")
Back to Journals » OncoTargets and Therapy » Volume 11
A retrospective examination of the US Food and Drug Administration’s clinical pharmacology reviews of oncology biologics for potential use of therapeutic drug monitoring
Authors Fleisher B, Ait-Oudhia S
Received 2 October 2017
Accepted for publication 3 December 2017
Published 27 December 2017 Volume 2018:11 Pages 113—121
DOI https://doi.org/10.2147/OTT.S153056
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Brett Fleisher, Sihem Ait-Oudhia
Center for Pharmacometrics and Systems Pharmacology, Department of Pharmaceutics, College of Pharmacy, University of Florida, Orlando, FL, USA
Background: Biologics have gained traction for use in oncology, but have demonstrate clinical variability for efficacy and safety. Therapeutic drug monitoring (TDM) can benefit patients’ outcomes from a biologic therapy when the latter has a defined therapeutic window. A clinically relevant therapeutic window may exist for biologics with established exposure-response (E–R) relationships for efficacy and/or safety and a documented maximum tolerated dose (MTD). Additionally, the inter-individual variability (IIV) on the clearance (CL) parameter could determine risks for patients falling outside the proposed therapeutic window.
Materials and methods: The US Food and Drug Administration (FDA)-approved oncology biologics between 2005–2016 were reviewed via FDA “Purple Book” (FDA-repository for licensed biologics). Data were extracted from biologics’ pharmacokinetic models available on the clinical pharmacology reviews published on the FDA-Approved Drug Products website. Evaluated features for biologics with established E–R relationships for efficacy and/or safety and MTD include an IIV for the CL and various other covariates including demographic factors, disease factors, blood chemistry, or immunogenicity.
Results: Five therapies were identified with documented E–R relationships for both efficacy and safety including, Yervoy®(ipilimumab), Zaltrap® (ziv-aflibercept), Portrazza® (necitumumab), Adcetris® (brentuximab-vedotin), and Blincyto® (blinatumomab). The corresponding IIV on CL were: 34%, 33%, 29%, 47%, and 97%, respectively. Among the five therapies, only three had defined MTD including, brentuximab-vedotin, necitumumab, and blinatumomab.
Conclusion: Of the medications examined, blinatumomab was identified as the anticancer drug with the most available information for the establishment of TDM, and hence, may benefit through the use of TDM to optimize effectiveness and minimize patients’ toxicity . The approach used here may provide a generalizable framework to retrospectively identify anticancer biologics with high IIV that may benefit from TDM to improve patients’ clinical outcome.
Keywords: therapeutic drug monitoring, oncology biologics, blinatumomab
Introduction
Biologics are complex molecules produced in living organisms. Over the last few decades, they have gained lot of attention in oncology medicine due to the greater understanding of the molecular biology of biomarkers associated to cancer progression. The US Food and Drug Administration (FDA)-approved biologics used in oncology fall into four categories: 1) monoclonal antibodies (mAbs) which include Vectibix® (panitumumab [Amgen, Thousand Oaks, CA, USA]), Arzerra® (ofatumumab [GlaxoSmithKline, Brentford, UK]), Yervoy® (ipilimumab [Bristol Myers Squibb, New York, NY, USA]), Perjeta® (pertuzumab [Genentech, San Francisco, CA, USA]), Gazyva® (obinutuzumab [Genentech]), Cyramza® (ramucirumab [Eli Lilly, Indianapolis, IN, USA]), Keytruda® (pembrolizumab [Merck, Kenilworth, NJ, USA]), Opdivo® (nivolumab [Bristol Myers Squibb]), Unituxin® (dinutuximab [United Therapeutics, Silver Springs, MD, USA]), Portrazza® (necitumumab [Eli Lilly]), and Tecentriq® (atezolizumab [Genentech]); 2) antibody–drug conjugates (ADCs) including Adcetris® (brentuximab vedotin [Seattle Genetics, Bothell, WA, USA]) and Kadcyla® (ado-trastuzumab emtansine [Genentech]); 3) fusion protein Zaltrap® (ziv-aflibercept [Sanofi Aventis, Paris, France]); and 4) bispecific T-cell engager (BiTE®) Blincyto (blinatumomab [Amgen]). These biologics have distinct mechanisms of action toward the cancer cell. For example, mAbs, large glycoproteins produced by B-cells exposed to a specific antigen,1 act by either inducing the immune system to attack tumor cells or improving tumor recognition by the immune system. The ADCs ado-trastuzumab emtansine and brentuximab vedotin are antibodies conjugated to potent chemotherapeutic agents (payloads [PLs]) through linkers which allow for the PL to be specifically delivered to the cancer cell bearing the specific antigen.2 The fusion protein, ziv-aflibercept, is made from a fusion gene of two dissimilar genes for inhibiting binding and activation of cognate receptors responsible for tumor survival.3 The novel BiTE blinatumomab transiently links CD3-positive T cells to CD19-positive B cells, resulting in induction of T-cell–mediated serial lysis of acute lymphocytic B-cells and concomitant T-cell proliferation.4
Biologics have great promise in oncology because of their adequate benefit to risk profile. Yet, biologics are not without variability in patient response5 or class-related adverse drug reactions (ADRs).6 Therapeutic drug monitoring (TDM) is a method of controlling for both inadequate response and ADRs through measuring plasma/blood trough concentrations during treatment and making necessary dosage adjustments for plasma/blood drug concentration to optimize dosing.7 Development and proactive utilization of TDM has demonstrated improved clinical application for several drugs8 including the biologic, infliximab9,10 used for the treatment of inflammatory bowel disease (IBS). Plasma trough levels of infliximab are positively associated with IBS treatment efficacy11 in addition to an increase in ADRs such as life-threatening infusion reactions.12 Combined with the significant inter-individual variability (IIV) of the pharmacokinetics (PK; how the body absorbs, distributes, metabolizes, and excretes drugs13) of infliximab (leading to varied patient response), TDM for infliximab was poised for successful clinical implementation.9 By using the profile of infliximab, characteristics may prove useful in determining which biologics may benefit from developing TDM for that agent.
