, Tianjin, People’s Republic of China; 4Harvard Medical School, Boston, MA, USA
Introduction: Uveitis, a leading cause of preventable blindness around the world, is a critically underserved disease in regard to the medications approved for use. Multiple immunomodulatory therapy (IMT) drugs are appropriate for uveitis therapy but are still off-label. These IMT agents, including antimetabolites, calcineurin inhibitors, alkylating agents, and biologic agents, have been designated as “orphan drugs” and are widely used for systemic autoimmune diseases or organ transplantation.
Area covered: The purpose of this paper is to comprehensively review and summarize the approved orphan drugs and biologics that are being used to treat systemic diseases and to discuss drugs that have not yet received approval as an “orphan drug for treating uveitis” by the US Food and Drug Administration (FDA).
Our perspective: IMT, as a steroid-sparing agent for uveitis patients, has shown promising clinical results. Refractory and recurrent uveitis requires combination IMT agents. IMT is continued for a period of 2 years while the patient is in remission before considering tapering medication. Our current goals include developing further assessments regarding the efficacy, optimal dose, and safety in efforts to achieve FDA approval for “on-label” use of current IMT agents and biologics more quickly and to facilitate insurance coverage and expand access to the products for this orphan disease.
Keywords: immunomodulatory, orphan drug, steroid sparing, uveitis
Introduction
Uveitis is a major cause of severe visual impairment. It can occur either alone or as part of a systemic syndrome (systemic disease-associated autoimmune uveitis), such as one of the spondyloarthritides (including those complicating inflammatory bowel disorders and juvenile idiopathic arthritis [JIA]), Adamantiades–Behcet’s disease (ABD), Vogt–Koyanagi–Harada (VKH) syndrome, systemic lupus erythematosus, sarcoidosis, autoimmune hepatitis, and multiple sclerosis, in which the eye is one of several organs involved.1 Autoimmune-mediated uveitis treatment is divided into acute phase and maintenance therapy. The acute stage can be controlled with corticosteroid therapy.
The Standardization of Uveitis Nomenclature Working Group Guidelines recommend the use of corticosteroids as first-line therapy for patients with active uveitis.2 However, long-term corticosteroid treatment can cause serious systemic and ocular side effects, such as hypertension, diabetes, cataract, and glaucoma. Alternatively, immunomodulatory therapy (IMT) drugs are given as steroid-sparing agents and have shown good clinical results for both systemic diseases and ocular inflammatory diseases.3,4 Given the side effects of chronic corticosteroid therapy and better understanding of the mechanisms of autoimmune-mediated uveitis, the aim of the treatment for patients with noninfectious uveitis is steroid-free remission with IMT. A stepladder approach is a common practice in immune-mediated uveitis: nonsteroidal anti-inflammatory drugs and conventional immunomudulatory agents are usually used before proceeding with biologic response modifiers.
IMT agents include the antimetabolites methotrexate, azathioprine, and mycophenolate mofetil; the calcineurin inhibitors that include cyclosporine, tacrolimus, and sirolimus; alkylating agents that include cyclophosphamide and chlorambucil; biologic response modifiers that include the tumor necrosis factor (TNF)-α inhibitors infliximab, adalimumab, etanercept, golimumab, and certolizumab; lymphocyte inhibitors that include daclizumab, rituximab, abatacept, and basiliximab; specific receptor antagonists that include anakinra, canakinumab, gevokizumab, tocilizumab, alemtuzumab, efalizumab, secukinumab, and ustekinumab; and interferon (INF) treatments. Refractory and recurrent uveitis requires immunomodulatory monotherapy or a combination protocol to control the inflammation. Continuing evidence shows that second-line agents, including antimetabolites, T-cell inhibitors, and alkylating agents, and biologics are effective in many patients, allowing reduction in steroid dose and preservation of visual function. Aggressive treatment may result in fewer complications and less recurrence.
Our team has reviewed IMT drugs approved by the US Food and Drug Administration (FDA) for use in uveitis.5 In this study, we review and summarize conventional IMT drugs or biologics that have not received approval as an “orphan drug” from the FDA for use in noninfectious uveitic patients, despite having been approved for use in treating other systemic diseases or organ transplantation.
Orphan drug status in ocular inflammatory diseases
The Orphan Drug Designation program provides orphan status to drugs and biologics that are defined as those intended for the safe and effective treatment, diagnosis, or prevention of rare diseases/disorders that affect fewer than 200,000 people in the USA, or that affect >200,000 persons but are not expected to recover the costs of developing and marketing a treatment drug.6
Although not approved as orphan agents for uveitis, there are many randomized clinical trials evaluating various IMT drugs and biologics used for ocular inflammatory diseases. These treatments have been approved as orphan drugs by the FDA to be used in malignancies, autoimmune disorders, and/or organ transplantation (Table 1). Table 2 shows the use and dosage of these drugs for the treatment of uveitis.
