")
Back to Journals » Journal of Experimental Pharmacology » Volume 12
A Recent Achievement In the Discovery and Development of Novel Targets for the Treatment of Type-2 Diabetes Mellitus
Authors Belete TM
Received 6 August 2019
Accepted for publication 13 December 2019
Published 10 January 2020 Volume 2020:12 Pages 1—15
DOI https://doi.org/10.2147/JEP.S226113
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bal Lokeshwar
Tafere Mulaw Belete
Department of Pharmacology, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
Correspondence: Tafere Mulaw Belete Tel +251 918045943
Email [email protected]
Abstract: Type 2 diabetes (T2DM) is a chronic metabolic disorder. Impaired insulin secretion, enhanced hepatic glucose production, and suppressed peripheral glucose use are the main defects responsible for developing the disease. Besides, the pathophysiology of T2DM also includes enhanced glucagon secretion, decreased incretin secretion, increased renal glucose reabsorption, and adipocyte, and brain insulin resistance. The increasing prevalence of T2DM in the world beseeches an urgent need for better treatment options. The antidiabetic drugs focus on control of blood glucose concentration, but the future treatment goal is to delay disease progression and treatment failure, which causes poorer glycemic regulation. Recent treatment approaches target on several novel pathophysiological defects present in T2DM. Some of the promising novel targets being under clinical development include those that increase insulin sensitization (antagonists of glucocorticoids receptor), decreasing hepatic glucose production (glucagon receptor antagonist, inhibitors of glycogen phosphorylase and fructose-1,6-biphosphatase). This review summarizes studies that are available on novel targets being studied to treat T2DM with an emphasis on the small molecule drug design. The experience gathered from earlier studies and knowledge of T2DM pathways can guide the anti-diabetic drug development toward the discovery of drugs essential to treat T2DM.
Keywords: Type-II diabetes mellitus, antihyperglycemic, insulin resistance, novel drug target
Background
Type 2 Diabetes (T2DM) is the most common heterogeneous metabolic disease, characterized by hyperglycemia accompanied by disorder in the metabolism of proteins, lipids, and carbohydrates. Enhanced hepatic glucose synthesis, impaired insulin release, and effect on peripheral tissues are the main contributing factor for developing T2DM. T2DM is one of the oldest and most prevalent diseases consist of about 90–95% of all diabetes cases.1 T2DM is becoming more prevalent due to the increased obesity in the population. In 2017, about 425 million people worldwide have diabetes mellitus; this figure expected to rise more than 629 million by the year 2045. The increasing prevalence of diabetes is of concern because of the morbidity and mortality associated with the disease. Besides, in 2017, diabetes mellitus prevalence in Ethiopia was 5.2%.1,2
Insulin is an anabolic hormone synthesized by β- cells of islets of the pancreas and has a key role in keeping glucose metabolism in our body. Insulin suppresses the blood glucose level by suppressing its release from tissue and facilitates glucose uptake through hexose transporters into different tissue, especially the liver, muscle, and adipose tissue.3 Insulin exerts its activity by activating insulin receptors, which stimulate glucose transporters (GLUT4) insertion from the intracellular site to the cell membrane, making the cell efficiently uptake glucose. Besides, insulin facilitates the lipid, protein and glycogen synthesis, and block proteolysis, lipolysis, and glycogenolysis.2,4
In T2DM tissue sensitivity to insulin decreases. The proposed mechanism is the downregulation of insulin receptors and signals cascade malfunction that links receptor activation with corresponding cellular effects. Several biochemical pathways show the relationship between hyperglycemia and vascular complications.5 Uncontrolled diabetes causes acute complications like diabetic ketoacidosis and nonketotic hyperosmolar coma. Chronic complications consist of several tissue damages that result in stroke, foot ulcers, impaired immunity, renal failure, cardiovascular disease, and eye damage.6 Blood glucose level regulation from an early stage decreases a diabetic complication that causes a decreased quality of life. So, the search for new orally active insulin-mimetic agents with improved benefit-risk profiles continues.
Several options of drugs available to treat T2DM including glucose-lowering agents like which suppress hepatic glucose production and increase glucose uptake, insulin secretagogues, like sulphonylureas and meglitinides, which enhance insulin secretion from pancreatic β-cells, peroxisome proliferator-activated receptor-γ (PPAR-γ) activator-like thiazolidinediones, which enhance insulin sensitivity and α-glucosidase inhibitors which block glucose production in the gut, Sodium-Dependent Glucose Co-Transporter 2 (SGLT2) Inhibitors, glucagon-like peptide 1 (GLP-1) Receptor agonist and DPP-4 inhibitors.7 Those drugs act by different pharmacological actions; enhance insulin secretion, increase insulin sensitivity, suppress hepatic glucose production, and inhibit glucose reabsorption by the kidney.8 Unfortunately, none of these anti-diabetic agents used to manage hyperglycemia do not stop or reverse disease progression and even may have severe side effects and comorbidities. For example, treatment of T2DM patients with insulin, meglitinides, and sulfonylureas associated with weight gain, hypoglycemia, and treatment with thiazolidinediones may cause osteoporosis, increased risk of heart failure, fluid retention, urinary bladder cancer, and hepatotoxicity. Besides, the Anti-diabetic agents like metformin, sulfonylureas, and GLP1 agonists lose their efficacy.9
Several studies demonstrated that approximately half of the T2DM patients need a new drug that lacks the side effects of conventional anti-diabetic drugs and can regulate long-lasting blood glucose concentration.10 This necessitates pharmaceutical companies continue searching to identify a novel therapeutic agent that acts at a new target site with a novel mechanism for glycemic control and several new drugs are under clinical trials. There is optimism that in the next few years, novel classes of anti-diabetic drugs, which are under clinical trial, will offer extra control over blood glucose amount via complementary mechanisms of action.11 Understanding of mechanisms of actions of anti-diabetic drugs, signaling pathways and therapeutic targets of T2DM can help to develop crucial anti-diabetic agents.10,11
The present review summarizes a variety types of promising therapeutic targets and their role in T2DM including protein tyrosine phosphatase 1 B (PTP-1B) inhibitor, free fatty acid receptors 1 (FFAR1), aldose reductase, glycogen phosphorylase (GP), fructose-1, 6-bisphosphatase (FBPase), glucagon receptor antagonist and phosphoenolpyruvate carboxykinase. This review also gives a medium on which future drug design and development toward diabetes management may be model, as many of the drug candidates highlighted show promise as effective anti-diabetic chemotherapy. This information may use to plan the therapeutic strategies to manage T2DM. The anti-diabetic agents with their side effects presented in Table 1.