Drug characteristics are traditionally examined in the context of PK and pharmacodynamics (PD; the effect of the drug on the body). The interplay between PD, PK, and clinical response can be assessed through exposure–response (E–R) relationships14 which examine the association between the drug’s serum concentrations or total exposure through the area under the curve (AUC) and clinical outcomes (efficacy and/or safety).15 Some oncology mAbs, such as alemtuzumab, demonstrate a positive E–R relationship for efficacy, meaning that an increase in therapeutic protein AUC correlates to a higher probability in influencing cancer response.16 A drug with a positive E–R relationship for efficacy warrants a larger exposure of the drug compared to one without an established E–R relationship, which is especially true with anti-oncologic medication given the inherent high risk associated with cancer. In addition to effectiveness, toxicity must also be considered because many (but not all) biologics have documented a maximum tolerated dose (MTD; the highest clinically tested dose with acceptable toxicity).17 If a drug has a positive E–R relationship of safety (which correlates an increase in AUC with an increased ADR risk) and an established MTD, it is plausible to assume that the drug will also have an upper limit to an acceptable plasma/blood exposure level to the biologic. In this sense, biologics with a positive E–R relationship of safety and efficacy with an established MTD have an optimal plasma level which balances cancer killing with acceptable toxicity.
PK is defined by clearance (CL), volume of distribution in the central compartment (Vc), and volume of distribution in the peripheral compartments (Vp). In a previous review, Oude Munnick et al10 made the argument that, of these three variables, CL appears to play the most significant role in mAb serum concentrations at the end of the dosing interval. Biologics require a minimum effective concentration to demonstrate adequate efficacy, and variations in CL can affect plasma drug levels, thereby impacting the effectiveness of these agents. If a therapeutic window exists for an oncologic biologic, patients with rapid CL are at risk of a suboptimal tumor control, while a low CL increases the risk of plasma levels putting patients at greater risk in experiencing ADRs. Conceivably, biologics with a positive E–R relationship for safety and efficacy, in addition to a large IIV of CL have the greatest chance of experiencing fluctuations in serum/plasma concentration and are at risk of falling outside of an optimal plasma concentration of the drug.
In assessing which biologics have the most potential for TDM, three assumptions were made in choosing the parameters set forth in this retrospective examination of FDA reviews: 1) the presence of a positive E–R relationship of efficacy indicates a correlation between drug effectiveness and AUC, therefore warranting a higher dose; 2) a positive E–R of safety with an established MTD indicates a correlation between AUC and ADR risk with a clinically set dose-limiting toxicity; and 3) adjusted IIV of CL indicates increased risk in AUC fluctuations resulting in increased risk of subtherapeutic plasma levels or increased risk of ADRs. Factors that may adjust for confounding in PK analysis (demographic factors, disease factors, blood chemistry, and immunogenicity)10 were also examined. By using these assumptions, this study sought biologics with an optimal therapeutic window of activity as evidence by the presence of positive E–R relationships of safety/efficacy and an established MTD. Furthermore, risk of deviating from the window was examined by utilizing adjusted IIV of CL. By examining recently approved biologics that meet these criteria, this study hopes to find biologics for oncology, which may benefit most as well as quickly address the barriers in TDM development and implementation.
Materials and methods
The biologic therapies for oncology approved between 2005 and 2016 were reviewed according to the FDA’s “Purple Book” (the list of biologic products and biosimilars licensed by the FDA).18 Data on these drugs were extracted from clinical pharmacology and biopharmaceutics reviews published on the FDA’s Approved Drug Products website.19 The evaluated features of each drug include whether the drug had established E–R relationships for efficacy and safety, a defined minimum effective concentration, and MTD. Adjusted IIV of PK data was pulled from population PK (popPK) analyses published in the FDA reviews. Factors influencing IIV, such as demographic factors (weight, body surface area, age, renal function, hepatic function, sex, race), disease factors (Eastern Cooperative Oncology Group, tumor burden, target expression, target in circulation), blood chemistry (albumin, alkaline phosphatase), and immunogenicity (presence of anti-drug antibody),10 were also included.
Results
As per the FDA’s Purple Book, 15 oncology biologics were approved between 2005 and 2016. These drugs are summarized in Table 1 and include panitumumab, ofatumumab, ipilimumab, brentuximab vedotin, pertuzumab, ziv-aflibercept, ado-trastuzumab emtansine, obinutuzumab, ramucirumab, pembrolizumab, blinatumomab, givolumab, dinutuximab, necitumumab, and atezolizumab.3,18,20–33 Data summarized in Table 1 include the number of subjects (n) in the final popPK analyses published in the FDA’s Clinical Pharmacology Review, the presence/absence of E–R relationship for efficacy or for safety, unexplained IIV for PK parameters (CL, Vc, and Vp), and covariates of PK IIV. All therapies had defined minimum therapeutic concentrations. Five therapies had documented E–R relationships for both efficacy and safety: ipilimumab, brentuximab vedotin, ziv-aflibercept, blinatumomab, and necitumumab. Of the five with efficacy and safety E–R relationships, three had defined MTD: brentuximab vedotin, necitumumab, and blinatumomab. Blinatumomab had the greatest adjusted IIV for the CL at 97%, while the adjusted IIV in CL for brentuximab vedotin, necitumumab, and ado-trastuzumab emtansine was 47%, 29%, and 19%, respectively (Figure 1).