 | Table 1 Orphan drugs approved by the US Food and Drug Administration for use in systemic disease/organ transplantation that have not received orphan drug approval for use in uveitis |
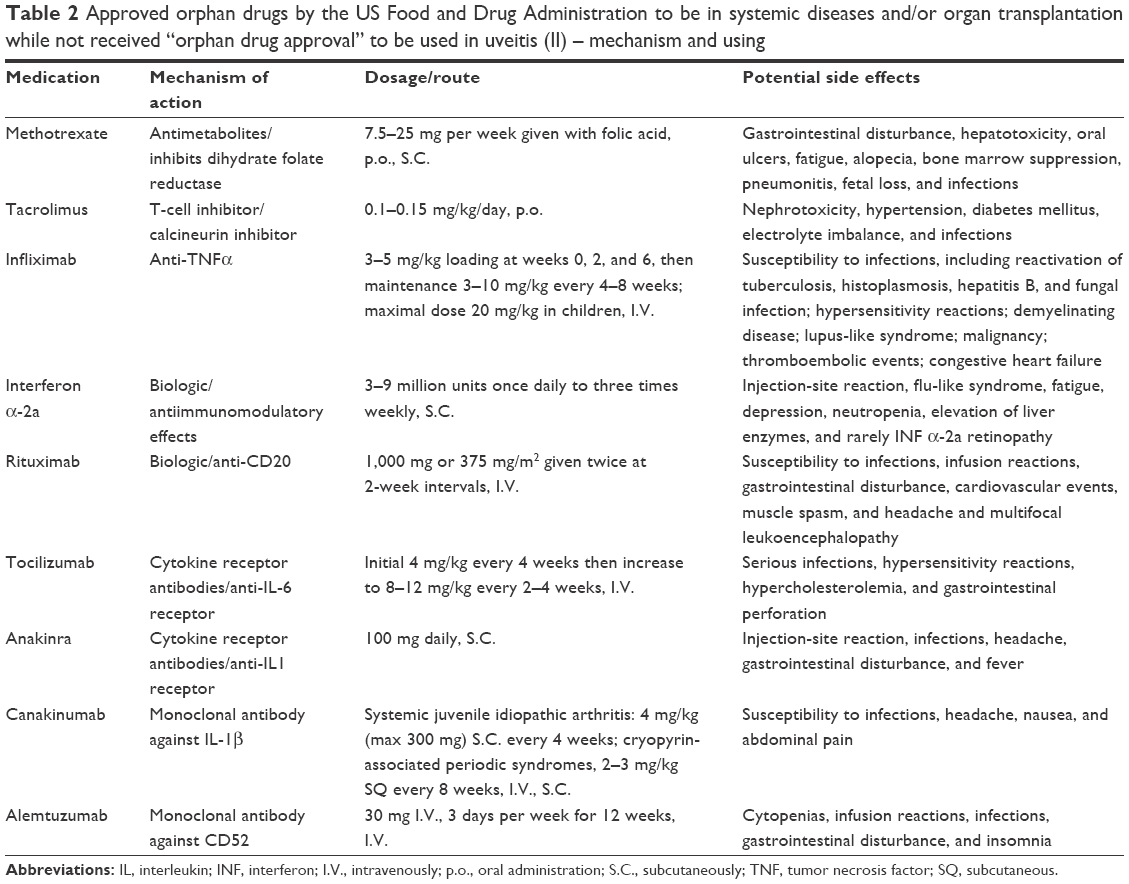 | Table 2 Approved orphan drugs by the US Food and Drug Administration to be in systemic diseases and/or organ transplantation while not received “orphan drug approval” to be used in uveitis (II) – mechanism and using |
Orphan drugs in noninfectious uveitis
Methotrexate
As an antimetabolite and antifolate drug, methotrexate (formerly known as amethopterin) is a folic acid analog that irreversibly, competitively binds and inactivates the enzyme dihydrofolate reductase. Methotrexate also partially, reversibly, competitively inhibits thymidylate synthetase. Ultimately, DNA synthesis, DNA repair, RNA synthesis, and cell division (S-phase cell cycle specific) are inhibited.7 Methotrexate suppresses both B and T cells. At low doses, it has little effect on cell-mediated immunity, but has been shown to depress acute-phase reactants. Therefore, it is suspected that the action of methotrexate is more likely anti-inflammatory than immunosuppressive.8,9 Potential side effects of methotrexate include nausea, diarrhea, fatigue, minimal hair loss, mouth ulcers, hepatitis, eosinophilic pneumonitis or inflammatory lung disease, increased risk of sunburn, increased risk of infection, and rare mood swings in children. In general, methotrexate is mostly well tolerated and side effects improve with reducing dosage.
Methotrexate is used to treat leukemia, cancers (including breast, head and neck, leukemia, lymphoma, lung, osteosarcoma, bladder, and trophoblastic neoplasms), autoimmune diseases (including rheumatoid arthritis, juvenile dermatomyositis, psoriasis, psoriatic arthritis, lupus, sarcoidosis, Crohn’s disease, eczema, and many forms of vasculitis), ectopic pregnancy, and for the induction of medical abortions.10,11 Methotrexate is a widely used agent for ocular inflammation because of its ease of administration and long track record. It is often regarded as the first-line agent when starting a patient with uveitis on IMT.
