")
Back to Journals » Journal of Blood Medicine » Volume 12
A Rare Association of Thrombotic Thrombocytopenic Purpura with Systemic Lupus Erythematosus in a Sudanese Woman: Case Report
Authors Ibn Idris Rodwan AA, Ahmed Elmansour OK, Ahmed AFE, Elagib EM, Ahmed Eltahir NI, Hassan A, El-Sadig SM , Mohammed AGAA , Awadalla HH, Mohammed AA, Adam Essa ME
Received 19 August 2021
Accepted for publication 13 October 2021
Published 6 November 2021 Volume 2021:12 Pages 945—949
DOI https://doi.org/10.2147/JBM.S334689
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Martin H Bluth
Amel Awad Ibn Idris Rodwan,1 Osama Khder Ahmed Elmansour,1,2 Amar F Eldow Ahmed,3 Elnour Mohammed Elagib,1,4 Noha Ibrahim Ahmed Eltahir,4,5 Abubaker Hassan,6 Sara M El-Sadig,7 Abdel Gaffar Abdel Allah Mohammed,8 Huyam H Awadalla,9 Abubakr Abdalwahab Mohammed,5 Mohammed Elmujtba Adam Essa6,10
1Department of Rheumatology, Sudan Medical Specialization Council, Khartoum, Sudan; 2Department of Rheumatology, Faculty of Medicine, Shendi University, Shendi, Sudan; 3Department of Internal Medicine, Wayne State University, Detroit, MI, USA; 4Department of Rheumatology, Omdurman Military Hospital, Khartoum, Sudan; 5Department of Medicine, Faculty of Medicine, Karary University, Khartoum, Sudan; 6Department of Clinical Medicine, Medical and Cancer Research Institute, Nyala, Sudan; 7Department of Medicine, Faculty of Medicine, University of Khartoum, Khartoum, Sudan; 8Rheumatology Department, Security Forces Hospital, Makkah, Saudi Arabia; 9Department of Internal Medicine, Detroit Medical Centre, Detroit, MI, USA; 10Faculty of Medicine, Al Fashir University, Al Fashir, Sudan
Correspondence: Mohammed Elmujtba Adam Essa
Department of Clinical Medicine, Medical and Cancer Research Institute, Nyala, Sudan
Tel +249-90-700-938
Email [email protected]
Abstract: Thrombotic thrombocytopenic purpura (TTP) is an uncommon life-threatening condition characterized by hemolytic disorders. The coexistence of systemic lupus erythematosus (SLE) with TTP is extremely rare, although Africans are at increased risk due to inherited risk factors. This report describes a rare clinical manifestation of TTP associated with SLE in a Sudanese patient. A 41-year-old Sudanese woman presented to the emergency department with symptoms and features that were suggestive of malaria, for which she had been treated accordingly. However, a few days later she complained of fever, and was found to have a body temperature of 39.5°C, jaundice, anemia, and thrombocytopenia. Soon after admission, she developed confusion and unrecordable blood pressure. After the patient had stabilized, clinical assessment, immune-system investigation (antinuclear antibody profile, complements, blood panel), and imaging revealed a diagnosis of TTP associated with SLE. The patient received imipenem 500 mg, five sessions of plasmapheresis (60 mL/kg), methylprednisolone 1 g pulse for 3 days, and rituximab 375 mg/week. Three weeks later, the patient was discharged after her condition had improved, and she is now on regular follow-up.
Keywords: TTP, SLE, African female, rituximab and plasmapheresis
Introduction
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease characterized by the presence of autoantibodies directed against nuclear antigens.1 The disease can presents with different features such as fever, skin changes, and hair loss. In severe cases of SLE patients can present with renal, hematological or central nervous system involvements.2
Thrombotic thrombocytopenic purpura (TTP) is a life-threatening condition characterized by affecting the capillaries and arterioles of multiple organs, mainly caused by deficiency in ADAMTS13, also known as von Willebrand factor–cleaving protease (VWFCP).3 VWFCP is significant in inhibiting spontaneous microvascular platelet clumping, an essential pathophysiological finding in TTP. In most TTP patients, ADAMTS13, the main regulator of VWF size, is severely deficient.4
TTP can occur through autoimmune inhibitors in acquired cases, such as infections, SLE, and neoplasms, or hereditarily through mutation of the ADAMTS13 gene in the plasma. TTP in SLE patients is extremely rare — <0.5%.5 Surprisingly, connective tissue diseases, including SLE, may occur with low ADAMTS13 levels, suggesting probable pathophysiology for the coexistence of TTP and SLE.6 The aim of this report was to describe a rare occurrence of TTP as the first presentation of SLE in Sudanese patients.