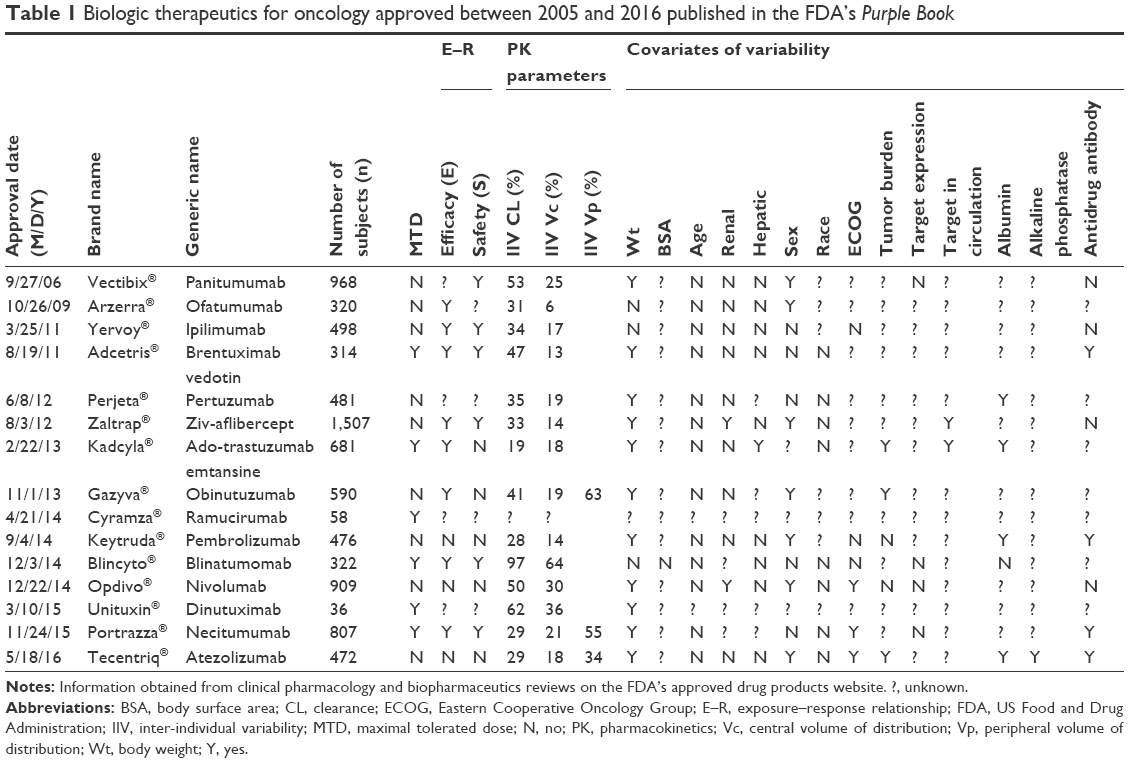 | Table 1 Biologic therapeutics for oncology approved between 2005 and 2016 published in the FDA’s Purple Book |
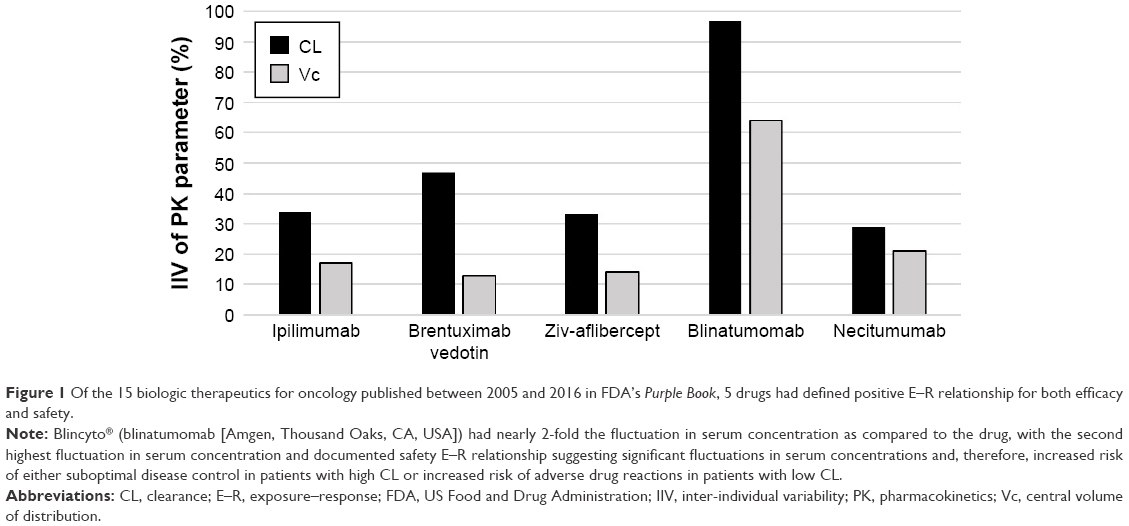 | Figure 1 Of the 15 biologic therapeutics for oncology published between 2005 and 2016 in FDA’s Purple Book, 5 drugs had defined positive E–R relationship for both efficacy and safety. |
Discussion
This retrospective examination of the FDA clinical pharmacology reviews of oncology biologics assumed that proposed oncology biologics that may benefit from TDM optimization include agents with an established E–R relationship for efficacy and safety, an established MTD, and IIV of CL. These criteria were inspired from a review published by Oude Munnick et al,10 which explored the rational for utilizing TDM in mAb therapy, focusing on characteristics of infliximab that influence plasma levels at the end of dosing intervals. Oude Munnick et al concluded that biologics which may be improved through TDM include Campath® (alemtuzumab), obinutuzumab, Rituxan® (rituximab), Erbitux® (cetuximab), Herceptin® (trastuzumab), and ado-trastuzumab emtansine. Of these agents, alemtuzumab, rituximab, cetuximab, and trastuzumab were not included in our study because they were licensed before the arbitrary cutoff year we set for the publication, that is, 2005. The dates of licensure for these products were 05/07/01, 11/26/97, 02/12/04, and 09/25/98, respectively.