Methotrexate has been reported to be effective in treating ocular inflammation in several uncontrolled retrospective case studies. In the retrospective Systemic Immunosuppression for Eye Disease Cohort Study (SITE), which included 384 patients, methotrexate was found to be moderately effective with an overall success of 66% at 12 months for sustained control and 58.4% for corticosteroid-sparing control (≤10 mg). From the data, it appeared to be more successful for the treatment of anterior disease; however, the authors caution that this observed effect may be related to the severity of the disease. It was found to be safe, with just 16% patients discontinuing therapy due to side effects, which were all reversible.3 Methotrexate was reported to be one of the agents of choice for recurrent anterior uveitis, as it was preferentially concentrated in the aqueous humor.12 Galor et al reported 90 patients with inflammatory eye disease, including 21 patients with scleritis, 17 patients with intermediate uveitis, 43 patients with posterior/panuveitis, and 9 patients with other types, who were treated with methotrexate. They reported that the median time to treatment success was 6.5 months. The proportion of patients able to discontinue prednisone after 6 months of antimetabolite therapy was 6% in methotrexate groups.13
Methotrexate is also safe, effective, and the most commonly used immunosuppressive agent in children with JIA-associated uveitis and chronic anterior or intermediate uveitis. In the literature on methotrexate for anterior uveitis associated with the seronegative spondyloarthropathies and JIA, methotrexate permitted adequate immunosuppression and was steroid sparing while maintaining disease control.14 In a large series of 465 patients with ocular sarcoidosis, including 365 patients treated with methotrexate, most cases respond well to IMT. This drug was both efficient and well tolerated; as 281 (77%) patients continued on methotrexate therapy and only 14 (3.8%) had to discontinue it due to drug toxicity.15
Recently, intravitreal methotrexate has shown success in eyes with uveitic cystoid macular edema (CME).16,17 For the treatment of 15 eyes with uveitic CME, statistically significant visual improvement of 4 and 4.5 lines occurred at 3 and 6 months with no statistically significant difference in vision when compared to previous treatment with corticosteroids or intravitreal triamcinolone acetonide. Relapse occurred at a median of 4 months, but reinjection had similar efficacy.18 Following intravitreal corticosteroids, methotrexate is the most widely used intravitreal immunosuppressive agent. The dose administrated is 400 μg/0.1 mL, being effective in different inflammatory ocular disorders as well as in masquerade syndromes due to intraocular lymphoma.
Tacrolimus
Tacrolimus is a macrolide antibiotic that was isolated from the fermentation broth of Streptomyces tsukubaensis. Tacrolimus is capable of inhibiting humoral and cellular immunity and alloantigen-driven proliferation across strong histocompatibility barriers.19
In 1994, a multicenter open clinical trial in Japan conducted on 16 patients with noninfectious uveitis studied the clinical effects of tacrolimus. The study included eight patients with Behcet’s disease, five patients with VKH, one patient with sympathetic ophthalmia, one patient with retinal vasculitis, and one patient with sarcoidosis. Results demonstrated that tacrolimus was effective in treating noninfectious uveitis but emphasized the necessity to closely monitor patients for the occurrence of adverse effects.20 Tacrolimus also showed efficacy and an excellent cardiovascular risk profile when treating 62 patients with noninfectious uveitis (4.8% anterior uveitis, 25.8% intermediate uveitis, 37.1% posterior uveitis, and 32.3% panuveitis) for 4 consecutive years.21 Another publication evaluated the efficacy of tacrolimus as monotherapy versus tacrolimus in combination with systemic corticosteroid therapies to achieve remission of inflammation in noninfectious posterior uveitis. This study showed that corticosteroid therapy can be reduced and/or withdrawn in patients treated with tacrolimus who are able to achieve control of posterior segment intraocular inflammation.22
Tacrolimus therapy showed similar efficacy and a more favorable safety profile compared to cyclosporine regarding control of posterior segment intraocular inflammation.23 Furthermore, the use of topical tacrolimus ointment is effective in controlling refractory inflammatory ocular surface disease and can reduce the need for steroid use while reducing inflammation recurrence.24
Infliximab
Infliximab, a chimeric monoclonal antibody that binds both circulating and membrane-bound TNF-α, has been reported to be effective for the treatment of uveitis associated with multiple uveitic diseases, such as human leukocyte antigen (HLA)-B27-related anterior uveitis, pars planitis, VKH, birdshot chorioretinopathy, recalcitrant uveitic CME, multifocal choroiditis, sympathetic ophthalmia, serpiginous choroidopathy, and idiopathic uveitis.25–27
In addition, infliximab has been described to be a rapid and very effective therapy for the treatment of ABD-related panuveitis and retinal vasculitis.28,29 Sfikakis et al reported in a prospective study of 25 patients with ABD-related uveitis that >90% of patients demonstrated the resolution of vitritis, CME, retinitis, and retinal vasculitis within 4 weeks after initiating infliximab therapy and benefit often occurred in under 1 week.29 It was shown to be more effective than conventional immunotherapy for ABD-related retinal vasculitis.30
Infliximab also showed efficacy for JIA uveitis, with the majority of patients experiencing rapid control of uveitis after the second infusion.31,32 However, multiple case reports and series suggest that TNF-α inhibitors may cause sarcoidosis-like conditions. TNF inhibitors analogously are effective for psoriasis, but they have also been reported to cause psoriasiform skin disease and developing drug-induced lupus.33,34 Furthermore, TNF-α inhibitors should be used with caution in patients with serpiginous choroidopathy despite previously negative QuantiFERON test.35
Interferon
INF-α and INF-β are cytokines that have an important role in treating severe sight-threatening uveitis. INF-α, which is naturally secreted in response to viral infection, is categorized into INF-α 2a (Roferon-A®) and INF-α2b (Intron-A®). INF therapy can cause severe adverse effects including sarcoidosis with or without uveitis.36,37 Thus, this therapy is not recommended for the treatment of sarcoid-related uveitis.