Case Presentation
A 41-year-old Sudanese woman presented to the emergency department with a 2-day history of high-grade fever (39.5°C) associated with fatigue, nausea, and vomiting. Clinical evaluation and a blood test revealed a diagnosis of malaria, for which treatment was administered. During the next few days, her symptoms worsened, in addition to yellow sclera and low hemoglobin (5.5 g/dL) and platelets (43 cells/mL). The patient was transferred to our rheumatology department at Omdurman Military Hospital for further evaluation. Clinical findings were normal except for jaundice (bilirubin 3.2 mg/dL). She indicated that her sister had Behçet’s disease. Upon admission, she developed a severe headache associated with confusion and fever, her blood pressure was unrecordable, and her Glasgow Coma Scale score was 9/15. The rapid-response team was called, and she was admitted to a high-dependency unit, where she received six units of fresh frozen plasma, two pools red blood cells, and kept under close observation. After her condition had stabilized, laboratory investigations were requested (Table 1). A diagnosis of SLE and TTP was made, and she was given imipenem 500 mg, acyclovir 75 mg, electrolyte correction, IV normal saline 0.9%, skin and bladder care (five sessions of plasmapheresis), and methylprednisolone 1 g pulse for 3 days, followed by prednisolone 1 mg/kg in a tapering manner. A few days later, after the infection had been controlled, the patient received four doses of rituximab 375 mg/week with caution. Two weeks later, she was discharged in good general condition, with normal results on complete blood count and renal function testing. She was discharged on hydroxychloroquine 200 mg, azathioprine 50 mg, prednisolone 10 mg, pantoprazole 40 mg, calcium supplement 500 mg, and omega 3. On review at two weeks, she was completely well without any complaints.
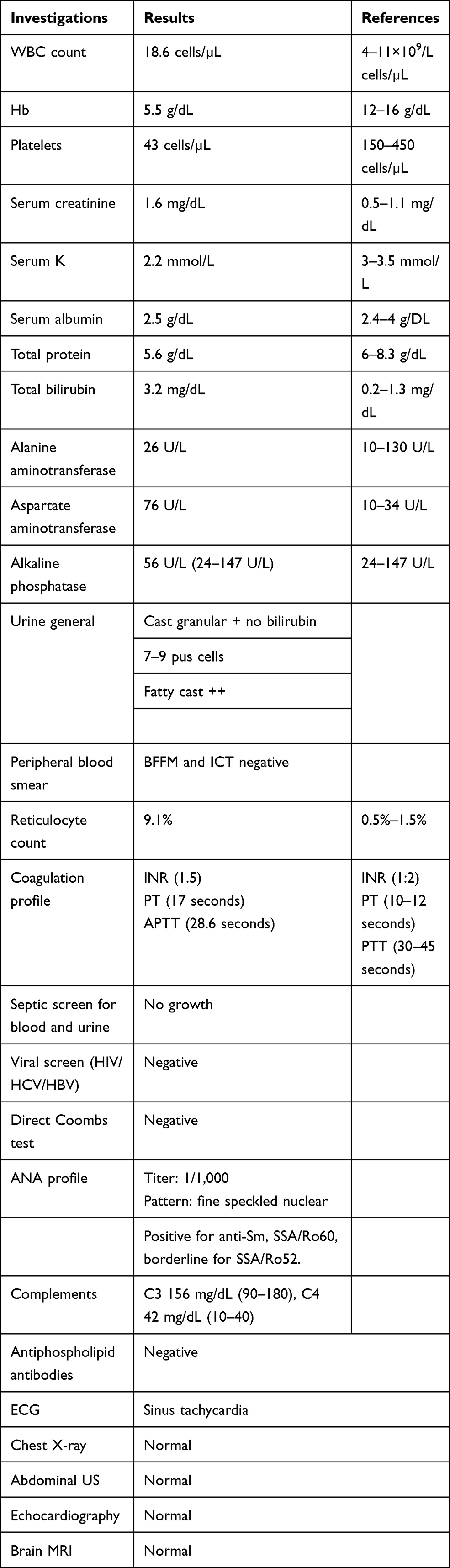 |
Table 1 Lab results |
Discussion
TTP is an uncommon life-threatening condition, and occurs in <0.5% of SLE patients.7 It was first reported in 1924, but the pentad of microangiopathic hemolytic anemia, severe thrombocytopenia, renal insufficiency, neurological abnormalities, and fever was first published in 1966.8 SLE is a chronic autoimmune disease that affects mainly women of reproductive age,9 and is characterized by the production of various autoantibodies that target different organs, resulting in many clinical symptoms and signs, such as malaise, fever, headache, myalgia, arthralgia, and loss of weight.10,11 In this case, the patient presented with clinical features of TTP and exhibited a progressive deteriorating course. The enormous potential for misrecognizing this disorder may be due to the complex picture of SLE presenting as TTP.12 The differential diagnosis for this case could be Evans syndrome, as it also occurs with fever, fatigue, mucosal bleeding, pallor, cytopenia, and jaundice,13 which are the same features that presented in this patient. However, Evans syndrome is characterized by autoimmune hemolytic anemia and a positive direct Coombs test result,13,14 in contrast to anemia in TTP, which is microangiopathic anemia due to fragmented red blood cells and a negative direct Coombs test result, as in this patient. Other main differentials include disseminated intravascular coagulopathy (DIC), which is also characterized by cytopenic anemia and hemolysis, but in contrast to Evans syndrome and TTP, coagulation in DIC is deranged. An absence of coagulopathy is the key differential, as all coagulation, fibrinolysis, and platelet systems are activated in DIC.15