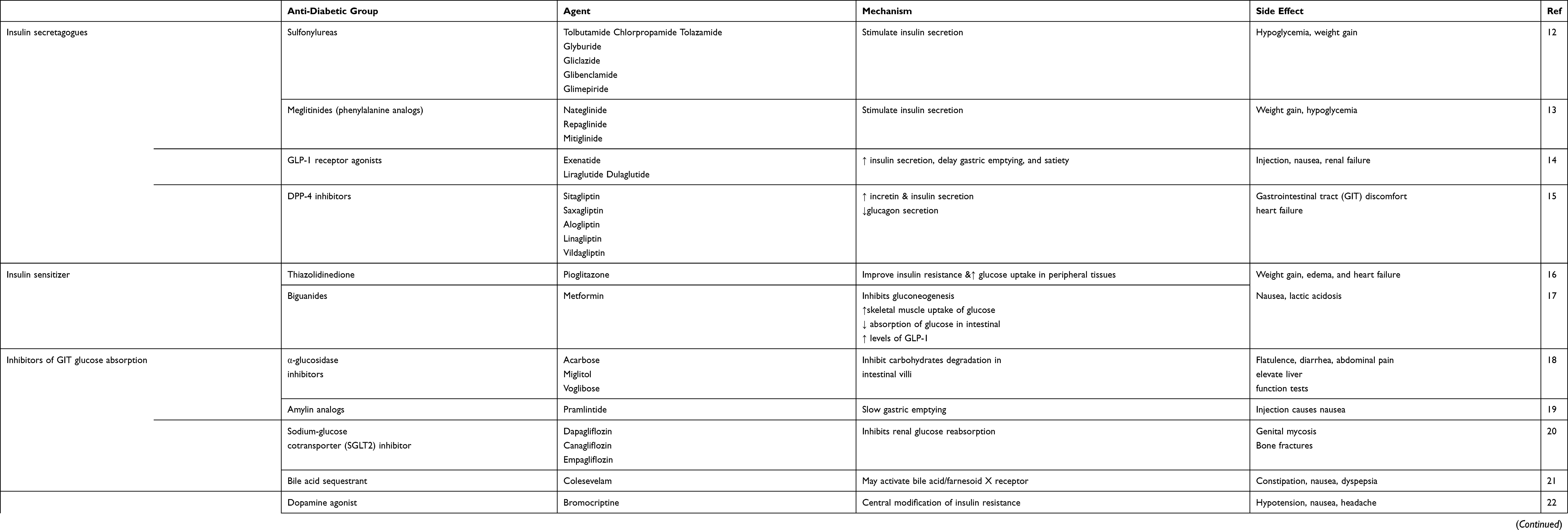 |
Table 1 Summary of the Anti-Diabetic Agents |
Insulin Secretagogues
Free Fatty Acid Receptor Agonist
G-protein coupled receptors (GPCRs) viewed as a source of most drug targets. Nowadays, about 40% of drugs used in the clinical practice act on GPCRs and regulate their activity either positively or negatively.24 Greater than 30 GPCRs identified which contribute to β-cell dysfunction, insulin resistance, and T2D. Only glucagon-like peptide 1 receptor (GLP-1R) targeted anti-diabetic agents approved to treat T2D.25 Free fatty acids (FFAs) activates several GPCRs expressed by human islets. These are GPR119, GPR132, GPR84, GPR120, GPR43 (FFAR2), GPR40 (FFAR1), and GPR41 (FFAR3). FFAs produce several essential activities like keeping the immune system by controlling T cell stimulation, and NF-kB signaling. Each Free fatty acid receptor expressed in several essential tissues, like pancreatic β-cells, adiposities, and immune cells.26
FFAR1 expressed in pancreatic β cells, intestinal L, and K cells, which secrete the glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide, and the effect of FFAs in these cells significantly suppressed after the loss of FFAR1 role. Thus, FFAR1 agonists may act as a promising novel target, which facilitates GLP-1 secretion, which subsequently enhances insulin secretion. Thus, identification of GPCRs for FFAs is a promising strategy to develop a drug based on targeting these receptors. Several Studies using FFAR1 agonists showed better glycemic control.27 Currently, several agents are under in the preclinical phase, and the clinical stage and some discounted from a study like fasiglifam, which terminated in Phase III due to hepatotoxicity. New FFAR1 agonists developed by Eli Lilly, including LY2922470, P11187, SHR0534, and LY2881835 are in Phase I clinical practice. Besides, DS-1558, CNX-011-67, BMS-986118, and SAR1 are in the preclinical phase, suggesting these compounds may still prove beneficial to treat T2DM.28 The GLP-1 secreting effect of FFA4 in enteroendocrine cells showed using FFA4 agonists, such as TUG-891, GSK137647A, and REMD-477. In Table 2, some of the GPCR agonists presented with their stage of development and company.
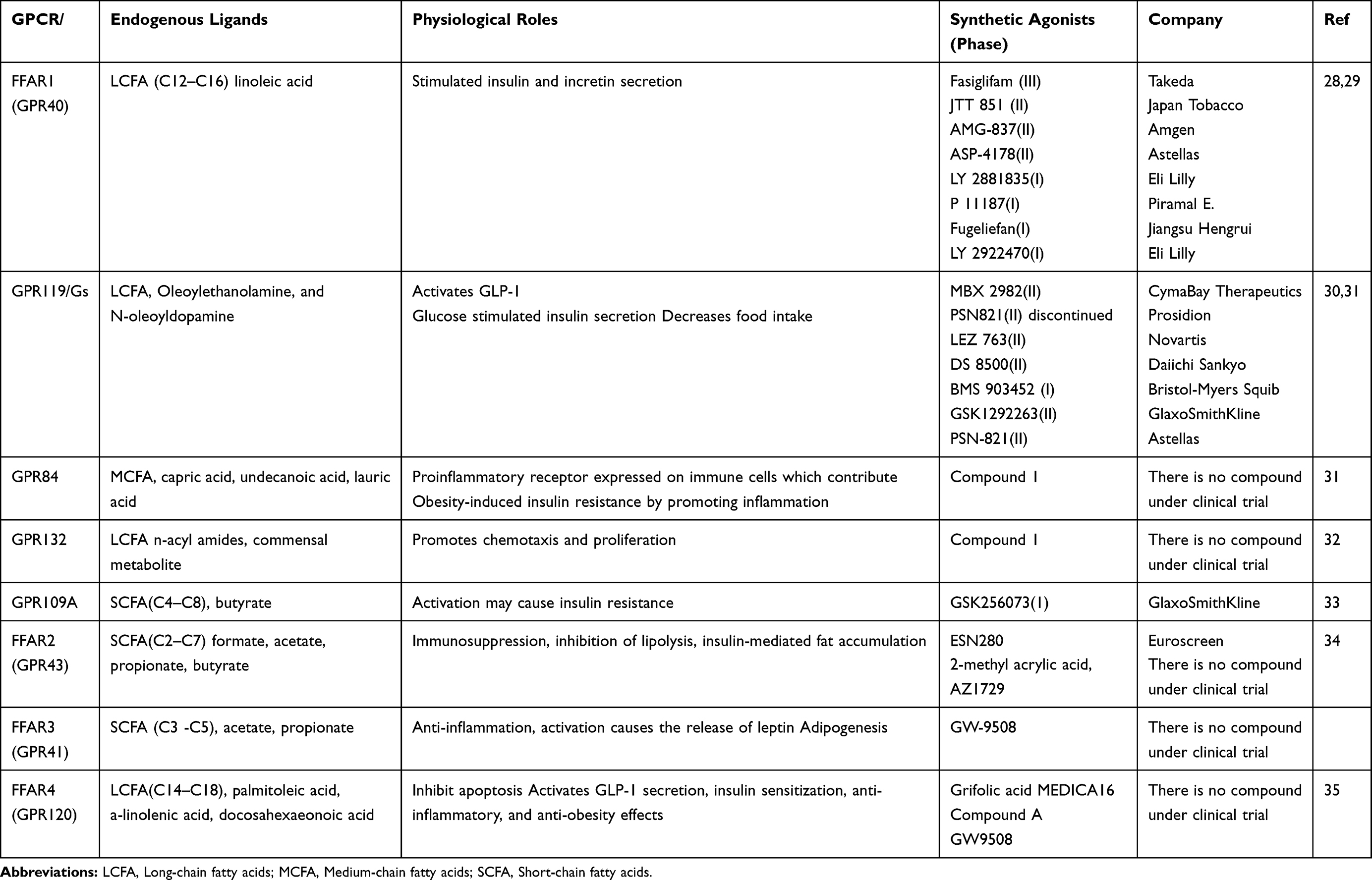 |
Table 2 G-Protein Receptors for Free Fatty Acids Endogenous and Synthetic Agonists |
Inhibitor of Hepatic Glucose Production
Even if there is a time of feeding and fasting, a healthy person’s blood glucose level kept constant. Several enzymes and hormones regulate the amount of glucose in the blood either by the breakdown of glycogen, Gluconeogenesis or absorption from GIT.36
Glucagon Receptor Blocker
Glucagon is a hormone synthesized by pancreas α-cells and released when blood glucose amounts lowered during fasting. Glucagon facilitates hepatic glucose production by glycogenolysis and gluconeogenesis by activating glucagon receptor. Glucagon also inhibits glycogen synthesis but facilitates lipolysis. It also antagonizes insulin effect to inhibit these processes.37 The enhanced glucagon activity and the decreased insulin-mediated suppression of glucagon activity contribute to hyperglycemia and ketoacidosis in T2DM patients. Thus, either suppression of glucagon level by immunoneutralization or inhibiting glucagon receptors, which are the GPCR family, represent an attractive strategy for regulating fasting blood glucose amount in T2DM patients.38 A study on glucagon receptor gene knockout mice with streptozotocin-induced diabetic displayed enhanced glucose tolerance, increased GLP-1 amount, and less pancreatic β-cell injuries. Regulating blood glucose concentration in these mice occurs due to either enhanced GLP-1 amount or decreased glucagon activity.39
Immunoneutralization of glucagon in animal models displayed decreased blood glucose amount and glycosylated hemoglobin A1c levels. It is clear from the studies that the immunoneutralization of glucagon is a novel target for the management of T2DM.37,38
Glucagon receptor blockers inhibit the effects of glucagon at its receptor. Several studies identified glucagon derived glucagon receptor blocker by the change of some amino acid sequences. Glucagon derived agent isolated include Trp25, Asp21, Ser16, Asp15, Tyr13, Lys12, Ser11, Tyr10, Ser8, Phe6, His1and bicyclic 19-residue peptide BI- 32169, Des-His(1)-[Glu(9)]-glucagon amide.40
Monoclonal antibodies against the glucagon receptor including, LY2786890 (Lilly), PF-06293620 (Pfizer), and REMD-477 (REMD Biotherapeutics) are in the clinical trial. Besides, several oral glucagon receptor antagonists identified including, NNC25-0926, Skyrin, Bicyclic 19-residue peptide BI-32169, Des-His (1)-[Glu(9)]- glucagon amide, Spiro-urea, and Benzimidazole. Agents like MK-0893(Merck), MK-3577(Merck), Bay27-9955(Bayer), and LY2409021 (Lilly) terminated clinical trial in Phase II. Agents like PF-06291874 (Pfizer) in Phase I, LGD-6972 (Ligand Pharmaceuticals) in Phase II, and RVT-1502 (Metavant Sciences) in Phase II are in a clinical trial.41
Glucokinase Activators
Glucokinase (GK), also known as hexokinase IV, is one of the Hexokinase families, which has a key role in glucose metabolism. In the liver, GK facilitates glucose uptake and glycogen synthesis. GK phosphorylates glucose to glucose-6-phosphate, which may enter the mitochondrial to produce pyruvate or use as a substrate for glycolysis. These processes facilitate glucose clearance.42 In pancreatic β cells, GK regulates glycolytic and oxidative ATP synthesis. GK increases ATP/ADP ratio, which closes the K+ channel, and makes the cell depolarized resulting in insulin secretion in Figure 1. Elevated blood glucose level induces GK expression in pancreatic β cells and sensitizes them to glucose-stimulated insulin synthesis and secretion.43
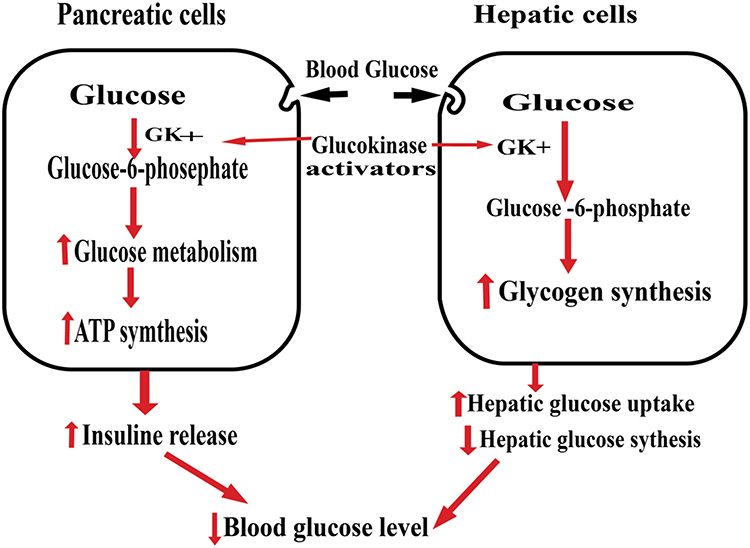 |
Figure 1 The role of Glucokinase activators. |
GK is a promising attractive target for developing an anti-diabetic agent due to its glucose sensor effect in pancreatic β-cells. GK limits the rate of glucose clearance, and glycogen production, all these processes are a malfunction in T2DM. Mice with Hepato-selective GK over-expression have better glucose tolerance and that do not express GK fail to secrete insulin and die within days with diabetes.44 In humans, heterozygous mutations in the GK gene lead to stable chronic hyperglycemia, called MODY2 (maturity-onset diabetes of the young), which lead to defective in insulin release.45
GK activators promote glucose metabolism in the liver and enhance insulin secretion. GK activators also increase the growth of pancreatic β cells and inhibit apoptosis due to glucotoxicity and oxidative stress. In the liver GK activity controlled by GKRP (GK regulatory protein), which forms a complex with GK in the presence of fructose-6-phosphate (Fru-6-P). Fructose-1-phosphate (Fru-1-P) blocks the inhibitory effect of GKRP by the displacing Fru-6-P, facilitating GK to move to the cytoplasm in an active state where it enhances glucose metabolism. Several studies were undertaken to identify novel agent Fru-1-P mimetics to inhibit GKRP.46