INF-α 2a and INF-α 2b have been used for the treatment of posterior uveitis.38 INF-α 2a has been studied mostly in patients with ABD.39,40 It has also been reported to treat sympathetic ophthalmia, VKH, birdshot retinochoroidopathy, intermediate uveitis, and idiopathic panuveitis.39,41 In a prospective study on patients with sight-threatening uveitis, subcutaneous daily use of human INF-α 2b showed a favorable response in 83% of the patients.38 INF-α 2b has been found to be an effective option in the treatment of refractory CME secondary to uveitis as well.42 Finally, INF-β has been successfully used in treating intermediate uveitis associated with multiple sclerosis, choroiditis, and choroidal neovascularization in chronic recurrent punctate inner choroidopathy.43,44
Rituximab
Rituximab (Rituxan®; Genentech, Inc., South San Francisco, CA, USA) is a murine-human chimeric monoclonal antibody against CD20 molecule that is expressed on B lymphocytes. It is FDA approved for both malignant and nonmalignant B-cell-dependent diseases. It has been used in the treatment of lymphoma, leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.45 Side effects of rituximab include infusion reaction, infection especially in combination with other immunosuppressive medications, and progressive multifocal leukoencephalopathy.
Current information about the use of rituximab in ophthalmology is limited. Rituximab has been used successfully in the treatment of JIA-associated uveitis, particularly in oligoarthritic (rather than polyarthritic) JIA.46,47 In concurrence, Miserocchi et al observed long-term remission of uveitis in patients with JIA who were treated with rituximab.45 The efficacy of rituximab in refractory ocular involvement of systemic lupus erythematosus,48 ABD,49 birdshot retinochoroidopathy,50 and type II essential cryoglobulinemia refractory to steroid treated with plasmapheresis has also been shown, although ABD is known to be a predominantly T-cell-mediated disease.51
Tocilizumab
Tocilizumab (Actemra®; Genentech, Inc.) is a monoclonal antibody against interleukin (IL)-6 receptors. It has been approved for the treatment of moderate-to-severe rheumatoid arthritis and polyarticular and systemic JIA refractory to other biologic response modifiers.
Tocilizumab has been successfully used in refractory JIA-associated uveitis, birdshot retinochoroidopathy, ABD, and refractory idiopathic uveitis.52–57 Hirano et al described a case of severe ABD-associated posterior uveitis resistant to conventional immunosuppression and infliximab infusions who experienced complete remission after tocilizumab monotherapy.58 Tocilizumab was also used for a case of multicentric Castleman disease with the resolution of uveitis and perivascular leakage on fluorescein angiography at 3 months after treatment.59 The efficacy of tocilizumab in noninfectious intermediate, posterior, or panuveitis and in JIA-associated uveitis is currently being examined in two separate ongoing phase I/II clinical trials.60 Adán et al demonstrated the effectiveness of tocilizumab in treating uveitic CME refractory to conventional chemotherapy.55 Tocilizumab was also reported to improve visual acuity and decrease in macular thickness in optical coherence tomography in birdshot retinochoroidopathy, JIA, and idiopathic panuveitis-associated CME.56,57
Although tocilizumab therapy is employed to improve rheumatologic symptoms, there are two case reports in regard to possible paradoxical inflammatory responses in patients treated with tocilizumab. The first patient (HLA-B27-positive ankylosing spondylitis) developed uveitis for the first time after treatment, and the second patient (rheumatoid arthritis) developed peripheral ulcerative keratitis for the first time 11 months after tocilizumab treatment.61
Anakinra
Anakinra (Kineret®; Swedish Orphan Biovitrum AB, Stockholm, Sweden) is a short-acting recombinant naturally occurring human IL-1 receptor antagonist, which blocks IL-1α and IL-β binding to the IL-1 receptor. In a case series of nine adult patients with ABD, three of four patients with recurrent uveitis showed a complete resolution of intraocular inflammation. Patient inflammation relapsed in an average period of 24 weeks. Side effects were observed in three patients that included itchy rash at the site of injection and was controlled with topical steroids.62 There are currently three case reports demonstrating the efficacy of anakinra in infantile posterior uveitis63,64 and one report showing efficacy in a patient having childhood uveitis associated with Blau syndrome.65
Canakinumab
Canakinumab (Ilaris®; Novartis Pharmaceuticals Corporation, Basel, Switzerland) is a human immunoglobulin G1 anti-IL-1β monoclonal antibody, which is indicated for systemic JIA and cryopyrin-associated periodic syndrome in adults and children aged ≥4 years. Its mode of action is based on the neutralization of 1β signaling, resulting in the suppression of inflammation in patients with disorders of autoimmune origin. Reports from clinical trials suggest that canakinumab is well tolerated in most patients, and no serious adverse effects have been reported. It is administered subcutaneously or intravenously every 4–8 weeks. Its effectiveness was demonstrated in a refractory case of Behcet’s syndrome-associated uveitis with a 150-mg single infusion.