Although the criteria published in the review by Oude Munnick et al were used to guide the examined biologics in this study, our study disagrees with the chosen biologics to focus on. Two biologics examined by both this study and the review by Oude Munnick et al10 include ado-trastuzumab emtansine and obinutuzumab. Data on ado-trastuzumab emtansine were collected from a pooled analysis of five clinical trials, and after E–R for efficacy was conducted, a correlation was found between the trough concentrations and median survival.34 However, the E–R relationship for safety revealed an inverse trend for Grade 3, or worse hepatotoxicity and no significant trend for thrombocytopenia.30 Similarly, popPK data of obinutuzumab from four clinical trials found a positive association between AUC and clinical response (change in tumor size, greater overall response, and prolonged progression-free survival), but E–R analysis for safety found no association between drug exposure and ADRs.35 Oude Munnick et al makes the valid argument that the presence of a positive E–R relationship for efficacy without an E–R for safety mitigates toxicity concerns during dose intensification for ado-trastuzumab emtansine and obinutuzumab.10 However, if one is to focus on agents with a positive E–R relationship for efficacy without a positive E–R relationship for safety, it could be argued that the drug should be given at as high a dose as possible since greater cancer killing effects are seen at higher doses regardless of the ADR risk profile. The lack of a defined E–R relationship calls into question the presence of a therapeutic window for these agents and the need for TDM.
E–R for safety was included in our examination because TDM has been classically reserved to monitor drugs with a narrow therapeutic window which includes monitoring drugs that can cause significant ADRs with increasing concentrations.7 Because of this observation, our study focused on biologics that not only demonstrated increased efficacy with increasing plasma exposure but also increased risk of ADRs. As mentioned in the “Introduction” section, the inclusion of both an E–R for efficacy as well as safety suggests the presence of an optimized window in which tumor killing efficacy can be balanced with ADRs. Five therapies approved between 2005 and 2016 had documented E–R relationships for both efficacy and safety (ipilimumab, ziv-aflibercept, necitumumab, brentuximab vedotin, and blinatumomab).
The first two biologics in our list include ipilimumab and ziv-aflibercept. The steady-state concentration of ipilimumab, a mAb approved for advanced melanoma, is associated with improved overall survival as well as increase in immune-related adverse events (including rash, diarrhea, colitis).24,36 Similarly, steady-state exposure of ziv-aflibercept, a fusion protein approved for metastatic oxaliplatin-resistant colorectal cancer, was related to overall survival and incidence of hypertension and hemorrhage. Biologics have a relatively large therapeutic window,10 and an indicator that could hint at clinical cutoff to upper level of therapeutic window is helpful in determining if an upper level to plasma level of biologic exists for these agents. MTD (an indicator for upper level of acceptable toxicity during Phase I dose-escalation studies for selecting a Phase II dose)17 was assumed to indicate the clinical cutoff to upper level of acceptable dosing and upper limit to tolerable plasma levels. Neither ipilimumab nor ziv-aflibercept reached MTD in Phase I dose-escalation studies. Although the exact reason why these agents failed to meet MTD was unclear, it has been postulated that these agents deplete key immunological targets responsible for the cell killing, thereby inadvertently changing the apparent acceptable toxicity profile.37,38 For instance, ipilimumab targets CTLA-4 antibody on cytotoxic T-cells, eliciting an immune-based attack of melanoma,39 depleting T-cells, and potentially altering immune-related adverse event risk in the process. Since neither ipilimumab nor ziv-aflibercept has a defined MTD, it is unclear whether the lack of MTD indicates a lack of a clinically reasonable upper limit to the therapeutic window or whether the lack of MTD is due to PD mechanisms.40
Necitumumab, brentuximab vedotin, and blinatumomab did meet the established MTDs during Phase I clinical trials, suggesting that an upper limit to therapeutic window can be reached during clinical dosing. Necitumumab is an IgG1 mAb approved for locally advanced or metastatic squamous non-small cell lung cancer in combination with gemcitabine and cisplatin with an MTD at 800 mg. After E–R analysis was conducted, plasma levels were found to be related to both overall survival as well as hypomagnesemia.28 Brentuximab vedotin, an ADC comprising a CD30-targeted mAb linked to an antimitotic PL, monomethyl auristatin E, was approved for use in both treatment of Hodgkin’s lymphoma and anaplastic large cell lymphoma with a defined MTD at 1.8 mg/kg.41 Efficacy (overall response rate) and safety (grade 2+ peripheral neuropathy and grade 3/4 neutropenia) were found to be related to an increase in average steady-state ADC trough levels.20 Blinatumomab is a novel BiTE approved for chromosome-negative (Ph-) acute lymphoblastic leukemia (ALL) in adult patients. Blinatumomab has a defined MTD at 60 μg/m2/day, a positive E-R relationship for both efficacy (CR/CRh; complete response with partial hematologic recovery) and safety (encephalopathy, aphasia, seizures, and cytokine release syndrome).21,42