Several GK activators identified during the past decades. Agents like R1511 or GK3, PF 04937319, TMG-123, HMS5552, TTP3999, Dorzagliatin, and PF-04937319 effectively control blood glucose level in clinical trials. There are also other agents undergoing the preclinical trial including, R1440 GKA2, GKA 50, YH GKA, PSN 010, MK-0941, ZYGK1, Ro-28-1675. Some agents like AZD6370, piragliatin, DS-7309, and ARRY-403 also terminated their drug development due to toxicity and loose of effectiveness with long-term administration.47–51 Some examples of the GK activator presented in Table 3 with their stage of development and company.
 |
Table 3 Glucokinase Activators in Clinical Development |
Fructose 1, 6-Bisphosphatase Inhibitors
Fructose-1, 6-bisphosphatase (FBPase) is a crucial rate-limiting enzyme in the hepatic glucose synthesis. It catalyzes the hydrolysis of fructose-1, 6-bisphosphate to fructose-6-phosphate and phosphate. Several studies demonstrated that gluconeogenesis is a key contributor to enhanced blood glucose concentration during fasting in T2DM patients.52 Thus, the isolation of the FBPase-1 blocker is a promising novel target for effective fasting blood glycemic regulation. Inhibitors of the FBPase bring a new way of treatment because most of the present anti-diabetic drugs act by facilitating the insulin release or decreasing insulin resistance and do not inhibit gluconeogenesis.53
The importance of the FBPase enzyme as a target to develop a new drug-related to its site in the gluconeogenesis as displayed in Figure 2, which makes inhibition of gluconeogenesis from all substrates while avoiding their effects on glycogenolysis, glycolysis, and the tricarboxylic acid cycle.54
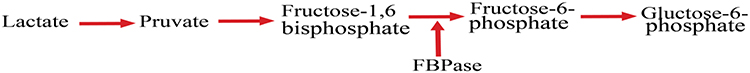 |
Figure 2 Hepatic gluconeogenesis pathway and site of FBPase inhibitor. |
FBPase inhibited by natural occurring metabolites like fructose-2, 6-bisphosphate, and AMP. These metabolites suppress glucose concentration without hypoglycemia. There are other inhibitors including, MB07803 (Phase II), Anilinoquinazolines, VK0612 (Phase II), and managlinat dialanetil (Phase II) are in clinical development.55
Glutamine Fructose-6-Phosphate Amidotransferase (GFAT) Inhibitor
T2DM characterized by insulin resistance, decrease insulin secretion and increased glucose production. Several studies have shown hyperglycemia contributes to developing insulin resistance and vascular complication, even though the biochemical mechanisms causing these effects have not yet understood.56
The hexosamine pathway proposed as one of the biochemical factors that may contribute to the enhanced glucose synthesis, glucose-induced insulin desensitization, decreased insulin release, glucose-induced growth factor induction, diabetes-associated macrovascular complications, and glomerulosclerosis.56,57 The pathway involves the synthesis of UDP-GlcNAc. Glucose metabolized to fructose-6 phosphate. Then fructose-6 phosphate metabolized by glutamine: fructose-6 phosphate amidotransferase (GFAT) into glucosamine-6 phosphate (GlcN-6-P) as shown in Figure 3. Finally GlcN-6-P metabolized to UDP-N-acetylglucosamine (UDP-GlcNAc) which uses to synthesis glycosyl side chains of proteoglycans, glycolipids, glycoproteins, and gangliosides. GlcN-6-P proposed to contribute to the glucotoxicity in diabetes.57
GFAT is the rate-limiting enzyme in the hexosamine pathway. Inhibiting GFAT enzymatic activity results in suppressed glucose levels in diabetes. Thus, inhibition of GFAT helps in the prevention of hyperglycemia-induced TGF-α and TGF-β1 induction, insulin-resistance, and diabetes-associated macrovascular complications. GFAT inhibitor azaserine prevented the expression of active TGF-β1. These findings show the effect of the hexosamine pathway in regulating TGF-β1 expression.58
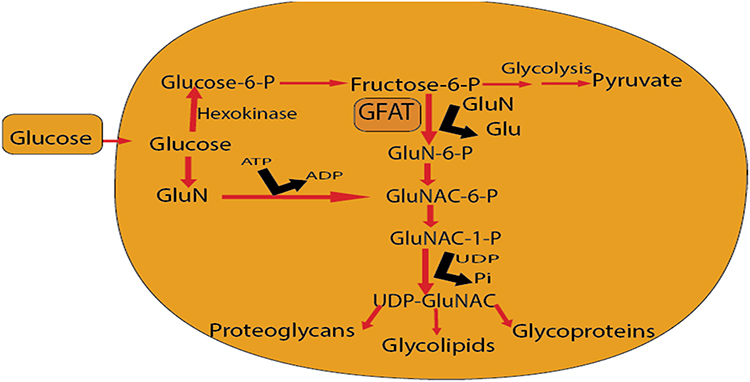 |
Figure 3 Hexosamine synthesis pathway. |
Pyruvate Dehydrogenase Kinase Inhibitors
Pyruvate dehydrogenase complex (PDC), which is a crucial enzyme in reducing blood glucose levels in a well-fed state, which directs the entrance of glycolytic products into the citric acid cycle by catalyzing the decarboxylation of pyruvate to acetyl- CoA and CO2 as shown in Figure 4. During fasting state, inhibition of PDC helps to keep blood glucose amount by conserving three-carbon substances (pyruvate, alanine, and lactate) for gluconeogenesis. PDC activity controlled by pyruvate dehydrogenase kinases (PDHK) which phosphorylate to inactivate PDC and dephosphorylation by the opposing pyruvate dehydrogenase phosphatases help to reactivated PDC.59
Enhanced hepatic gluconeogenesis causes hyperglycemia in T2DM with fasting blood glucose concentration. Suppressing the level of gluconeogenic precursors, by facilitating the oxidation of pyruvate in peripheral tissue, is a promising approach for decreasing excessive gluconeogenesis.60 Pyruvate is a precursor for the synthesis of glucose, fatty acids, glycerol, and nonessential amino acids. The up-regulation of PDK4 occurs in humans with T2DM. Inhibition of PDHK in muscle enhances glucose utilization by increasing pyruvate oxidation, decreases the level of compound (alanine, lactate) for gluconeogenesis in the liver.59,60 Now a day several PDHK inhibitors are in a clinical trial including, JTT-251, AZD 2545, and leelamine which have proven effective in lowering blood glucose levels in diabetic rodent models.61
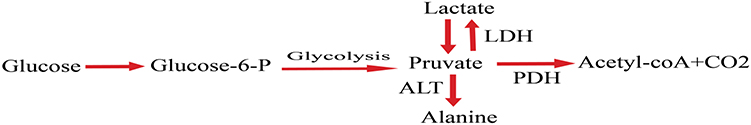 |
Figure 4 Cellular metabolism of glucose. |
Aldose Reductase Inhibitor
Aldose reductase catalyzes the reduction of reactive oxygen species-toxic aldehydes to inactive alcohols, but if hyperglycemia occurs, it reduces glucose to sorbitol, the first and rate-limiting step in the polyol pathway of glucose metabolism, which later oxidized to fructose.62 Reducing glucose to sorbitol, depletes NADPH, resulting in decreased glutathione levels, which lead to oxidative stress. Sorbitol is hydrophilic alcohol, which accumulates in cells, resulting in osmotic stress. Osmotic and oxidative stresses are the main causes of complications of diabetes. Thus, reduction of the polyol pathway presented in Figure 5 by aldose reductase inhibitor proposed as a promising therapeutic target in the treatment and prevention of diabetic complications.62,63
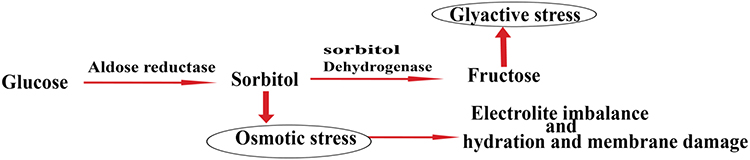 |
Figure 5 The polyol pathway of glucose metabolism. |