Vitale et al reported a case series of three patients (two of three patients had ocular involvement, one anterior uveitis, and one panuveitis) with Behcet’s disease refractory to steroid and multiple immunosuppressive therapies, who responded to 150 mg of subcutaneous canakinumab monotherapy every 6 weeks. One patient’s uveitis was controlled by canakinumab and the other patient achieved remission on anakinra and remained in remission on canakinumab after stopping anakinra due to side effects.66 Ugurlu et al described a case of a 16-year-old female with severe bilateral panuveitis with hypopyon and retinal vasculitis associated with ABD. The patient was intolerant to INF therapy and refractory to conventional immunosuppressive and several biologic agents, including infliximab, adalimumab, and anakinra. The inflammation resolved with improved visual acuity, which was sustained for at least 8 weeks after a single infusion of 150 mg canakinumab. However, there were no long-term outcomes presented.67 Simonini et al reported on the clinical response to canakinumab in a 4-year-old boy with sporadic nucleotide-binding oligomerization domain-containing protein 2-associated pediatric granulomatous arthritis and severe resistant panuveitis, macular edema, and retinal detachment. In this case, canakinumab was given 2 mg/kg each month, which led to rapid control of uveitis for at least 6 months.68
Alemtuzumab
Alemtuzumab (Campath®; Genzyme Corporation, Cambridge, MA, USA) is a human monoclonal antibody against CD52 protein on all types of lymphocytes. Alemtuzumab has not received FDA approval, thus it is not commercially available but is obtainable through a restricted distribution program by the manufacturer. Adverse effects of alemtuzumab include significant lymphopenia; secondary autoimmunity, specifically Graves’ disease; and idiopathic thrombocytopenic purpura.69
Isaacs et al described a case of a 36-year-old male who presented with severe panuveitis with choroiditis and retinal vasculitis resistant to immunosuppressive agents, including cyclophosphamide and intravenous immunoglobulin. The patient had significant systemic symptoms, presumably from chronic corticosteroid therapy. He was given intravenous alemtuzumab with brief improvement of ocular inflammation. However, the patient subsequently died with myocardial infarction.70 Dick et al reported a series of 10 patients with various ocular inflammatory diseases treated with intravenous alemtuzumab (four patients with retinal vasculitis, one with ABD-related uveitis, and one with sympathetic ophthalmia). The first five patients had clinical improvement, but the latter with sympathetic ophthalmia had stable inflammation without deterioration. Half of the patients flared up during follow-up period.71 There is a report of four cases of adult patients with Behcet’s disease-associated uveitis, who were treated with a single course, at the escalating regimen of 4, 10, 40, 40, and 40 mg intravenously daily, for 5 consecutive days, without concomitant immunosuppressive agents. They reported complete remission in two cases and partial remission in two other cases at 6-month follow-up. However, long-term remission was observed in only one patient.72
In conclusion, continuing evidence shows that IMT agents and biologics orphan drugs approved by the FDA for the treatment of systemic diseases are effective in treating refractory and recurrent uveitis. These treatments allow for reduction in steroid dose and preservation of visual function. However, due to the lack of evidence from large randomized controlled clinical trials and rarity and heterogeneity of uveitis, the use of these drugs still remains “off-label” for noninfectious uveitis. We hope that these restrictions and limitations will be further addressed in near future.
Our perspective
Uveitis constitutes a sight-threatening disease that should be managed according to the severity. Given our current understanding of the immune mechanisms of uveitis, IMT drugs can greatly reduce inflammation and minimize the use of topical and systemic steroids. There are a number of IMT options available that have improved outcomes of uveitic patients when used in specific situations. These IMT orphan drugs serve as the foundation to modern corticosteroid-sparing therapy.
Conventional IMT agents are the first steroid-sparing options for uveitic patients with refractive active inflammation and/or sight-threatening complications. Using IMTs in treating autoimmune uveitis follows one of the following scenarios: 1) as a steroid-sparing agent; 2) when corticosteroids fail to control the inflammation: persistent, recurrent, or progressing in the same eye or bilaterally; 3) when uveitis is associated with autoimmune systemic diseases, IMTs are used to control both; and 4) with frequent recurrences and/or the development of ocular or steroid-induced nonophthalmic complications.
Previous experience indicates that an alternative and/or combined IMT regimen is the best treatment choice in cases of worsening inflammation. In these worsening patients, a physician can either change or add other conventional IMT drugs and/or a biologic agent. Medications in the same group can be replaced with each other from more aggressive treatments, such as infusions to less aggressive ones such as subcutaneous injections. Moreover, the failure of a biologic response modifier in a group does not necessarily mean that similar agents in the same group will also be ineffective. Corticosteroids are usually modulated with IMT drugs according to the degree of intraocular inflammation and the possible occurrence of complications and/or relapses. In cases of ocular inflammation improvement, corticosteroids should be gradually tapered until final withdrawal, together with a subsequent reduction and/or withdrawal of the use of IMT drugs.
If remission is achieved, there is no consensus on when to stop IMT. In our experience, IMT maintenance (without corticosteroids) while the patient is in remission should be prolonged for a period of 2 years before considering tapering the medication, in order to achieve stable control of the disease and to reduce the probability of recurrence. The end point of IMT is also determined by the presence of the underlying systemic autoimmune disease that is being controlled by the same IMT, which often requires coordination between the ophthalmologist and the rheumatologist. Additional controlled trials are needed to assess long-term remission outcomes after IMT tapering and develop proper end points of IMT regimens.
Lack of randomized controlled clinical trials, rarity and heterogeneity of uveitis, and the high cost of these agents make using IMTs and biologics challenging in ophthalmology. Among the studies referenced in this study, most are small retrospective clinical trials, which have no standard treatment regimen, definition of success or remission, and little consensus on IMT end points. Nowadays, the number of IMT therapeutic options in uveitis has expanded vastly. It remains imperative for a clinician to diagnose the disease early and institute aggressive therapy. Furthermore, selection, administration, and monitoring of IMT sometimes need coordination with a rheumatologist and/or a chemotherapist to avoid complications.
There are many other biologic response modifiers that have been employed for the treatment of systemic autoimmune diseases and experimental autoimmune uveitis that are promising for the treatment of idiopathic noninfectious uveitis. The development of monoclonal antibodies that recognize two or more targets at the same time, and the development of a safe and effective local sustained-release device delivering biologic response modifiers may be promising approaches for treatment in the future.