Marked PK variability is also a hallmark for drugs that benefit from TDM;7 so, the fundamental aspects of PK (absorption, distribution, metabolism, and elimination) must also be considered.10 Drug absorption can be affected by the administration method used for the drug, but the five biologics with proposed therapeutic window are administered via intravenous administration, which assumes 100% absorption of the drug.43 Drug distribution is presumed to play a minimal role in variability in biologics because the large size of the molecules limits diffusion from the blood to peripheral compartments.43 Hepatic metabolism is not expected to play as vital a role with biologics as with other oncology drugs because, aside from brentuximab vedotin, which has a monomethyl auristatin E PL that becomes hepatically metabolized,20 biologic agents are primarily metabolized via proteolytic degradation into amino acids.43 Elimination appears to play the biggest role in PK processes because of how biologic agents are cleared from the body. mAbs bind to neonatal Fc-receptor (FcRn) found on the phagocytic cells responsible for metabolizing the biologic agents from the body. FcRn acts as a salvage receptor, delaying phagocytic degradation and prolonging the circulation time through FcRn-mediated recycling.44 Of the PK covariates, CL appears to play the most significant role in serum concentrations at the end of dosing intervals.10 Mechanisms of elimination and the role of CL are most apparent in blinatumomab compared to the other therapeutic proteins. Blinatumomab lacks the FcRn, and protein degradation is not delayed through the salvage pathway.4 While most biologics have long half-lives (up to 4 weeks),43 blinatumomab has a very short half-life of 2 hours.45
Upon examination of popPK models published in the Clinical Pharmacology Review, blinatumomab had nearly double the IIV in CL compared to the other agents. The blinatumomab popPK analysis data from six clinical trials demonstrated a linear PK profile dosing between 5 and 90 μg/m2/day in adult patients. The PK profile was independent of time, body weight, and most other factors.21
PD factors were examined to account for PK variability in blinatumomab therapy, but the complex interplay between blinatumomab and patient’s immune system proves singling out variables difficult. Immunodynamics was recently proposed as a method to quantify the variations in biologic agent–immune system interactions through quantifying biomarkers. Biomarkers from Cancer Immunotherapy Trials Network include target expression in tumor microenvironment, target antigen expression, tumor infiltrating immune response, and systemic cytokine response.40 For instance, patients started on blinatumomab may rapidly lose peripheral CD19-positive B and CD3-positive T cells, but no association was found between the effect of drug target-related factors (eg, CD19-positive B cells, CD3-positive T cells, and B-cell/T-cell ratio) and CL.4,46,47 Patients on blinatumomab may also experience a transient, dose-related spike in cytokines (interleukin-6, interleukin-10, and interferon-gamma) during the first cycle of treatment, which tends to decline to baseline within 48 hours after discontinuing therapy. Patients with rapid cytokine release may be at higher risk for cytokine release syndrome or “cytokine storm”,42,48 a condition that can lead to multiorgan failure and cardiac failure.49–51 Cytokine elevation variability is high (100%–300%), putting patients with higher levels of cytokine elevations at greater risk for immune-related toxicity during the initial stages of therapy,4 which leads to the creation of blinatumomab “step-up” therapy (cycle 1 with 9 μg/day continuous intravenous infusion [cIV] for 1 week followed by 28 μg/day cIV for 3 weeks and 28 μg/day cIV for 4 weeks in subsequent cycles).21 It is unclear if cytokine elevations directly influence PK variability or what patient-specific factors influence cytokine variability.
Immunogenicity, the process of creating ADAs, was also examined, as it may alter the PK of biologics used in oncology.10,52 Once bound to the therapeutic protein, the ADA–protein complex triggers a regular endogenous elimination process where phagocytic cells degrade the ADA–protein complex, thereby increasing CL of the drug.53 ADAs come in two general categories: non-neutralizing antibodies (non-nAb or “binding Ab”) and neutralizing antibodies (nAb). The non-nAbs bind to the therapeutic protein without affecting the ability of the drug to bind to the therapeutic target (creating no change in PD), while the nAbs bind to the drug and prevent target binding (altering the PD).52 In the published blinatumomab popPK analysis, four patients (<1%) tested positive for both non-nAb and nAb. As per the authors, the low incidence of the non-Ab and nAb combination did not raise concerns about ADA-related immunogenicity;4 however, blinatumomab exposure was shown to decrease in the presence of both non-nAb and nAb.4 Considering ADA risk increases after 4 weeks therapy53,54 (which also coincides with the length of cIV blinatumomab therapy),21 variability in CL from either non-nAb or nAb formation should not be ruled out.
