")
Back to Journals » International Journal of Nanomedicine » Volume 12
A quality by design approach on polymeric nanocarrier delivery of gefitinib: formulation, in vitro, and in vivo characterization
Authors Kola Srinivas NS, Verma R, Pai Kulyadi G, Kumar L
Received 19 September 2016
Accepted for publication 8 November 2016
Published 16 December 2016 Volume 2017:12 Pages 15—28
DOI https://doi.org/10.2147/IJN.S122729
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Navya Sree Kola Srinivas,1 Ruchi Verma,2 Girish Pai Kulyadi,1 Lalit Kumar1
1Department of Pharmaceutics, 2Department of Pharmaceutical Chemistry, Manipal College of Pharmaceutical Sciences, Manipal University, Manipal, Karnataka, India
Abstract: Gefitinib is an anticancer agent which acts by inhibiting epidermal growth factor receptor tyrosine kinase receptors. The aim of the present study was to prepare gefitinib nanosuspension. Gefitinib was encapsulated in Eudragit® RL100 and then dispersed in stabilizer solution, polyvinyl alcohol, and polyvinylpyrrolidone K30. Nanosuspension was prepared by using homogenization and ultrasonication techniques. The quality by design approach was also used in the study to understand the effect of critical material attributes (CMAs) and critical processing parameters (CPPs) on critical quality attributes and to improve the quality and safety of formulation. To study the effect of CMAs and CPPs, 23 full factorial design was applied. The particle size, polydispersity index, and zeta potential of the optimized solution were 248.20 nm, 0.391, and -5.62 mV, respectively. Drug content of the optimized nanoformulation was found to be 87.74%±1.19%. Atomic force microscopy studies of the optimized formulation confirmed that the prepared nanoparticles are smooth and spherical in nature. In vitro cytotoxicity studies of the nanosuspension on Vero cell line revealed that the formulation is nontoxic. The gefitinib nanosuspension released 60.03%±4.09% drug over a period of 84 h, whereas standard drug dispersion released only 10.39%±3.37% drug in the same duration. From the pharmacokinetic studies, half-life, Cmax, and Tmax of the drug of an optimized nanosuspension were found to be 8.65±1.99 h, 46,211.04±5,805.97 ng/mL, and 6.67±1.77 h, respectively. A 1.812-fold increase in relative bioavailability of nanosuspension was found, which confirmed that the present formulation is suitable to enhance the oral bioavailability of gefitinib.
Keywords: gefitinib, cancer, epidermal growth factor receptor tyrosine kinase receptors inhibitor, bioavailability, Eudragit® RL100, PVP K30, PVA, Biopharmaceutical Classification System class II, QbD, design of experiment, full factorial design
Introduction
Gefitinib is a drug which is used for the treatment of certain cancers such as colon, lung, ovarian, and breast cancer.1–3 It acts by inhibiting epidermal growth factor receptor tyrosine kinase4 and belongs to Biopharmaceutical Classification System class II. It is sparingly soluble at pH 1 which further decreases with respect to the increase in pH; its solubility decreases sharply in the upper gastric range especially between pH 4 and 6. Its low solubility in upper gastric fluid affects the onset of action, bioavailability, and therapeutic activity. The log P-value of gefitinib is 4.15, which shows that gefitinib is highly hydrophobic.5 The daily oral dose and bioavailability of gefitinib are 250 mg and <44%, respectively. The most common adverse drug reactions reported with 250 mg dose of gefitinib are hepatobiliary disorder (elevations in alanine aminotransferase), gastrointestinal disorders (diarrhea, nausea, vomiting, and stomatitis), metabolism and nutrition disorder (anorexia), skin and subcutaneous disorders (skin reactions, pustular rash, and itching with dry skin), and so on.6 All the aforementioned problems clearly indicate that there is a need to reduce the daily oral dose and to improve the oral bioavailability of gefitinib. The nanoparticulate drug delivery system holds abundant potential to overcome these problems. Hence, in the present study, Eudragit® RL100 nanosuspension of gefitinib was prepared to improve its oral bioavailability. The reasons for the selection of Eudragit® RL100 to prepare the nanosuspension were its high physicochemical stability,7,8 high permeability profile, mucoadhesiveness, drug targeting to the gastrointestinal tract, high gastrointestinal retention time,9–12 and nontoxic nature.8,13
The major challenges in the preparation of nanoparticles are manufacturing variability, due to lack of understanding of the effect of critical material attributes (CMAs) and critical processing parameters (CPPs) to attain small size, narrow polydispersity index (PDI), and so on. This lack of understanding and manufacturing variabilities increase the cost of nanoparticulate drug delivery system.14 Nanoparticulate drug delivery system is also known for its toxicities due to the fast onset of action, increase in solubility, permeability, and bioavailability. Hence, the aim of the present study was to prepare the gefitinib nanosuspension with the help of quality by design (QbD) approach to understand the effect of CMAs and CPPs on critical quality attributes (CQAs) to reduce the manufacturing variability, control the manufacturing cost, and to improve the formulation safety and quality.