As our understanding of autoimmune diseases expands, new targets and approaches to treatment are becoming available. We emphasize the need for the FDA to approve “on-label” use of current orphan IMT agents and biologics more quickly and to facilitate insurance coverage and expanded access to uveitis patients. Further assessment of current IMT agents and new IMT regimens is needed to provide evidence confirming the efficacy, optimal dose, regimen, maintenance, and end point for the treatment of uveitis.
Disclosure
C Stephen Foster discloses the following: Consultancies with Aldeyra Therapeutics (Lexington, MA), Baush & Lomb Surgical, Inc (Rancho Cucamonga, CA), Eyegate Pharma (Waltham, MA), Novartis (Cambridge, MA), pSivida (Watertown, MA), and Xoma (Berkeley, CA). Grants or grants pending with Alcon (Aliso Viejo, CA), Aldeyra Therapeutics (Lexington, MA), Bausch & Lomb (Bridgewater, NJ), Clearside Biomedical (Alpharetta, GA), Dompé pharmacetical (Milan, Italy), Eyegate Pharma (Waltham, MA), Mallinckrodt pharmaceuticals (Dublin, Ireland), Novartis Pharmaceuticals (Cambridge, MA), pSivida (Watertown, MA), Santen (Osaka, Japan). Payment for lectures including service on speaking bureaus: Alcon (Aliso Viejo, CA), Allergan (Dublin, Ireland). Stock or Stock Options: Eyegate Pharama (Waltham, MA).
There was no funding or support received for this manuscript. The authors report no other conflicts of interest in this work.
References
Barisani-Asenbauer T, Maca SM, Mejdoubi L, Emminger W, Machold K, Auer H. Uveitis–a rare disease often associated with systemic diseases and infections – a systematic review of 2619 patients. Orphanet J Rare Dis. 2012;7:57. | ||
Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140(3):509–516. | ||
Gangaputra S, Newcomb CW, Liesegang TL, et al. Systemic Immunosuppressive Therapy for Eye Diseases Cohort Study. Methotrexate for ocular inflammatory diseases. Ophthalmology. 2009;116(11):2188–2198. | ||
Levy RA, de Andrade FA, Foeldvari I. Cutting-edge issues in autoimmune uveitis. Clin Rev Allergy Immunol. 2011;41(2):214–223. | ||
Metzinger JL, Foster CS. ‘Approved for use in uveitis’: drug approval for an orphan disease. Expert Opin Orphan Drugs. 2015;3(7):799–807. | ||
Developing Products for Rare Diseases & Conditions. Designation an Orphan Product: Drug and Biological Products: Searchable Databases for Orphan Designated and or Approved Products. U.S. Food and Drug Administration; 2016. Available from: http://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm Accessed January 27, 2016. | ||
Gilman AG, Rall TW, Nies AS, Taylor P. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. New York, NY: Pergamon Press; 1990. | ||
Weinblatt ME, Coblyn JS, Fox DA, et al. Efficacy of low dose methotrexate in rheumatoid arthritis. N Engl J Med. 1985;312(13):818–822. | ||
Shah SS, Lowder CY, Schmitt MA, Wilke WS, Kosmorsky GS, Meisler DM. Low-dose methotrexate therapy for ocular inflammatory disease. Ophthalmology. 1992;99(9):1419–1423. | ||
Rossi S. Australian Medicines Handbook. Adelaide, Australia: The Australian Medicines Handbook Unit Trust; 2013. | ||
Joint Formulary Committee. British National Formulary (BNF) 65th ed. London, UK: Pharmaceutical Press; 2013. | ||
Lima BR, Nussenblatt RB, Sen HN. Pharmacogenetics of drugs used in the treatment of ocular inflammatory diseases. Expert Opin Drug Metab Toxicol. 2013;9(7):875–882. | ||
Galor A, Jabs DA, Leder HA, et al. Comparison of antimetabolite drugs as corticosteroid-sparing therapy for noninfectious ocular inflammation. Ophthalmology. 2008;115(10):1826–1832. | ||
Imrie FR, Dick AD. Nonsteroidal drugs for the treatment of noninfectious posterior and intermediate uveitis. Curr Opin Ophthalmol. 2007;18(3):212–219. | ||
Baughman RP, Lower EE, Ingledue R, Kaufman AH. Management of ocular sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2012;29(1):26–33. | ||
Larson T, Nussenblatt RB, Sen HN. Emerging drugs for uveitis. Expert Opin Emerg Drugs. 2011;16(2):309–322. | ||
Bach JH. The Mode of Action of Immunosuppressive Drugs. Amsterdam, the Netherlands: Elsevier; 1975. | ||
Taylor SR, Habot-Wilner Z, Pacheco P, Lightman SL. Intraocular methotrexate in the treatment of uveitis and uveitic cystoid macular edema. Ophthalmology. 2009;116(4):797–801. | ||
Kino T, Hatanaka H, Hashimoto M, et al. FK-506, a novel immunosuppressant isolated from a Streptomyces. I. Fermentation, isolation, and physico-chemical and biological characteristics. J Antibiot (Tokyo). 1987;40(9):1249–1255. | ||
Ishioka M, Ohno S, Nakamura S, et al. FK506 treatment of noninfectious uveitis. Am J Ophthalmol. 1994;118(6):723–729. | ||