Another possibility for the apparent lack of covariates for blinatumomab PK variability may be due to the data used to build the E–R analysis. Clinical data were collected from the Phase II single-arm study, MT103-211. Because MT103-211 lacked a control arm, E–R analysis of clinical data may have appeared falsely steep,55 decreasing the likelihood of teasing out baseline risk factors that influence efficacy or safety.21,56 In addition, patients enrolled in MT103-211 had poor prognostic factors, including early first relapse and later lines of salvage therapy57 making it difficult to determine if PK variability or the lack of apparent covariates in this study is directly comparable to all adult patients with Ph− ALL.
Assuming the IIV of blinatumomab PK is a product of PD changes in all adult patients with Ph− ALL rather than data imbalances, teasing out immunodynamic factors that contribute to PK variability is still a challenge. Immunodynamic interactions as well as technologies that measure the individual components of immune system are still in their infancy compared to other measurement tools used in medicine. If covariates for blinatumomab therapy cannot currently be used to correct for the significant IIV during initial blinatumomab therapy, then perhaps developing TDM is a rational approach to monitor plasma levels to improve safety and optimize efficacy. To do this, a minimum effective plasma trough concentration must be first identified.9,58 The minimum plasma trough concentration of blinatumomab used in the New Drug Application was determined from preclinical studies using a CD19-positive pre-B lymphoma cell line (NALM-6) and CD8-positive T-cells. The addition of blinatumomab increased cell contact between T- and NALM-6 cells, inducing increased apoptosis and lysis of NALM-6 cells with a determined minimum inhibitor concentration (in vitro IC90) of 470 pg/mL.46,59 Average clinical steady-state concentration ranged from around 500 to 700 pg/mL when patients received blinatumomab 28 μg/day cIV.60 However, patients are placed on a significantly lower, subtherapeutic dose (9 μg/day) during the first week of therapy21,60 because of the potential for life-threatening reactions during the initiation of therapy.49–51 In the case of ALL, where ALL mortality can be months when untreated,61 optimized dosing should be put in place as soon as possible.
Recent clinical evidence has shown increased benefit for infliximab TDM-guided dosing during the initial stages of therapy,62 suggesting that TDM for initial therapy may be applicable to other biologics, including blinatumomab. Infliximab TDM is completed by testing free infliximab serum concentrations via a variety of enzyme-linked immunosorbent assay (ELISA) technologies (eg, SHIKARI Q-Inflixi [Matriks Biotek, Ankara, Turkey], LISA-Tracker Duo Infliximab [Theradiag, Croissy-Beaubourg, France], or RIDASCREEN IFX [R-Biopharm, Darmstadt, Germany])63 to determine trough levels and guide dosing.9,58 A standardized assay, such as ELISA, would need to be developed to provide a quick turnaround time for clinical TDM.64 Ideally, information from the ELISA would be able to feed into a decision-support tool (dashboard) for practitioners to optimize therapy,65 which takes input data from TDM to update underlying models and provides clinicians with appropriate blinatumomab dose adjustments.
In summary, using criteria published in a previous review,10 blinatumomab appears to have the most potential in TDM-guided dosing out of the 15 oncology biologics examined. Blinatumomab has a therapeutic window within narrow enough parameters to be defined through TDM, as suggested by the published E–R relationships for efficacy and safety in addition to a defined minimum therapeutic concentration and an MTD. It is uncertain if patients will fall outside of the therapeutic window during initial therapy, as suggested by the high IIV of CL. While immunodynamic markers have potential in identifying patients with high variability (as evident by variation in cytokines), more work will need to be done in assessing how to translate PD profiles into optimizing dosing. Blinatumomab is a novel valuable tool in the arsenal against hematologic oncology, which has steadily been gaining momentum in treating B-cell hematologic cancers. To bypass the uncertainty of how covariates influence the variability in PK in addition to the complexities that come with immunodynamic interactions, a logical goal would be to identify an optimal plasma level that provides patients with the highest dose possible without developing life-threatening ADRs. Once identified, fast assay technologies and patient-friendly dashboards should be developed to quickly identify plasma blinatumomab levels in order to reach the optimized plasma level. The hope is that by optimizing blinatumomab levels and developing a TDM process of blinatumomab, efficacy and safety may be improved during the initial stages of therapy.
Acknowledgments
The authors would like to thank Dr Rushang Patel at Florida Hospital for reviewing this manuscript and sharing his knowledge on the clinical application of biologics used in oncology and Dr Yow-Ming Wang at the FDA for providing feedback on oncology biologic therapies and the exposure–response relationships.
Disclosure
The authors report no conflicts of interest in this work.