Materials and methods
Materials
Gefitinib and Eudragit® RL100 were obtained as gift samples from Neon Laboratories Ltd. (Mumbai, India) and Dr Reddy’s Laboratories Ltd. (Hyderabad, India), respectively. Polyvinyl alcohol (PVA) with molecular weight 13,000 was obtained from National Chemicals (Gujarat, India). Polyvinylpyrrolidone (PVP) K30 with molecular weight 40,000 was procured from HiMedia® Laboratories Pvt. Ltd. (Mumbai, India). Methanol high-performance liquid chromatography (HPLC) grade and dimethyl sulfoxide (DMSO) were purchased from Finar Ltd. (Ahmedabad, India). Highly pure water (18.2 MΩ cm resistivity, Milli Q) was produced in the laboratory. Dulbecco’s Modified Eagle’s Media (DMEM) and trypsin ethylenediaminetetraacetic acid (EDTA) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Fetal bovine serum (FBS), sterile phosphate-buffered saline (PBS), 3-(4,5-dimethyl thiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT), and antibiotic solution (a mixture of penicillin, streptomycin, and clarithromycin) were obtained from HiMedia® Laboratories Pvt. Ltd. All other chemicals were purchased from Merck Ltd. (Mumbai, India).
Methods
Preparation of gefitinib nanosuspension
Gefitinib (100 mg) was dissolved in DMSO (1 mL) and Eudragit® RL100 (200 mg) was dissolved in methanol (1 mL) separately. Then, both were mixed together in a 2 mL microcentrifuge tube. PVA as a stabilizer solution (20 mL) was taken in a 50 mL glass beaker and was kept in an ice bath. The above organic solution of drug and polymer was added dropwise into the stabilizer solution using a syringe drop by drop with continuous homogenization (Polytron PT-MR 3100; Kinematica AG, Luzern, Switzerland) at 15,000 rpm for 10 min. Then, it was further added drop by drop into PVP as a co-stabilizer solution (20 mL) kept in an ice bath to maintain low temperature with continuous homogenization at 15,000 rpm for 10 min. After homogenization, the preparation was sonicated immediately (Ultrasonic Processor, VC 130; Sonics & Materials Inc., Newtown, CT, USA) at 36±1 W for specified time using 10 s pulses. Then the prepared nanoformulation was subjected to magnetic stirring (3 h) at room temperature for the evaporation of organic solvent.
Quality target product profile (QTPP)
In order to determine the QTPP, regulatory, practical aspects, scientific aspects, risk, and other factors were considered. Target product profile and target product quality profile (TPQP) were defined as the goal (QTPP) of the study. Control space (QTPP) also helped to establish the region of operability. CMAs and CPPs were selected in this study to achieve the predefined target (QTPP). The identified QTPP, CMAs, and CPPs are given in Table 1.14–22
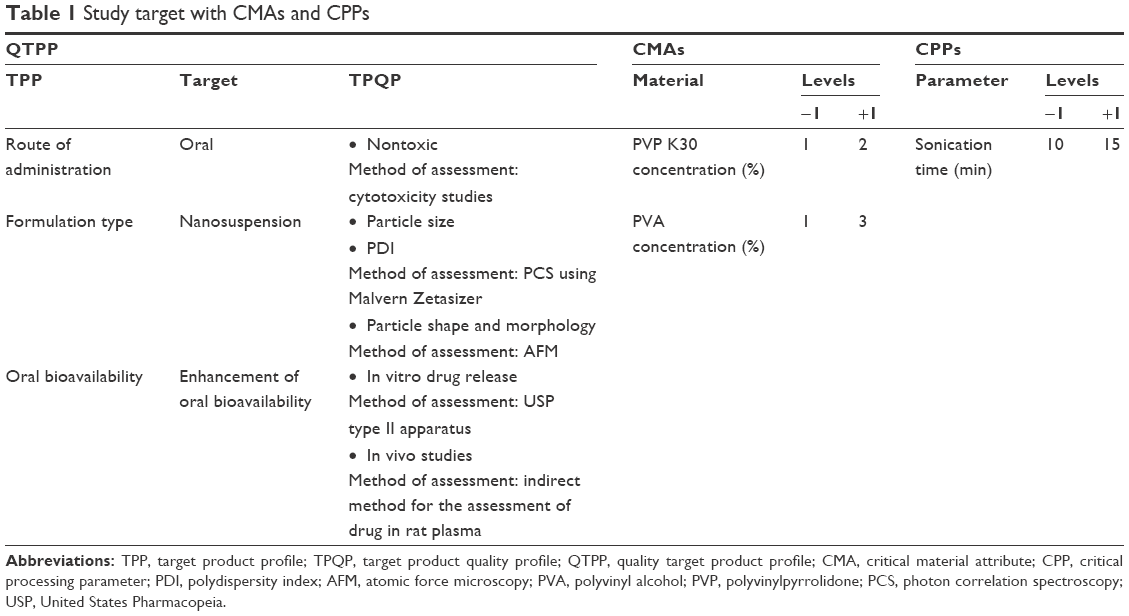 | Table 1 Study target with CMAs and CPPs |
Risk assessment of CQAs