Hogan AC, McAvoy CE, Dick AD, Lee RW. Long-term efficacy and tolerance of tacrolimus for the treatment of uveitis. Ophthalmology. 2007;114(5):1000–1006. | ||
Lee RW, Greenwood R, Taylor H, et al. A randomized trial of tacrolimus versus tacrolimus and prednisone for the maintenance of disease remission in noninfectious uveitis. Ophthalmology. 2012;119(6):1223–1230. | ||
Murphy CC, Greiner K, Plskova J, et al. Cyclosporine vs tacrolimus therapy for posterior and intermediate uveitis. Arch Ophthalmol. 2005;123(5):634–641. | ||
Lee YJ, Kim SW, Seo KY. Application for tacrolimus ointment in treating refractory inflammatory ocular surface diseases. Am J Ophthalmol. 2013;155(5):804–813. | ||
Lindstedt EW, Baarsma GS, Kuijpers RW, van Hagen PM. Anti-TNF-alpha therapy for sight threatening uveitis. Br J Ophthalmol. 2005;89(5):533–536. | ||
Suhler EB, Smith JR, Giles TR, et al. Infliximab therapy for refractory uveitis: 2-year results of a prospective trial. Arch Ophthalmol. 2009;127(6):819–822. | ||
Arida A, Fragiadaki K, Giavri E, Sfikakis PP. Anti-TNF agents for Behcet’s disease: analysis of published data on 369 patients. Semin Arthritis Rheum. 2011;41(1):61–70. | ||
Suhler EB, Smith JR, Wertheim MS, et al. A prospective trial of infliximab therapy for refractory uveitis: preliminary safety and efficacy outcomes. Arch Ophthalmol. 2005;123(7):903–912. | ||
Sfikakis PP, Kaklamanis PH, Elezoglou A, et al. Infliximab for recurrent, sight-threatening ocular inflammation in Adamantiades-Behcet disease. Ann Intern Med. 2004;140(5):404–406. | ||
Tabbara KF, Al-Hemidan AI. Infliximab effects compared to conventional therapy in the management of retinal vasculitis in Behcet disease. Am J Ophthalmol. 2008;146(6):845.e1–850.e1. | ||
Richards JC, Tay-Kearney ML, Murray K, Manners P. Infliximab for juvenile idiopathic arthritis-associated uveitis. Clin Experiment Ophthalmol. 2005;33(5):461–468. | ||
Tynjala P, Lindahl P, Honkanen V, Lahdenne P, Kotaniemi K. Infliximab and etanercept in the treatment of chronic uveitis associated with refractory juvenile idiopathic arthritis. Ann Rheum Dis. 2007;66(4):548–550. | ||
Clementine RR, Lyman J, Zakem J, Mallepalli J, Lindsey S, Quinet R. Tumor necrosis factor-alpha antagonist-induced sarcoidosis. J Clin Rheumatol. 2010;16(6):274–279. | ||
Olivier A, Gilson B, Lafontaine S, Pautot JX, Bindi P. Pulmonary and renal involvement in a TNFα antagonist drug-induced sarcoidosis. Rev Med Interne. 2012;33(5):e25–e27. | ||
Cordero-Coma M, Benito MF, Hernandez AM, Antolin SC, Ruiz JM. Serpiginous choroiditis. Ophthalmology. 2008;115(9):1633, 1633.e1–e2.36. | ||
Doycheva D, Deuter C, Stuebiger N, Zierhut M. Interferon-alpha-associated presumed ocular sarcoidosis. Graefes Arch Clin Exp Ophthalmol. 2009;247(5):675–680. | ||
Alazemi S, Campos MA. Interferon-induced sarcoidosis. Int J Clin Pract. 2006;60(2):201–211. | ||
Plskova J, Greiner K, Forrester JV. Interferon-alpha as an effective treatment for noninfectious posterior uveitis and panuveitis. Am J Ophthalmol. 2007;144(1):55–61. | ||
Plskova J, Greiner K, Muckersie E, Duncan L, Forrester JV. Interferon-alpha: a key factor in autoimmune disease? Invest Ophthalmol Vis Sci. 2006;47(9):3946–3950. | ||
Tugal-Tutkun I, Guney-Tefekli E, Urgancioglu M. Results of interferon-alfa therapy in patients with Behcet uveitis. Graefes Arch Clin Exp Ophthalmol. 2006;244(12):1692–1695. | ||
Bodaghi B, Gendron G, Wechsler B, et al. Efficacy of interferon alpha in the treatment of refractory and sight threatening uveitis: a retrospective monocentric study of 45 patients. Br J Ophthalmol. 2007;91(3):335–339. | ||
Bulter NJ, Suhler EB, Rosenbaum JT. Interferon alpha 2b in the treatment of cystoid macular edema. Ocul Immunol Inflamm. 2012;20(2):86–90. | ||
Becker MD, Heiligenhaus A, Hudde T, et al. Interferon as a treatment for uveitis associated with multiple sclerosis. Br J Ophthalmol. 2005;89(10):1254–1257. | ||
Cirino AC, Mathura JR Jr, Jampol LM. Resolution of activity (choroiditis and choroidal neovascularization) of chronic recurrent punctate inner choroidopathy after treatment with interferon B-1A. Retina. 2006;26(9):1091–1092. | ||
Miserocchi E, Modorati G, Berchicci L, Pontikaki I, Meroni P, Gerloni V. Long-term treatment with rituximab in severe juvenile idiopathic arthritis-associated uveitis. Br J Ophthalmol. 2016;100(6):782–786. | ||
Miserocchi E, Pontikaki I, Modorati G, Bandello F, Meroni PL, Gerloni V. Rituximab for uveitis. Ophthalmology. 2011;118(1):223–224. | ||
Heiligenhaus A, Miserocchi E, Heinz C, Gerloni V, Kotaniemi K. Treatment of severe uveitis associated with juvenile idiopathic arthritis with anti-CD20 monoclonal antibody (rituximab). Rheumatology (Oxford). 2011;50(8):1390–1394. | ||