Despite the fact that many patients with TTP in sub-Saharan Africa are at greater risk of mortality due to the absence of prompt diagnostic facilities and access to adequate therapy, an African patient with SLE and TTP may be at even greater risk. The clinical presentation of a TTP patient may not essentially compel a clinician to undertake workup for a primary connective tissue disease, such as SLE, as both conditions can have overlapping features. In our case, TTP was the first presentation of SLE, contrary to previous reports of TTP usually presenting in patients previously managed for SLE for years, with a high SLE Disease Activity Index score and coexisting nephritis.12,16 Children with hematological conditions, such as venous thromboembolism, are managed by heparin, which may cause by what is known as heparin‐associated thrombocytopenia. This may present similarly to TTP.17
Arthritis, polyarthralgia, hair loss, fever, osteonecrosis, and myopathy are common presentations reported in previous studies of TTP patients with SLE.18 However, in addition to hair loss and fever, our patient also presented with confusion, jaundice, anemia, and low platelet count, which led to a high index of suspicion. We thus undertook a complement assay, and immunological findings reinforced the diagnosis of SLE. Furthermore, the occurrence of neurological complications in SLE patients of African descent may cause difficulty in diagnosis and proper management.
The clinical overlap between these two diseases has been described more commonly in young black female patients, and since certain severe manifestations have been found in SLE patients of African descent, TTP may constitute another such manifestation with notable links to genetic attributes.19
Our patient exhibited clinical and laboratory features for the diagnosis of SLE, which included skin changes, such as malar rash, positive immunological markers like anti-Smith antibodies and anti-dsDNA, abnormal antinuclear antibody profile, and high antinuclear antibody titers.
Plasma exchange continues to be the mainstay of treatment in patients with TTP, even with concomitant SLE and showing a low response20,21 (Table 2), emphasizing the significance of looking out for this association, early diagnosis, and aggressive management with plasma exchange and immunosuppression, which is life-saving. Although there was no significant improvement in platelet count, after hepatitis infection had been ruled out, four doses of IV rituximab 375 mg/m2 once weekly led to dramatic improvement in the patient’s condition and normalization of the platelet count.22 Critical care specialists need to develop a multidisciplinary approach urgently, emphasizing early recognition and initiation of treatment to ensure better outcomes in this scenario.