An Ishikawa diagram was constructed to identify the potential risks and corresponding causes that have the greatest possibility of producing product failure.14,20,22–24 This approach helps to enhance safety, efficacy, and quality of the pharmaceutical product.17 On the basis of prior knowledge, experiment trials, literature review, and the application of failure mode effect analysis method, the major quality attributes (or most dependent response parameters) of the nanoparticles such as particle size, PDI, and zeta potential were selected for risk assessment and analysis of CQAs.14,24,25 The response parameters were considered as the CQAs and were analyzed within the pre-decided knowledge space. Figure 1 shows the Ishikawa diagram illustrating the effect of CMAs and CPPs on TPQP.
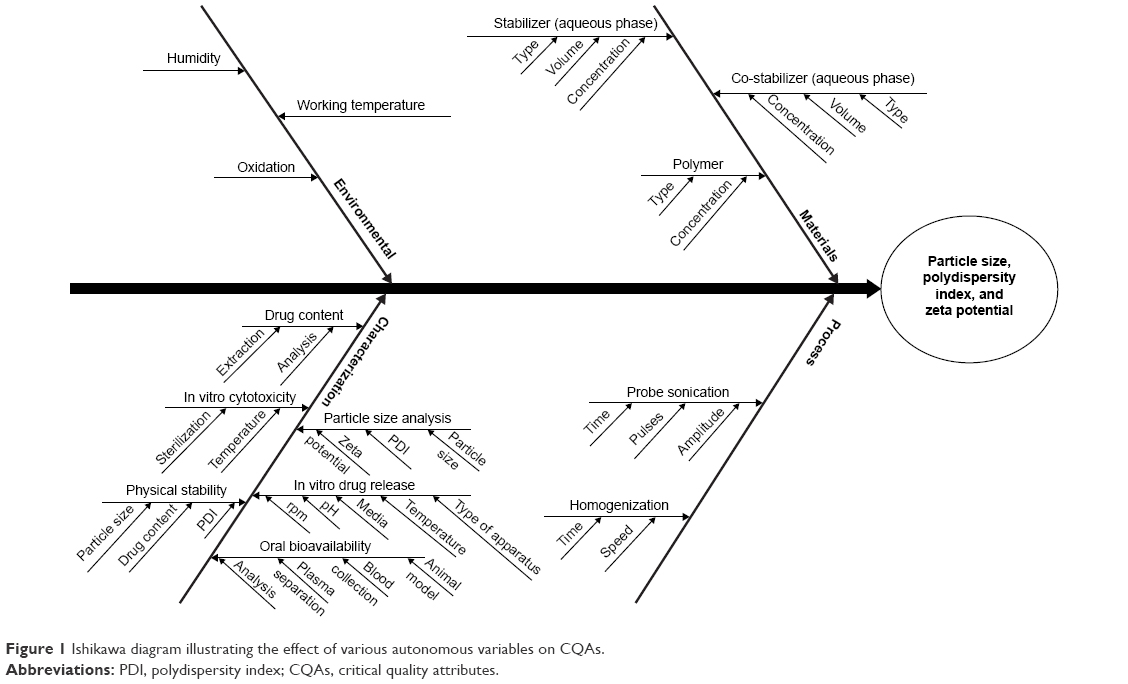 | Figure 1 Ishikawa diagram illustrating the effect of various autonomous variables on CQAs. |
Design of experiment for optimization of CMAs and CPPs
The selection of the optimization design is based upon the number of autonomous variables and their levels. The formulation (or processing) design is a part of region of operability, and in this study, it was customized on the basis of the decided CMAs and CPPs with their different levels. The CQAs are mainly influenced by CMAs and CPPs. Hence, these should be scrutinized and organized as a part of knowledge space to produce the obligatory quality of the product.25,26 In the present study, CMAs and CPPs were considered as autonomous variables, namely concentration of PVP K30 (X1), concentration of PVA (X2), and sonication time (X3) with two different levels of each (as given in Table 1). Similarly, three CQAs (or responses) such as particle size (Y1), PDI (Y2), and zeta potential (Y3) were considered for optimization purpose. The polynomial equations were constructed by Design Expert® software and also reviewed for statistical significance to develop the models. Formulation was optimized to establish the region of interest by means of considering the desirability values of CQAs. Optimized formulation was prepared again and reevaluated with respect to particle size, PDI, and zeta potential. Then, the residual values were calculated from actual obtained results and software predicted results of CQAs to establish the design space. The residual values help in the verification of feasibility and validation of statistical models. On the basis of design space and/or residual values, the control strategy was adopted for pharmaceutical development and quality system17,20,26 to achieve the target. Three-dimensional (3D) response surface plots were also constructed using Design Expert® software for better understanding of the effect of CMAs and CPPs on CQAs.14 Eight experimental runs were carried out in the knowledge space on the basis of 23 full factorial design.