References
Buss NA, Henderson SJ, McFarlane M, Shenton JM, de Haan L. Monoclonal antibody therapeutics: history and future. Curr Opin Pharmacol. 2012;12(5):615–622. | ||
Diamantis N, Banerji U. Antibody-drug conjugates an emerging class of cancer treatment. Br J Cancer. 2016;114(4):362–367. | ||
Zaltrap® (Ziv-Aflibercept), application number 125418Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Sanofi Aventis US; 2012. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/125418Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Zhu M, Wu B, Brandl C, et al. Blinatumomab, a Bispecific T-cell Engager (BiTE(®)) for CD-19 targeted cancer immunotherapy: clinical pharmacology and its implications. Clin Pharmacokinet. 2016;55(10):1271–1288. | ||
Rosenzweig SA. Acquired resistance to drugs targeting receptor tyrosine kinases. Biochem Pharmacol. 2012;83(8):1041–1048. | ||
Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278–287. | ||
Kang JS, Lee MH. Overview of therapeutic drug monitoring. Korean J Intern Med. 2009;24(1):1–10. | ||
Herviou P, Thivat E, Richard D, et al. Therapeutic drug monitoring and tyrosine kinase inhibitors. Oncol Lett. 2016;12(2):1223–1232. | ||
Papamichael K, Chachu KA, Vajravelu RK, et al. Improved long-term outcomes of patients with inflammatory bowel disease receiving proactive compared with reactive monitoring of serum concentrations of infliximab. Clin Gastroenterol Hepatol. 2017;15(10):1580.e3–1583.e3. | ||
Oude Munnink TH, Henstra MJ, Segerink LI, Movig KL, Brummelhuis-Visser P. Therapeutic drug monitoring of monoclonal antibodies in inflammatory and malignant disease: translating TNF-alpha experience to oncology. Clin Pharmacol Ther. 2016;99(4):419–431. | ||
Adedokun OJ, Sandborn WJ, Feagan BG, et al. Association between serum concentration of infliximab and efficacy in adult patients with ulcerative colitis. Gastroenterology. 2014;147(6):1296.e5–1307.e5. | ||
Ben-Horin S, Chowers Y. Review article: loss of response to anti-TNF treatments in Crohn’s disease. Aliment Pharmacol Ther. 2011;33(9):987–995. | ||
Eser A, Primas C, Reinisch W. Drug monitoring of biologics in inflammatory bowel disease. Curr Opin Gastroenterol. 2013;29(4):391–396. | ||
Sandritter TL, McLaughlin M, Artman M, Lowry J. The interplay between pharmacokinetics and pharmacodynamics. Pediatr Rev. 2017;38(5):195–206. | ||
Overgaard RV, Ingwersen SH, Tornoe CW. Establishing good practices for exposure-response analysis of clinical endpoints in drug development. CPT Pharmacometrics Syst Pharmacol. 2015;4(10):565–575. | ||
Mould DR, Baumann A, Kuhlmann J, et al. Population pharmacokinetics–pharmacodynamics of alemtuzumab (Campath®) in patients with chronic lymphocytic leukaemia and its link to treatment response. Br J Clin Pharmacol. 2007;64(3):278–291. | ||
Minasian L, Rosen O, Auclair D, Rahman A, Pazdur R, Schilsky RL. Optimizing dosing of oncology drugs. Clin Pharmacol Ther. 2014;96(5):572–579. | ||
FDA.gov [homepage on the Internet]. List of Licensed Biological Products with (1) Reference Product Exclusivity and (2) Biosimilarity or Interchangeability Evaluations to Date [updated September 15, 2017]. Available from: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm411418.htm. Accessed April 30, 2017. | ||
FDA.gov [homepage on the Internet]. Administration USFD. Drugs@FDA: FDA Approved Drug Products. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/. Accessed April 30, 2017. | ||
Adecentris® (Brentuximab Vedotin), application number 125388Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Seattle Genetics; 2011. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125388Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Blincyto® (Blinatumomab), application number 125557Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Amgen; 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125557Orig1s000ClinPharmRedt.pdf. Accessed April 20, 2017. | ||
Vectibix® (Panitumumab), application number 125147/0 [Clinical Pharmacology and Biopharmaceutics Review]. Amgen; 2006. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/125147s0000_ClinPharmR.pdf. Accessed April 20, 2017. | ||
Arzerra® (Ofatumumab) Injection, application number 125326 [Clinical Pharmacology and Biopharmaceutics Review]. Glaxo GRP LTD; 2009. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/1,25,326s000_ClinPharmR.pdf. Accessed April 20, 2017. | ||
Yervoy® (Ipilimumab), application number 125377Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Bristol Myers Squibb; 2011. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/1,25,377Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Perjeta® (Pertuzumab), application number 125409Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Genentech; 2012. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/125409Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Gazyva® (Obinutuzumab), application number 125486Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Genentech; 2013. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/125486Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Cyramza® (Ramucirumab), application number 125477Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Eli Lilly and Co; 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125477Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Portrazza® (Necitumumab), application number 125547Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Eli Lilly and Co; 2015. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/125547Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Tecentriq® (Atezolizumab), application number 761034Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Genentech; 2016. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761034Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Kadcyla® (Trastuzumab Emtansine), application number 125427Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Genentech; 2012. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/125427Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Keytruda® (Pembrolizumab), application number 125514Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Merck Sharp Dohme; 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125514Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Opdivo® (Nivolumab), application number 125527Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. Bristol-Myers Squibb; 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/125527Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Unituxin® (Dinutuximab), application number 125516Orig1s000 [Clinical Pharmacology and Biopharmaceutics Review]. United Therap; 2015. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/125516Orig1s000ClinPharmR.pdf. Accessed April 20, 2017. | ||