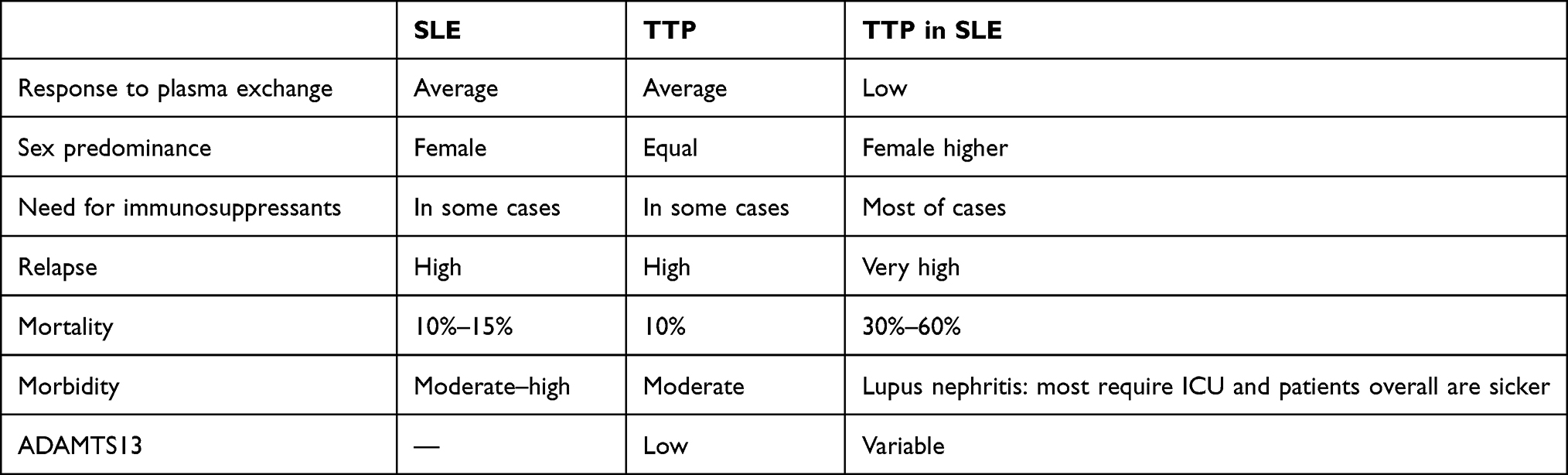 |
Table 2 Clinical manifestations and treatment |
Limitation
ADAMTS13-antibody testing, one of the investigations to confirm acquired TTP, was not done on the patient, as it is not available in Sudan.
Conclusion
A middle-aged Sudanese woman presented with fever, jaundice, confusion, unrecordable blood pressure, anemia, and thrombocytopenia. Immunological studies confirmed a diagnosis of TTP on top of SLE, which is a very rare association. The patient received antibiotics, plasmapheresis, methylprednisolone, and rituximab. Her condition improved well, and she was discharged and followed up. Initial diagnosis can overlap with malaria, as happened in this case, as severe malaria can cause thrombocytopenia. Therefore, it is important to consider TTP in patients presenting with hemolytic anemia and thrombocytopenia, as SLE and TTP can occur concomitantly and share similar hematological manifestations.
Data Sharing Statement
All the data used in the study are available from the first and corresponding authors on reasonable request.
Consent
Signed consent was obtained from the patient for publication of the case details.
Ethics Approval and Consent to Publish
Obtained from the Sudan Federal Ministry of Health.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Manson JJ, Rahman A. Systemic lupus erythematosus. Orphanet J Rare Dis. 2006;1:6. PubMed PMID: 16722594. Pubmed Central PMCID: PMC1459118. doi:10.1186/1750-1172-1-6
2. Adelowo OO, Oguntona SA. Pattern of systemic lupus erythematosus among Nigerians. Clin Rheumatol. 2009;28(6):699–703. PubMed PMID: 19242770. doi:10.1007/s10067-009-1139-6
3. Fujimura Y, Isonishi A. [Pathophysiology of thrombotic thrombocytopenic purpura]. Nihon Jinzo Gakkai Shi. 2014;56(7):1043–1051. Japanese. PubMed PMID: 25420404.
4. Kremer Hovinga JA, Lammle B. Role of ADAMTS13 in the pathogenesis, diagnosis, and treatment of thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program. 2012;2012:610–616. PubMed PMID: 23233642. doi:10.1182/asheducation.V2012.1.610.3798654
5. Ali Z, Zafar MU, Wolfe Z, Akbar F, Lash B. Thrombotic thrombocytopenic purpura induced by immune checkpoint inhibitiors: a case report and review of the literature. Cureus. 2020;12(10):e11246. PubMed PMID: 33274128. Pubmed Central PMCID: PMC7707147.