Many aldose reductase inhibitors are available as drug candidates for the treatment and prevention of diabetic complications. Epalrestat approved in Japan, China, and India for the treatment of diabetic neuropathy. Alrestatin the first aldose reductase inhibitor for diabetic cataract was effective in reducing the swelling of diabetic lenses in glucose medium. It decreased the accumulation of sorbitol in the lenses and sciatic nerves of rats with streptozotocin-induced diabetes leading to suppression of cataract formation.64 Sorbinil, Fidarestat, Minalrestat, Fifarestat, Imirestat, Rubrolid, zenarestat, Ponalrestat, kinostat, and Ranirestat prevented the development of cataract formation in the diabetic rat lens.65
Increase Insulin Sensitization
Protein-Tyrosine Phosphatase 1B Inhibitor
Insulin resistance occurs in most T2DM patients and forms obesity linked to metabolic syndrome. Increasing insulin sensitivity decreases abnormal glucose metabolism. The process of insulin signal transduction involves tyrosine phosphorylation in the insulin-receptor activation pathway. This process controlled by Protein-tyrosine phosphatase 1B (PTP-1B) by the dephosphorylating insulin receptor. The role of PTP-1B on insulin signaling cascades acts as a negative regulator. Hence, the inhibition of PTP-1B gives a new promising approach as a class of insulin-sensitizing agents in regulating T2DM.66 Reducing the PTP-1B level not only increases insulin sensitivity and enhances glucose metabolism, but also protects obesity-induced high-fat feeding. Several agents have shown increased insulin signaling and glucose tolerance in preclinical models. PTP-1B knockout mice showed enhanced insulin sensitivity, better glycemic regulation, and resistant to diet-induced obesity. In cells administered with PTP-1B antibody, insulin activated receptor autophosphorylation increased.67
Besides, inhibition of PTP-1B in insulin receptor by using different compound decreases glucose-induced insulin resistance and increase insulin signaling. Several Agents like SF-5060, aquastatin A, Benzofuran, Benzothiophen, Maslinic acid, Vanadium complexes, and Ursolic acid have in vitro inhibitory activity against PTP-1B enzyme.68 Recent studies have identified different agents undergoing clinical trials. These are ertiprotafib (discontinued in Phase II), TTP814, Alexidine dihydrocloride, trivaric acid, BVT948, TCS401, ISIS-PTP1BRx (Phase II), KQ-791(Phase I), Trodusquemine (Phase I), and IONIS-PTP1BRx (Phase II).69–71
11-β-Hydroxysteroid Dehydrogenase-1 Inhibitors
Glucocorticoids control several biochemical pathways. Glucocorticoids enhance gluconeogenesis in the liver. It inhibits insulin secretion and peripheral glucose uptake. Glucocorticoids also increase insulin resistance, and induce inflammatory cytokines by activating the glucocorticoid receptor in adiposities. Thus, excess cortisol related to diabetes mellitus.72 Thus inhibiting glucocorticoid activity by using glucocorticoid receptor antagonists is one of the most promising treatment approaches. Agents like IONIS-GCCRRx in Phase II clinical trial. But, long-term use may lead to adrenal insufficiency and may stimulate increased cortisol secretion.73
Due to this reason, the current approach directed to tissue-specific glucocorticoid metabolism, by inhibiting 11b-hydroxysteroid dehydrogenase (11b-HSD), which catalyzes the conversion of cortisone, to the active form cortisol without affecting the pituitary-adrenal axis. Overexpression of 11b-HSD related to metabolic syndromes like T2DM and obesity. Thus, inhibition of 11b-HSD improves insulin sensitivity, hyperglycemia, and lipid profile.74 several classes of 11b-HSD inhibitors are in clinical trials. These are Arylsulfonamidothiazoles, Carbenoxolone, RO151, Adamantyltriazoles, Anilinothiazolone, MK-0736, RO838, BVT2733, PF-00915275 (Phase II), INCB 13739 (Phase II), Glycyrrhetinic acid (Phase II), and MK-0916 (Phase II).75
Inhibitors of 17β-Hydroxysteroid Dehydrogenase 1 (17β-HSD1)
17β-HSD1 plays a key role in the local synthesis of the most potent estrogen estradiol. Its expression is a characteristic marker for the diagnosis of patients with T2DM. The 17β-HSD1 inhibition is under consideration for T2DM treatment.76 several studies revealed that increased estrogen levels linked to increased insulin resistance. In estrogen target cells 17β -HSD1 catalyze the NADPH-dependent reduction of estrone to the potent 17β-estradiol, which leads to overexpression of 17β -HSD1 in breast tumor cells. In postmenopausal women, hormone proliferation led by increased levels of estrogen, thus it is a promising novel therapeutic target.77
Fibroblast Growth Factor-21
Fibroblast growth factor 21 (FGF21) is a metabolic regulator. Most portions synthesize by the liver. FGF21 reduces blood glucose concentration and improves the lipid profile in animal models of obesity and T2DM. It enhances insulin sensitivity and glucose uptake by upregulating GLUT1 expression in adiposities.78 FGF21 regulates body weight and has a key role during nutritional deprivation and ketogenic state. The target sites for its metabolic effect are adipose tissue, liver, and pancreas.79
Transgenic mice with FGF21-overexpression were resistant to diet-induced obesity. FGF-21 administration in obese and insulin-resistant mice and monkeys, increases energy expenditure, insulin sensitivity, weight loss, and makes normal glucose, triglyceride, insulin, glucagon, and lipid level. Hypoglycemia was not displayed during the study period.80 FGF21 activities are insulin-independent and give the additive effect of insulin-induced metabolic activities. While insulin acts by enhancing glucose uptake within minutes, FGF21 activity mediated through gene expression changes. In a recent study, FGF-21 analogs like BMS-986036, which is in Phase II, and LY 2405319 showed effective as glucose and lipid-lowering drug.81
Sirtuin-1 Activators
Aging is a risk factor for developing T2DM.82 Studies on calorie restriction (CR) without malnutrition retard aging in yeast, worms, flies, and rodent. The beneficial effects of CR involve the function of silent information regulator 1(sirtuin-1), which is NAD+-dependent protein deacetylase catalyzes acetyl groups removal of lysine residues which induced by CR in various tissues.83 Sirtuin-1 involved in inflammation, gene repression, metabolic regulation, cell survival and apoptosis, DNA repair, neuroprotection, and aging. Sirtuin-1 also controls glucose homeostasis by regulating insulin secretion and protecting pancreatic β-cells, improves insulin resistance by facilitating post insulin signaling, suppressing inflammation, and hepatic glucose production.82,83
The significance of sirtuin-1 on the effects of CR shown by using Sirtuin-1 transgenic mice, which displayed CR-like phenotype, reduced fasting glucose level, and greater glucose tolerance. Besides, a 25% reduction in calorie intake for 6 months in nonobese young adults led to the upregulation of sirtuin-1 accompanied by a decrease in visceral fat mass, insulin resistance, and oxidative stress. Thus, under the excess energy intake, decreased sirtuin-1 activity may contribute to developing insulin resistance and T2DM. Thus, sirtuin-1 activation as a CR mimetic is a promising therapeutic target to treat insulin-resistance T2DM.84