Process analytical technology (PAT) – particle size analysis
Zetasizer (ZEN 3600; Malvern Instruments, Malvern, UK) was used as PAT for the analysis of particle size, PDI, and zeta potential.
The prepared nanosuspension was diluted 10 times with Milli-Q water to produce the working concentration. The diluted nanosuspension (1 mL) was taken in a zeta cell, and the cell was fixed into the Zetasizer to record the zeta potential. Similarly, the diluted nanosuspension (2 mL) was placed in the transparent glass cell to record the particle size and PDI. Analysis of samples was carried out at 90° scattering angle. This experiment was performed at 25°C.27
Drug content
Nanosuspension was diluted with methanol in a ratio of 1:5% v/v and sonicated (Equitron; Medica Instrument Manufacturing Co., Mumbai, India) for 5 min. A 10 mL volume was obtained using mobile phase (acetonitrile and 40 mM ammonium formate buffer with pH 2.5 in the ratio of 30:70% v/v) (unpublished method) and resonicated for 10 min. The obtained solution was filtered using a syringe filter with pore size 0.22 μm. Presence of drug was determined using HPLC (LC-2010CHT; Shimadzu Corporation, Kyoto, Japan), and the percent drug content was calculated.
Atomic force microscopy (AFM)
Surface morphology analysis of the nanoparticles was performed using scanning probe microscopy method, namely AFM (Bruker Innova; Bruker Corporation, Billerica, MA, USA). AFM experiment was performed by depositing a drop of diluted nanosuspension (100 times dilution with Milli-Q water) on a glass coverslip followed by drying under vacuum over a period of ~10 h at 25°C and 15 bar pressure. AFM imaging was carried out under ambient conditions using the tapping mode with resonance frequencies of 260–340 kHz.28
In vitro drug release study
In vitro drug release of an optimized nanosuspension was carried out by taking the nanosuspension (4 mL) in a dialysis membrane (molecular cutoff 12–14 kDa; HiMedia® Laboratories Pvt. Ltd.) while closing the ends with dialysis clips and suspending in the dissolution medium. The study was carried out using United States Pharmacopeia (USP) type II dissolution apparatus. The same procedure was followed for the standard while taking the drug (9 mg) in 4 mL Milli-Q water. Study was performed in phosphate buffer pH 6.8. Volume of dissolution used in the study was 500 mL. The speed of the paddle and temperature of the medium were maintained at 50 rpm and 37°C±0.5°C, respectively. The samples (2 mL) were collected at predetermined time intervals and sink conditions were maintained throughout the experiment by replacing the same volume of the sample with blank phosphate buffer. The collected samples were filtered, diluted (with mobile phase), and analyzed using HPLC method.