Wang J, Song P, Schrieber S, et al. Exposure-response relationship of T-DM1: insight into dose optimization for patients with HER2-positive metastatic breast cancer. Clin Pharmacol Ther. 2014;95(5):558–564. | ||
Gibiansky E, Gibiansky L, Carlile DJ, Jamois C, Buchheit V, Frey N. Population pharmacokinetics of obinutuzumab (GA101) in chronic lymphocytic leukemia (CLL) and non-hodgkin’s lymphoma and exposure-response in CLL. CPT Pharmacometrics Syst Pharmacol. 2014;3:e144. | ||
Feng Y, Roy A, Masson E, Chen TT, Humphrey R, Weber JS. Exposure-response relationships of the efficacy and safety of ipilimumab in patients with advanced melanoma. Clin Cancer Res. 2013;19(14):3977–3986. | ||
Agrawal S, Feng Y, Roy A, Kollia G, Lestini B. Nivolumab dose selection: challenges, opportunities, and lessons learned for cancer immunotherapy. J Immunother Cancer. 2016;4:72. | ||
Nie L, Rubin EH, Mehrotra N, et al. Rendering the 3 + 3 design to rest: more efficient approaches to oncology dose-finding trials in the era of targeted therapy. Clin Cancer Res. 2016;22(11):2623–2629. | ||
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | ||
Kohrt HE, Tumeh PC, Benson D, et al; Cancer Immunotherapy Trials Network (CITN). Immunodynamics: a cancer immunotherapy trials network review of immune monitoring in immuno-oncology clinical trials. J Immunother Cancer. 2016;4(1):15. | ||
van de Donk NW, Dhimolea E. Brentuximab vedotin. MAbs. 2012;4(4):458–465. | ||
Benjamin JE, Stein AS. The role of blinatumomab in patients with relapsed/refractory acute lymphoblastic leukemia. Ther Adv Hematol. 2016;7(3):142–156. | ||
Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507. | ||
Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11(1):81–88. | ||
Wu B, Sun YN. Pharmacokinetics of Peptide-Fc fusion proteins. J Pharm Sci. 2014;103(1):53–64. | ||
Portell CA, Wenzell CM, Advani AS. Clinical and pharmacologic aspects of blinatumomab in the treatment of B-cell acute lymphoblastic leukemia. Clin Pharmacol. 2013;5(Suppl 1):5–11. | ||
Klinger M, Brandl C, Zugmaier G, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119(26):6226–6233. | ||
Nägele V, Kratzer A, Zugmaier G, et al. Changes in clinical laboratory parameters and pharmacodynamic markers in response to blinatumomab treatment of patients with relapsed/refractory ALL. Exp Hematol Oncol. 2017;6(1):14. | ||
Darvishi B, Farahmand L, Jalili N, Majidzadeh AK. Blinatumomab provoked fatal heart failure. Int Immunopharmacol. 2016;41:42–46. | ||
Huang M, Owen E, Myers S, Raj A. Cardiopulmonary failure requiring ECMO bypass resulting from leukemia cell lysis in a patient with childhood acute myelomonocytic leukemia. Case Rep Hematol. 2015;2015:640528. | ||
Adams PS, Shapiro R, Hilmi IA. Postoperative cardiac tamponade after kidney transplantation: a possible consequence of alemtuzumab-induced cytokine release syndrome. Transplantation. 2013;95(3):e18–e19. | ||
Gunn GR 3rd, Sealey DC, Jamali F, Meibohm B, Ghosh S, Shankar G. From the bench to clinical practice: understanding the challenges and uncertainties in immunogenicity testing for biopharmaceuticals. Clin Exp Immunol. 2016;184(2):137–146. | ||
Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012;14(2):296–302. | ||
Lanning DK, Knight KL. Somatic hypermutation: mutations 3′ of rabbit VDJ H-chain genes. J Immunol. 1997;159(9):4403–4407. | ||
Wang Y, Booth B, Rahman A, Kim G, Huang SM, Zineh I. Toward greater insights on pharmacokinetics and exposure-response relationships for therapeutic biologics in oncology drug development. Clin Pharmacol Ther. 2017;101(5):582–584. | ||
Yang J, Zhao H, Garnett C, et al. The combination of exposure-response and case-control analyses in regulatory decision making. J Clin Pharmacol. 2013;53(2):160–166. | ||
Gokbuget N, Kelsh M, Chia V, et al. Blinatumomab vs historical standard therapy of adult relapsed/refractory acute lymphoblastic leukemia. Blood Cancer J. 2016;6(9):e473. | ||
Vaughn BP, Martinez-Vazquez M, Patwardhan VR, Moss AC, Sandborn WJ, Cheifetz AS. Proactive therapeutic concentration monitoring of infliximab may improve outcomes for patients with inflammatory bowel disease: results from a pilot observational study. Inflamm Bowel Dis. 2014;20(11):1996–2003. | ||
Hoffmann P, Hofmeister R, Brischwein K, et al. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int J Cancer. 2005;115(1):98–104. | ||
Yuraszeck T, Kasichayanula S, Benjamin JE. Translation and clinical development of bispecific T-cell engaging antibodies for cancer treatment. Clin Pharmacol Ther. 2017;101(5):634–645. | ||
Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–847. | ||
Van Stappen T, Vande Casteele N, Van Assche G, Ferrante M, Vermeire S, Gils A. Clinical relevance of detecting anti-infliximab antibodies with a drug-tolerant assay: post hoc analysis of the TAXIT trial. Gut. Epub 2017 Apr 27. | ||
Malickova K, Duricova D, Bortlik M, et al. Serum trough infliximab levels: A comparison of three different immunoassays for the monitoring of CT-P13 (infliximab) treatment in patients with inflammatory bowel disease. Biologicals. 2016;44(1):33–36. | ||
Stroh M, Lum BL. Should therapeutic drug monitoring for monoclonal antibodies remain the exception or become the norm? Clin Pharmacol Ther. 2016;100(3):215–217. | ||
Mould DR. Why therapeutic drug monitoring is needed for monoclonal antibodies and how do we implement this? Clin Pharmacol Ther. 2016;99(4):351–354. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.