Resveratrol a natural polyphenolic substance extracted from grapes is a sirtuin-1 activator, which has a good blood controlling effect. Administration of several Synthetic sirtuin-1activating agents like SRT2379 (Phase I), SRT3025 (Phase I), SRT2104 (Phase I), and MHY2233 to high fat diet-induced obese mice improved insulin sensitivity and decreased plasma glucose level.85–87
Anti-Inflammatory Therapies
In T2DM patient adiposities are in a state of inflammation and release proinflammatory mediators like Tumor necrosis factor-alpha (TNF-α), and interleukin-1b (IL-1b). Studies showed that inflammation has a significant effect on the pathogenesis of T2DM, insulin resistance, impaired insulin secretion, and its complications. Several studies showed that drugs that reduce inflammation increase insulin sensitivity and improve glucose control in insulin-resistant patients with T2DM.88
IL-1 receptor antagonists like canakinumab decreased HbA1c, increased insulin sensitivity, and secretion but terminated their development at Phase III due to inadequate efficacy.89 Diacerein, which is in Phase III, inhibits the effect of IL-1b and down-regulates IL-1R.
Salsalate, a prodrug of salicylate, has shown beneficial effects on glycemia, and insulin sensitivity, through inhibition of the NF-κB pathway, which has a key role in the pathogenesis of T2DM and becomes promising therapeutic targets.90 TNF-α is a pro-inflammatory mediator secreted by macrophages that induce tissue-specific inflammation, impair insulin transduction, and affect glucose metabolism. Anti-TNF-α (etanercept, infliximab, adalimumab), reduced insulin resistance in T2DM. The Newer hypothesis proposed that inhibition of more than one inflammatory mediator might have a promising effect.91
Ghrelin Receptor Antagonism
Ghrelin (the hunger hormone) is an appetite-stimulating hormone secreted by the gastric mucosa and human islets, which acts at the ghrelin receptor (GHSR). Blood levels of ghrelin increase with fasting and decrease after a meal. It also has a variety of metabolic functions like blocking insulin release; activate glucagon secretion, impairing insulin sensitivity, and accelerates gastric emptying.92 GHSR antagonism is a promising strategy to treat diabesity. Reduced intake of food, weight loss and improved glucose tolerance displayed in mice treated with ghrelin antagonists like TZP- 102 (Phase II) and NOX-B11.93
Obestatin is an anorexigenic peptide that exists in the same neuroendocrine cells as ghrelin but antagonizes the effects of ghrelin on food intake, appetite, gastric emptying. obestatin protects diet-induced insulin resistance, increases β-cell proliferation and survival, and suppress inflammation-induced β-cell death. To translate this into a therapeutic concept, a ghrelin antagonist can combined with an obestatin agonist to simulate this tilt in ratios.94
Diacylglycerol O-acyltransferase-1 Inhibitors
Diacylglycerol O-acyltransferase-1(DGAT-1) inhibitors have emerged as a promising therapeutic agent against diabetes. It catalyzes the final step of triglyceride synthesis from diacylglycerol and acyl-CoA. Inhibition of DGAT-1 suppresses fat synthesis, increase incretin release, and insulin-induced glucose uptake.95 The combined effects from these actions may contribute to the beneficial effects including weight reduction, enhanced insulin resistance, and hyperlipidemia. DGAT1 knockout mice showed increased insulin sensitivity.96 Several DGAT1 inhibitors are in clinical development including, LCQ-908 (Phase II), AZD-7687(Phase I discontinued), P7435 (Phase II), and PF-4620110 (Phase I discontinued). Besides, companies like Abbott (A-922500), Roche (RO-6036), and AstraZeneca (AZD-3988) disclosed preclinical compounds.97
Chelating Therapy for Diabetic Complications
Many trace elements are essential for human metabolic processes. Oxidative stress contributes to the pathogenesis of T2DM.98 Metals generate oxidative stress, acts as a pro-oxidant and catalyzes free radical formation.99 Metals also involved in the formation of advanced glycation end products, protein oxidation, and lipoxygenation, which contribute to developing T2DM complications.100 several studies showed that high levels of iron and copper contributed to the incidence of T2DM and insulin resistance.101 Copper identified as a key player in several metabolic derangements.102
Chelating agents inhibit metal-catalyzed production of reactive oxygen species, thereby inhibiting chemical damage to proteins and subsequent pro-inflammatory cascades. Using copper and iron chelator is a promising treatment strategy to prevent/reverse organ damage in diabetes, heart failure, and neurodegenerative diseases.103 Triethylenetetramine is the first in a new class of copper chelator anti-diabetic agents, which suppresses copper-mediated oxidative stress and restores anti-oxidant defense. The drug facilitates urinary Copper excretion in patients with T2DM and rats with streptozotocin-induced diabetes and restores cardiac structure and function in both groups.104
Conclusion
The treatment of T2DM presents several management challenges, but treatment with a new drug that acts on novel targets are a welcome addition to complement the present drugs. Regulation of Blood glucose levels is crucial for the successful treatment of T2DM. Although the presence of several agents that act with a different mechanism, no one can reverse the disease progression or maintain normal glucose metabolism. Thus, developing newer agents with novel targets provides an opportunity for treating diabetes efficiently in the future. A range of novel targets with new agents has shown significant activity like the one GPR119 agonists have significant potential for meeting the needs of T2DM patients because of their safety profile, lack of weight gain, and beta-cell preservation effect.
The 11β-HSD-1 inhibitor may safe and efficacious agent that lowers fasting blood glucose, A1c and weight. The glycogen phosphorylase inhibitors showed rapid and safe blood glucose decreases in mice without the risk of hypoglycemia. GK inhibitors in clinical trials showed a significant decrease in glucose levels in insulin-resistant patients.
These compounds show promising effects and deserve as a novel candidate in the treatment of diabetes. Besides, these agents with promising activity and well-defined mechanisms considered as key candidates in the design and development of novel and more effective synthetic compound based modulators. Further studies will help to define these new and emerging antihyperglycemic agents’ roles in therapy.