6. Shah AA, Higgins JP, Chakravarty EF. Thrombotic microangiopathic hemolytic anemia in a patient with SLE: diagnostic difficulties. Nat Clin Pract Rheumatol. 2007;3(6):357–362. PubMed PMID: 17538567. doi:10.1038/ncprheum0511
7. Crawley JT, Scully MA. Thrombotic thrombocytopenic purpura: basic pathophysiology and therapeutic strategies. Hematology Am Soc Hematol Educ Program. 2013;2013:292–299. PubMed PMID: 24319194. doi:10.1182/asheducation-2013.1.292
8. Luken BM, Turenhout EA, Hulstein JJ, Van Mourik JA, Fijnheer R, Voorberg J. The spacer domain of ADAMTS13 contains a major binding site for antibodies in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 2005;93(2):267–274. PubMed PMID: 15711742. doi:10.1160/TH04-05-0301
9. Fanouriakis A, Tziolos N, Bertsias G, Boumpas DT. Update on the diagnosis and management of systemic lupus erythematosus. Ann Rheum Dis. 2021;80(1):14–25. PubMed PMID: 33051219. doi:10.1136/annrheumdis-2020-218272
10. Catalina MD, Owen KA, Labonte AC, Grammer AC, Lipsky PE. The pathogenesis of systemic lupus erythematosus: harnessing big data to understand the molecular basis of lupus. J Autoimmun. 2020;110:102359. PubMed PMID: 31806421. doi:10.1016/j.jaut.2019.102359
11. Cojocaru M, Cojocaru IM, Silosi I, Vrabie CD. Manifestations of systemic lupus erythematosus. Maedica. 2011;6(4):330–336. PubMed PMID: 22879850. Pubmed Central PMCID: PMC3391953.
12. Merayo-Chalico J, Demichelis-Gomez R, Rajme-Lopez S, et al. Risk factors and clinical profile of thrombotic thrombocytopenic purpura in systemic lupus erythematosus patients. Is this a distinctive clinical entity in the thrombotic microangiopathy spectrum?: a case control study. Thromb Res. 2014;134(5):1020–1027. PubMed PMID: 25257921. doi:10.1016/j.thromres.2014.09.005
13. Jaime-Perez JC, Aguilar-Calderon PE, Salazar-Cavazos L, Gomez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018;9:171–184. PubMed PMID: 30349415. Pubmed Central PMCID: 6190623. doi:10.2147/JBM.S176144
14. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114(15):3167–3172. PubMed PMID: 19638626. doi:10.1182/blood-2009-04-215368
15. Wada H, Matsumoto T, Suzuki K, et al. Differences and similarities between disseminated intravascular coagulation and thrombotic microangiopathy. Thromb J. 2018;16(1):14. doi:10.1186/s12959-018-0168-2
16. Kwok SK, Ju JH, Cho CS, Kim HY, Park SH. Thrombotic thrombocytopenic purpura in systemic lupus erythematosus: risk factors and clinical outcome: a single centre study. Lupus. 2009;18(1):16–21. PubMed PMID: 19074164. doi:10.1177/0961203308094360
17. Molinari AC, Banov L, Bertamino M, Barabino P, Lassandro G, Giordano P. A practical approach to the use of low molecular weight heparins in VTE treatment and prophylaxis in children and newborns. Pediatr Hematol Oncol. 2015;32(1):1–10. PubMed PMID: 25325764. Epub 2014/ 10/18.eng. doi:10.3109/08880018.2014.960119
18. Torrente-Segarra V, Monte TCS, Corominas H. Musculoskeletal involvement and ultrasonography update in systemic lupus erythematosus: new insights and review. Eur J Rheumatol. 2018;5(2):127–130. PubMed PMID: 30183613. Pubmed Central PMCID: PMC6072693. doi:10.5152/eurjrheum.2017.17198
19. Terrell DR, Vesely SK, Kremer Hovinga JA, Lammle B, George JN. Different disparities of gender and race among the thrombotic thrombocytopenic purpura and hemolytic-uremic syndromes. Am J Hematol. 2010;85(11):844–847. PubMed PMID: 20799358. Pubmed Central PMCID: PMC3420337. doi:10.1002/ajh.21833
20. George P, Das J, Pawar B, Kakkar N. Thrombotic thrombocytopenic purpura and systemic lupus erythematosus: successful management of a rare presentation. Indian J Crit Care Med. 2008;12(3):128–131. PubMed PMID: 19742252. Pubmed Central PMCID: PMC2738311. doi:10.4103/0972-5229.43682
21. Adam Z, Sokwala A, Shah J, Ali SK. A delay in diagnosis: thrombotic thrombocytopenia purpura occurring in systemic lupus erythematous. Pan Afr Med J. 2019;34:103. PubMed PMID: 31934246. Pubmed Central PMCID: 6945373. doi:10.11604/pamj.2019.34.103.20524
22. Aggarwal V, Singer Z, Ledingham D, Othman I. Refractory acquired thrombotic thrombocytopenic purpura in a patient with sickle cell trait successfully treated with caplacizumab. Hematology. 2021;26(1):590–593. PubMed PMID: 34396933. doi:10.1080/16078454.2021.1959984
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.