Hickman RA, Denniston AK, Yee CS, Toescu V, Murray PI, Gordon C. Bilateral retinal vasculitis in a patient with systemic lupus erythematodes and its remission with rituximab therapy. Lupus. 2010;19(3):327–329. | ||
Davatchi F, Shams H, Rezaipoor M, et al. Rituximab in intractable ocular lesions of Behcet’s disease; randomized single-blind control study (pilot study). Int J Rheum Dis. 2010;13(3):246–252. | ||
Tomkins-Netzer O, Taylor SR, Lightman S. Can rituximab induce long-term disease remission in patients with intra-ocular non-infectious inflammation? Ophthalmologica. 2013;230(3):109–115. | ||
Nicholson L, Sobrin L. Anterior uveitis secondary to type II essential cryoglobulinemia. J Ophthalmic Inflamm Infect. 2013;3(1):56. | ||
Tappeiner C, Heinz C, Ganser G, Heiligenhaus A. Is tocilizumab an effective option for treatment of refractory uveitis associated with juvenile idiopathic arthritis? J Rheumatol. 2012;39(6):1294–1295. | ||
Tsang AC, Roth J, Gottlieb C. Tocilizumab for severe chronic anterior uveitis associated with juvenile idiopathic arthritis in a pediatric patient. Ocul Immunol Inflamm. 2014;22(2):155–157. | ||
Mesquida M, Leszczynska A, Llorenc V, Adán A. Interleukin-6 blockade in ocular inflammatory diseases. Clin Exp Immunol. 2014;176(3):301–309. | ||
Adán A, Mesquida M, Llorenc V, et al. Tocilizumab treatment for refractory uveitis-related cystoid macular edema. Graefes Arch Clin Exp Ophthalmol. 2013;251(11):2627–2632. | ||
Mesquida M, Molins B, Llorenc V, Sainz de la Maza M, Adán A. Long-term effects of tocilizumab therapy for refractory uveitis-related macular edema. Ophthalmology. 2014;121(12):2380–2386. | ||
Papo M, Bielefeld P, Vallet H, et al. Tocilizumab in severe and refractory non-infectious uveitis. Clin Exp Rheumatol. 2014;32(4 Suppl 84):875–879. | ||
Hirano T, Ohguro N, Hohki S, et al. A case of Behcet’s disease treated with a humanized anti-interleukin-6 receptor antibody, tocilizumab. Mod Rheumatol. 2012;22(2):298–302. | ||
Oshitari T, Kajita F, Tobe A, et al. Refractory uveitis in patient with castleman disease successfully treated with tocilizumab. Case Rep Ophthalmol Med. 2012;2012:968180. | ||
Lin P. Targeting interleukin-6 for noninfectious uveitis. Clin Ophthalmol. 2015;9:1697–1702. | ||
Wendling D, Dernis E, Prati C, Frisch E, Delbosc B. Onset of inflammatory eye disease under tocilizumab treatment for rheumatologic conditions: a paradoxical effect? J Rheumatol. 2011;38(10):2284. | ||
Cantarini L, Vitale A, Scalini P, et al. Anakinra treatment in drug-resistant Behcet’s disease: a case series. Clin Rheumatol. 2015;34(7):1293–1301. | ||
Teoh SC, Sharma S, Hogan A, Lee R, Ramanan AV, Dick AD. Tailoring biological treatment: anakinra treatment of posterior uveitis associated with the CINCA syndrome. Br J Ophthalmol. 2007;91(2):263–264. | ||
Alexander T, Klotz O, Feist E, Rüther K, Burmester GR, Pleyer U. Successful treatment of acute visual loss in Muckle-Wells syndrome with interleukin 1 receptor antagonist. Ann Rheum Dis. 2005;64(8):1245–1246. | ||
Aróstegui JI, Arna C, Merino R, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin 1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56(11):3805–3813. | ||
Vitale A, Rigante D, Caso F, et al. Inhibition of interleukin-1 by canakinumab as a successful mono-drug strategy for the treatment of refractory Behçet’s disease: a case series. Dermatology. 2014;228(3):211–214. | ||
Ugurlu S, Ucar D, Seyahi E, Hatemi G, Yurdakul S. Canakinumab in a patient with juvenile Behcet’s syndrome with refractory eye disease. Ann Rheum Dis. 2012;71(9):1589–1591. | ||
Simonini G, Xu Z, Caputo R, et al. Clinical and transcriptional response to the long-acting interleukin-1 blocker canakinumab in Blau syndrome-related uveitis. Arthritis Rheum. 2013;65(2):513–518. | ||
Buggage RR, Levy-Clarke G, Sen HN, et al. A double-masked, randomized study to investigate the safety and efficacy of daclizumab to treat the ocular complications related to Behcet’s disease. Ocul Immunol Inflamm. 2007;15(2):63–70. | ||
Isaacs JD, Hale G, Waldmann H, et al. Monoclonal antibody therapy of chronic intraocular inflammation using Campath-1H. Br J Ophthalmol. 1995;79(11):1054–1055. | ||
Dick AD, Meyer P, James T, et al. Campath-1H therapy in refractory ocular inflammatory disease. Br J Ophthalmol. 2000;84(1):107–109. | ||
Lockwood CM, Hale G, Waldmann H, Jayne DR. Remission induction in Behcet’s disease following lymphocyte depletion by the anti-CD52 antibody CAMPATH-1H. Rheumatology (Oxford). 2003;42(12):1539–1544. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.