Abbreviations
DGAT-1, Diacylglycerol O-acyltransferase-1; FGF21, Fibroblast growth factor 21; FBPase, fructose-1,6-bisphosphatase; FFAs, Free fatty acids; GK, Glucokinase; GLP-1R, glucagon-like peptide 1 receptor; GPCRs, G-protein coupled receptors; PTP-1B, protein tyrosine phosphatase 1 B; PDC, pyruvate dehydrogenase complex; 11b-HSD, 11b-hydroxysteroid dehydrogenase; IL-1b, Interleukin-1b; T2DM, Type 2 Diabetes.
Acknowledgments
I would like to acknowledge the School of Pharmacy (University of Gondar) for providing the resources.
Disclosure
The author declares that he has no competing interests.
References
1. Zheng Y, Ley SH, Hu FB. Global etiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88. doi:10.1038/nrendo.2017.151
2. Aynalem SB, Zeleke AJ. Prevalence of diabetes mellitus and its risk factors among individuals aged 15 years and above in Mizan-Aman town, Southwest Ethiopia, 2016: a cross-sectional study. Int J Endocrinol. 2018;2018:1–7. doi:10.1155/2018/9317987
3. Xu Y, Rubin BR, Orme CM, et al. Dual-mode of insulin action controls GLUT4 vesicle exocytosis. J Cell Biol. 2011;193(4):643–653. doi:10.1083/jcb.201008135
4. Jaldin-Fincati JR, Pavarotti M, Frendo-Cumbo S, Bilan PJ, Klip A. Update on GLUT4 vesicle traffic: a cornerstone of insulin action. Trends Endocrinol Metab. 2017;28(8):597–611. doi:10.1016/j.tem.2017.05.002
5. Ormazabal V, Nair S, Elfeky O, Aguayo C, Salomon C, Zuñiga FA. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol. 2018;17(1):122. doi:10.1186/s12933-018-0762-4
6. Lotfy M, Adeghate J, Kalasz H, Singh J, Adeghate E. Chronic complications of diabetes mellitus: a mini-review. Curr Diabetes Rev. 2017;13(1):3–10. doi:10.2174/1573399812666151016101622
7. Moon MK, Hur KY, Ko SH, et al. Combination therapy of oral hypoglycemic agents in patients with type 2 diabetes mellitus. Diabetes Metab J. 2017;41(5):357–366. doi:10.4093/dmj.2017.41.5.357
8. Chaudhury A, Duvoor C, Dendi R, et al. Clinical review of anti-diabetic drugs: implications for type 2 diabetes mellitus management. Front Endocrinol (Lausanne). 2017;8:6. doi:10.3389/fendo.2017.00006
9. Bolen S, Feldman L, Vassy J, et al. Systematic review: comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann Intern Med. 2007;147(6):386–399. doi:10.7326/0003-4819-147-6-200709180-00178
10. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2012;55(6):1577–1596. doi:10.1007/s00125-012-2534-0
11. DeFronzo RA, Triplitt CL, Abdul-Ghani M, Cersosimo E. Novel agents for the treatment of type 2 diabetes. Diabetes Spectr. 2014;27(2):100–112. doi:10.2337/diaspect.27.2.100
12. Lapane KL, Yang S, Brown MJ, Jawahar R, Pagliasotti C. Sulfonylureas and risk of falls and fractures: a systematic review. Drugs Aging. 2013;30(7):527–547. doi:10.1007/s40266-013-0081-0
13. Guardado-Mendoza R, Prioletta A, Jiménez-Ceja LM, Sosale A, Folli F. The role of nateglinide and repaglinide, derivatives of meglitinide, in the treatment of type 2 diabetes mellitus. Arch Med Sci. 2013;9(5):936. doi:10.5114/aoms.2013.34991
14. Prasad-Reddy L, Isaacs D. A clinical review of GLP-1 receptor agonists: efficacy and safety in diabetes and beyond. Drugs Context. 2015;4.
15. Abbas AS, Dehbi HM, Ray KK. Cardiovascular and non-cardiovascular safety of dipeptidyl peptidase-4 inhibition: a meta-analysis of randomized controlled cardiovascular outcome trials. Diabetes Obes Metab. 2016;18(3):295–299. doi:10.1111/dom.12595
16. Nanjan MJ, Mohammed M, Kumar BP, Chandrasekar MJ. Thiazolidinediones as anti-diabetic agents: a critical review. Bioorg Chem. 2018;1(77):548–567. doi:10.1016/j.bioorg.2018.02.009
17. Sanchez-Rangel E, Inzucchi SE. Metformin: clinical use in type 2 diabetes. Diabetologia. 2017;60(9):1586–1593. doi:10.1007/s00125-017-4336-x
18. Hücking K, Kostic Z, Pox C, et al. α-Glucosidase inhibition (acarbose) fails to enhance secretion of glucagon-like peptide 1 (7–36 amide) and to delay gastric emptying in Type 2 diabetic patients. Diabet Med. 2005;22(4):470–476. doi:10.1111/dme.2005.22.issue-4
19. Jorsal T, Rungby J, Knop FK, Vilsbøll T. GLP-1 and amylin in the treatment of obesity. Curr Diab Rep. 2016;16(1):1. doi:10.1007/s11892-015-0693-3
20. Zaccardi F, Webb DR, Htike ZZ, Youssef D, Khunti K, Davies MJ. Efficacy and safety of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes mellitus: a systematic review and network meta-analysis. Diabetes Obes Metab. 2016;18(8):783–794. doi:10.1111/dom.12670
21. Fonseca VA, Handelsman Y, Staels B. Colesevelam lowers glucose and lipid levels in type 2 diabetes: the clinical evidence. Diabetes Obes Metab. 2010;12(5):384–392. doi:10.1111/dom.2010.12.issue-5
22. Lamos EM, Levitt DL, Munir KM. A review of dopamine agonist therapy in type 2 diabetes and effects on cardio-metabolic parameters. Prim Care Diabetes. 2016;10(1):60–65. doi:10.1016/j.pcd.2015.10.008
23. Mannucci E, Monami M, Marchionni N. Short-acting insulin analogues vs. regular human insulin in type 2 diabetes: a meta-analysis. Diabetes Obes Metab. 2009;11(1):53–59. doi:10.1111/dom.2009.11.issue-1
24. Brauner-Osborne H, Wellendorph P, Jensen AA. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr Drug Targets. 2007;8(1):169–184. doi:10.2174/138945007779315614
25. Ahren B. Islet G protein-coupled receptors as potential targets for the treatment of type 2 diabetes. Nat Rev Drug Discov. 2009;8(5):369. doi:10.1038/nrd2782
26. Lee Kennedy R, Vangaveti V, Jarrod G, Shashidhar V, Shashidhar V, Baune BT. Free fatty acid receptors: emerging targets for the treatment of diabetes and its complications. Ther Adv Endocrinol Metab. 2010;1(4):165–175. doi:10.1177/2042018810381066
27. Ichimura A, Hasegawa S, Kasubuchi M, Kimura I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front Pharmacol. 2014;6(5):236.
28. Li Z, Qiu Q, Geng X, Yang J, Huang W, Qian H. Free fatty acid receptor agonists for the treatment of type 2 diabetes: drugs in preclinical to phase II clinical development. Expert Opin Investig Drugs. 2016;25(8):871–890. doi:10.1080/13543784.2016.1189530
29. Defossa E, Wagner M. Recent developments in the discovery of FFA1 receptor agonists as a novel oral treatment for type 2 diabetes mellitus. Bioorg Med Chem Lett. 2014;24(14):2991–3000. doi:10.1016/j.bmcl.2014.05.019
30. Yang JW, Kim HS, Choi YW, Kim YM. Therapeutic application of GPR119 ligands in metabolic disorders. Diabetes Obes Metab. 2018;20(2):257–269. doi:10.1111/dom.13062
31. Liu Y, Zhang Q, Chen LH, et al. Design and synthesis of 2-alkylpyrimidine-4, 6-diol and 6-alkylpyridine-2, 4-diol as potent GPR84 agonists. ACS Med Chem Lett. 2016;7(6):579–583. doi:10.1021/acsmedchemlett.6b00025
32. Shehata MA, Christensen HB, Isberg V, et al. Identification of the first surrogate agonists for the G protein-coupled receptor GPR132. RSC Adv. 2015;5(60):48551–48557. doi:10.1039/C5RA04804D
33. Sprecher D, Maxwell M, Goodman J, et al. Discovery and characterization of GSK256073, a non-flushing hydroxy-carboxylic acid receptor 2 (HCA2) agonist. Eur J Pharmacol. 2015;756:1–7. doi:10.1016/j.ejphar.2015.01.051
34. Milligan G. G protein-coupled receptors not currently in the spotlight: free fatty acid receptor 2 and GPR35. Br J Pharmacol. 2018;175(13):2543–2553. doi:10.1111/bph.v175.13
35. Ou HY, Wu HT, Hung HC, Yang YC, Wu JS, Chang CJ. Multiple mechanisms of GW-9508, a selective G protein-coupled receptor 40 agonists, in the regulation of glucose homeostasis and insulin sensitivity. Am J Physiol Endocrinol Metab. 2013;304(6):E668–76. doi:10.1152/ajpendo.00419.2012
36. Wasserman DH. Four grams of glucose. Am J Physiol Endocrinol Metab. 2009;296(1):E11–E21. doi:10.1152/ajpendo.90563.2008
37. Müller TD, Finan B, Clemmensen C, DiMarchi RD, Tschöp MH. The new biology and pharmacology of glucagon. Physiol Rev. 2017;97(2):721–766. doi:10.1152/physrev.00025.2016
38. Ali S, Drucker DJ. Benefits and limitations of reducing glucagon action for the treatment of type 2 diabetes. Am J Physiol Endocrinol Metab. 2009;296(3):E415–21. doi:10.1152/ajpendo.90887.2008
39. Ouhilal S, Vuguin P, Cui L, et al. Hypoglycemia, hyperglucagonemia, and fetoplacental defects in glucagon receptor knockout mice: a role for glucagon action in pregnancy maintenance. Am J Physiol Endocrinol Metab. 2011;302(5):522–531. doi:10.1152/ajpendo.00420.2011
40. Lotfy M, Kalasz H, Szalai G, Singh J, Adeghate E. Recent progress in the use of glucagon and glucagon receptor antagonists in the treatment of diabetes mellitus. Open Med Chem J. 2014;8:28. doi:10.2174/1874104501408010028
41. Scheen AJ, Paquot N, Lefèbvre PJ. Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Expert Opin Investig Drugs. 2017;26(12):1373–1389. doi:10.1080/13543784.2017.1395020
42. Lei L, Liu S, Li Y, et al. The potential role of glucokinase activator SHP289-04 in anti-diabetes and hepatic protection. Eur J Pharmacol. 2018;826:17–23. doi:10.1016/j.ejphar.2018.02.036
43. Pal M. Recent advances in glucokinase activators for the treatment of type 2 diabetes. Drug Discov Today. 2009;14(15–16):784–792. doi:10.1016/j.drudis.2009.05.013
44. Nakamura A, Terauchi Y. Present status of clinical deployment of glucokinase activators. J Diabetes Investig. 2015;6(2):124–132. doi:10.1111/jdi.2015.6.issue-2
45. Anık A, Çatlı G, Abacı A, Böber E. Maturity-onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab. 2015;28(3–4):251–263. doi:10.1515/jpem-2014-0384
46. Bhuniya D, Kapkoti SG, Warrier SJ, Kukrejka G, Mavinahalli NJ, Palle PV Pyrrole-2-carboxamide derivatives as glucokinase activators, their process, and pharmaceutical application. United States patent US 8,299,115. 2012 Oct 30.
47. Scheen AJ. Investigational insulin secretagogues for type 2 diabetes. Expert Opin Investig Drugs. 2016;25(4):405–422. doi:10.1517/13543784.2016.1152260
48. Kamimura H, Ito S, Chijiwa H, et al. Simulation of human plasma concentration-time profiles of the partial glucokinase activator PF-04937319 and its disproportionate N-demethylated metabolite using humanized chimeric mice and semi-physiological pharmacokinetic modeling. Xenobiotica. 2017;47(5):382–393. doi:10.1080/00498254.2016.1199063
49. Zhu XX, Zhu DL, Li XY, et al. Dorzagliatin (HMS5552), a novel dual-acting glucokinase activator, improves glycaemic control and pancreatic β-cell function in patients with type 2 diabetes: a 28-day treatment study using biomarker-guided patient selection. Diabetes Obes Metab. 2018;20(9):2113–2120. doi:10.1111/dom.13338
50. Tsumura Y, Tsushima Y, Tamura A, et al. TMG-123, a novel glucokinase activator, exerts durable effects on hyperglycemia without increasing triglyceride in diabetic animal models. PLoS One. 2017;12(2):e0172252. doi:10.1371/journal.pone.0172252
51. Vella A, Freeman JL, Dunn I, Keller K, Buse JB, Valcarce C. Targeting hepatic glucokinase to treat diabetes with TTP399, a hepatoselective glucokinase activator. Sci Transl Med. 2019;11:475. doi:10.1126/scitranslmed.aau3441
52. Kubota K, Inaba SI, Nakano R, et al. Identification of activating enzymes of a novel FBPase inhibitor prodrug, CS-917. Pharmacol Res Perspect. 2015;3(3):e00138. doi:10.1002/prp2.138
53. Kaur R, Dahiya L, Kumar M. Fructose-1, 6-bisphosphatase inhibitors: a new valid approach for management of type 2 diabetes mellitus. Eur J Med Chem. 2017;141:473–505. doi:10.1016/j.ejmech.2017.09.029
54. Yip J, Geng X, Shen J, Ding Y. Cerebral gluconeogenesis and diseases. Front Pharmacol. 2017;4(7):521.
55. Gizak A, Duda P, Wisniewski J, Rakus D. Fructose-1, 6-bisphosphatase: from a glucose metabolism enzyme to the multifaceted regulator of cell fate. Adv Biol Regul. 2019;72:41–50. doi:10.1016/j.jbior.2019.03.001
56. Teo CF, Wollaston-Hayden EE, Wells L. Hexosamine flux, the O-GlcNAc modification, and the development of insulin resistance in adipocytes. Mol Cell Endocrinol. 2010;318(1–2):44–53. doi:10.1016/j.mce.2009.09.022
57. Nakaishi Y, Bando M, Shimizu H, et al. Structural analysis of human glutamine: fructose-6-phosphate amidotransferase, a key regulator in type 2 diabetes. FEBS Lett. 2009;583(1):163–167. doi:10.1016/j.febslet.2008.11.041
58. Buse MG. Hexosamines, insulin resistance, and the complications of diabetes: current status. Am J Physiol Endocrinol Metab. 2006;290(1):E1–E8. doi:10.1152/ajpendo.00329.2005
59. Jeong JY, Jeoung NH, Park KG, Lee IK. Transcriptional regulation of pyruvate dehydrogenase kinase. Diabetes Metab J. 2012;36(5):328–335. doi:10.4093/dmj.2012.36.5.328
60. Mayers RM, Leighton B, Kilgour E. PDH kinase inhibitors: a novel therapy for Type II diabetes? 2005:367–370.
61. Hasan N, Mujahid M, Badruddeen MK, et al. J Pharm Res. 2017;6(2).
62. Singh Grewal A, Bhardwaj S, Pandita D, Lather V, Singh Sekhon B. Updates on aldose reductase inhibitors for management of diabetic complications and non-diabetic diseases. Mini Rev Med Chem. 2015;16(2):120–162. doi:10.2174/1389557515666150909143737
63. Ramana KV. Aldose reductase: new insights for an old enzyme. Biomol Concepts. 2011;2(1–2):103–114. doi:10.1515/bmc.2011.002
64. Suzen S, Buyukbingol E. Recent studies of aldose reductase enzyme inhibition for diabetic complications. Curr Med Chem. 2003;10(15):1329–1352. doi:10.2174/0929867033457377
65. Iyer S, Murthy K, Parton Z, et al. Repurposing the aldose reductase inhibitor and diabetic neuropathy drug epalrestat for the congenital disorder of glycosylation PMM2-CDG. bioRxiv. 2019;Jan:626697.
66. Zhao C, Wu Y, Yang C, Liu B, Huang Y. Hypotensive, hypoglycaemic and hypolipidaemic effects of bioactive compounds from microalgae and marine micro-organisms. Int J Food Sci Technol. 2015;50(8):1705–1717. doi:10.1111/ijfs.2015.50.issue-8
67. Nieto-Vazquez I, Fernández-Veledo S, de Alvaro C, Rondinone CM, Valverde AM, Lorenzo M. Protein–tyrosine phosphatase 1B–deficient myocytes show increased insulin sensitivity and protection against tumor necrosis factor-α–induced insulin resistance. Diabetes. 2007;56(2):404–413. doi:10.2337/db06-0989
68. Møller NP, Iversen LF, Andersen HS, McCormack JG. Protein tyrosine phosphatases (PTPs) as drug targets: inhibitors of PTP-1B for the treatment of diabetes. Curr Opin Drug Discov Devel. 2000;3(5):527–540.
69. Luo J, Xu Q, Jiang B, et al. Selectivity, cell permeability and oral availability studies of novel bromophenol derivative HPN as protein tyrosine phosphatase 1B inhibitor. Br J Pharmacol. 2018;175(1):140–153. doi:10.1111/bph.v175.1
70. Limbocker R, Chia S, Ruggeri FS, et al. Trodusquemine enhances Aβ 42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat Commun. 2019;10(1):225. doi:10.1038/s41467-018-07699-5
71. Patil PD, Mahajan UB, Patil KR, et al. Past and current perspective on new therapeutic targets for Type-II diabetes. Drug Des Devel Ther. 2017;11:1567. doi:10.2147/DDDT.S133453
72. Di Dalmazi G, Pagotto U, Pasquali R, Vicennati V. Glucocorticoids and type 2 diabetes: from physiology to pathology. J Nutr Metab. 2012;2012:1–9. doi:10.1155/2012/525093
73. Bauerle KT, Harris C. Glucocorticoids and Diabetes. Mo Med. 2016;113(5):378.
74. Stewart PM, Tomlinson JW. Selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1 for patients with metabolic syndrome: is the target liver, fat, or both? Diabetes. 2009;58(1):14–15. doi:10.2337/db08-1404
75. Anagnostis P, Katsiki N, Adamidou F, et al. 11beta-Hydroxysteroid dehydrogenase type 1 inhibitors: novel agents for the treatment of metabolic syndrome and obesity-related disorders? Metabolism. 2013;62(1):21–33. doi:10.1016/j.metabol.2012.05.002
76. Nguyen ND, Le LT. Targeted proteins for diabetes drug design. Adv Nat Sci. 2012;3(1):013001.
77. Tiwari N, Thakur AK, Kumar V, Dey A, Kumar V. Therapeutic targets for diabetes mellitus: an update. Clin Pharmacol Biopharm. 2014;3(1):1. doi:10.4172/2167-065X.1000117
78. Liu JJ, Foo JP, Liu S, Lim SC. The role of fibroblast growth factor 21 in diabetes and its complications: a review from the clinical perspective. Diabetes Res Clin Pract. 2015;108(3):382–389. doi:10.1016/j.diabres.2015.02.032
79. Yuan D, Wu BJ, Henry A, Rye KA. Role of fibroblast growth factor 21 in gestational diabetes mellitus: a mini-review. Clin Endocrinol (Oxf). 2019;90(1):47–55. doi:10.1111/cen.13881
80. Domouzoglou EM, Maratos-Flier E. Fibroblast growth factor 21 is a metabolic regulator that plays a role in the adaptation to ketosis. Am J Clin Nutr. 2011;93(4):901S–5S. doi:10.3945/ajcn.110.001941
81. Kim JH, Bae KH, Choi YK, et al. Fibroblast growth factor 21 analogues LY2405319 lowers blood glucose in streptozotocin-induced insulin-deficient diabetic mice by restoring brown adipose tissue function. Diabetes Obes Metab. 2015;17(2):161–169. doi:10.1111/dom.12408
82. Kitada M, Koya D. SIRT1 in type 2 diabetes: mechanisms and therapeutic potential. Diabetes Metab J. 2013;37(5):315–325. doi:10.4093/dmj.2013.37.5.315
83. Bonkowski MS, Sinclair DA. Slowing aging by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol. 2016;17(11):679. doi:10.1038/nrm.2016.93
84. Huynh FK, Hershberger KA, Hirschey MD. Targeting sirtuins for the treatment of diabetes. Diabetes Manag (Lond). 2013;3(3):245. doi:10.2217/dmt.13.6
85. Milne JC, Lambert PD, Schenk S, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450(7170):712.
86. Nguyen LT, Chen H, Mak C, Zaky A, Pollock C, Saad S. SRT1720 attenuates obesity and insulin resistance but not liver damage in the offspring due to maternal and postnatal high-fat diet consumption. Am J Physiol Endocrinol Metab. 2018;315(2):E196–E203. doi:10.1152/ajpendo.00472.2017
87. Kim MJ, An HJ, Kim DH, et al. Novel SIRT1 activator MHY2233 improves glucose tolerance and reduces hepatic lipid accumulation in db/db mice. Bioorg Med Chem Lett. 2018;28(4):684–688. doi:10.1016/j.bmcl.2018.01.021
88. Rehman K, Akash MS. Mechanisms of inflammatory responses and development of insulin resistance: how are they interlinked? J Biomed Sci. 2016;23(1):87. doi:10.1186/s12929-016-0303-y
89. Everett BM, Donath MY, Pradhan AD, et al. Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. J Am Coll Cardiol. 2018;71(21):2392–2401. doi:10.1016/j.jacc.2018.03.002
90. Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov. 2014;13(6):465. doi:10.1038/nrd4275
91. Akash MS, Rehman K, Liaqat A. Tumor necrosis factor-alpha: role in the development of insulin resistance and pathogenesis of type 2 diabetes mellitus. J Cell Biochem. 2018;119(1):105–110. doi:10.1002/jcb.26174
92. Delporte C. Recent advances in the potential clinical application of ghrelin in obesity. J Obes. 2012;20:2012.
93. McCallum RW, Lembo A, Esfandyari T, et al. TZP-102 Phase 2b Study Group. Phase 2b, randomized, double-blind 12-week studies of TZP-102, a ghrelin receptor agonist for diabetic gastroparesis. Neurogastroenterol Motil. 2013;25(11):e705–17. doi:10.1111/nmo.2013.25.issue-11
94. Pradhan G, Wu CS, Lee JH, et al. Obestatin stimulates glucose-induced insulin secretion through ghrelin receptor GHS-R. Sci Rep. 2017;7(1):979. doi:10.1038/s41598-017-00888-0
95. Cao J, Zhou Y, Peng H, et al. Targeting Acyl-CoA: diacylglycerol acyltransferase 1 (DGAT1) with small molecule inhibitors for the treatment of metabolic diseases. J Biol Chem. 2011;286(48):41838–41851. doi:10.1074/jbc.M111.245456
96. Qian Y, Wertheimer SJ, Ahmad M, et al. Discovery of orally active carboxylic acid derivatives of 2-phenyl-5-trifluoromethyloxazole-4-carboxamide as potent diacylglycerol acyltransferase-1 inhibitors for the potential treatment of obesity and diabetes. J Med Chem. 2011;54(7):2433–2446. doi:10.1021/jm101580m
97. Waring MJ, Birch AM, Birtles S, et al. Optimisation of biphenyl acetic acid inhibitors of diacylglycerol acetyl transferase 1–the discovery of AZD2353. MedChemComm. 2013;4(1):159–164. doi:10.1039/C2MD20190A
98. Peguero JG, Arenas I, Lamas GA. Chelation therapy and cardiovascular disease: connecting scientific silos to benefit cardiac patients. Trends Cardiovasc Med. 2014;24(6):232–240. doi:10.1016/j.tcm.2014.06.002
99. Qiu Q, Zhang F, Zhu W, Wu J, Liang M. Copper in diabetes mellitus: a meta-analysis and systematic review of plasma and serum studies. Biol Trace Elem Res. 2017;177(1):53–63. doi:10.1007/s12011-016-0877-y
100. Frizzell N, Baynes JW. Chelation therapy for the management of diabetic complications: a hypothesis and a proposal for clinical laboratory assessment of metal ion homeostasis in plasma. Clin Chem Lab Med. 2014;52(1):69–75. doi:10.1515/cclm-2012-0881
101. Gong D, Lu J, Chen X, et al. A copper (II)-selective chelator ameliorates diabetes-evoked renal fibrosis and albuminuria, and suppresses pathogenic TGF-β activation in the kidneys of rats used as a model of diabetes. Diabetologia. 2008;51(9):1741–1751. doi:10.1007/s00125-008-1088-7
102. Tarantino G, Porcu C, Arciello M, Andreozzi P, Balsano C. Prediction of carotid intima–media thickness in obese patients with low prevalence of comorbidities by serum copper bioavailability. J Gastroenterol Hepatol. 2018;33(8):1511–1517. doi:10.1111/jgh.2018.33.issue-8
103. Lu J, Gong D, Choong SY, et al. Copper (II)-selective chelation improves function and antioxidant defences in cardiovascular tissues of rats as a model of diabetes: comparisons between triethylenetetramine and three less copper-selective transition-metal-targeted treatments. Diabetologia. 2010;53(6):1217–1226. doi:10.1007/s00125-010-1698-8
104. JS Cooper G. Selective divalent copper chelation for the treatment of diabetes mellitus. Curr Med Chem. 2012;19(17):2828–2860. doi:10.2174/092986712800609715
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.