In vitro cytotoxicity study
The Vero cell line (African green monkey kidney cells) was obtained from the National Centre for Cell Science, Pune, India. The Vero cells were cultured in DMEM containing 10% FBS and 1% penicillin–streptomycin. Prepared test samples (100 μL) were added in triplicates to the wells containing cell suspension in three different, sterile, 96-well tissue culture plates. Then, the tissue culture plate was incubated at 37°C for 24 and 48 h with 5% carbon dioxide supply. Negative control (DMEM without FBS), positive control (with standard pure drug), and DMSO in the same concentration (100 μL with 900 μL DMEM without FBS) were also maintained in triplicate in the tissue culture plate. Then, the samples were analyzed under a microscope for the effect of drug on cells or presence of infections. After 24 and 48 h, the supernatant was decanted from the wells of the tissue culture plate and each well was washed with 100 μL of PBS. In dark, MTT reagent (50 μL) was added to each well and again the tissue culture plate was incubated at 37°C with 5% carbon dioxide for 4 h. The solution was gently decanted from each well. Then, 100 μL of sterile DMSO (after filtration) was added to each well and vigorously shaken in order to ensure solubilization of the formazan crystals formed. Absorbance was recorded using an enzyme-linked immunosorbent assay microplate reader (ELx – 800MS absorbance reader; BioTek® Instruments, Inc., Winooski, VT, USA) at 540 nm wavelength. Plates were read within 30 min after the addition of DMSO.29–33 Percent cell viability and CC50 values were calculated.34 Percent cell viability and percent cell inhibition were calculated using the following formulae:
|
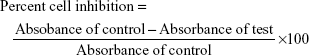
Percent cell viability = 100 − percent cell inhibition. |
Pharmacokinetic study
Animals
Pharmacokinetic study of the nanosuspension and standard drug dispersion (gefitinib in Milli-Q water) was performed on female Wistar rats (200–250 g) procured by Central Animal Research Facility, Manipal University, Manipal. The animal experiment protocol was reviewed and approved by KMC Manipal, Institutional Animal Ethics Committee (IAEC), Manipal University, Manipal (IAEC/KMC/95/2015) dated November 20, 2015. All Animal experiments were performed as per the Committee for the Purpose of Control and Supervision on Experiments on Animals (CPCSEA) and the animal care guidelines of KMC Manipal, IAEC.
Study design and blood sample collection
Before the experiment, the animals had free access to standard grub and water ad libitum. Throughout the experiment, all the animals were maintained at 12 h dark and 12 h light cycles. The 12 female Wistar rats were randomly divided into two groups as six animals in each group. The groups were marked as “standard” and “test”. The standard group received orally a pure drug dispersion prepared in 0.25% sodium carboxymethyl cellulose (NaCMC) at a dose of 5 mg per 250 g body weight. Similarly, the test group received orally a nanosuspension at the dose of 5 mg per 250 g body weight.
Blood sample (500 μL) was collected at 0, 0.25, 0.5, 1, 2, 4, 6, 10, 12, 24, 36, 48, 60, and 72 h time intervals from each rat by retro orbital plexus puncture method in a microcentrifuge tube containing 10 μL dipotassium EDTA solution (10% w/v). Then, the plasma was separated from collected blood samples using a cooling centrifuge (3K30; Sigma® Laborzentrifugen, Osterode am Harz, Germany) at 15,000 rpm for 15 min at 4°C. The separated plasma samples were stored at −20°C until further analysis.35–37
Drug analysis in plasma samples
The presence of drug in rat plasma was determined using HPLC (unpublished data). Chromatographic conditions such as HyperClone C18 column (250×4.6 mm id, 5 μm, BDS 130 Å) as a stationary phase, a mixture of acetonitrile: 40 mM ammonium formate buffer with pH 2.5 (28:72% v/v) as a mobile phase, flow rate: 0.8 mL/min, UV detector with wavelength 248 nm, column temperature maintained at 25°C, and injection volume: 80 μL were used. The developed method was validated as per USFDA guidelines.
A plasma sample (200 μL) was taken in the microcentrifuge tube. Sodium hydroxide solution (NaOH, 0.1 M, 40 μL) was added and vortexed (Spinix™ MC-01, Vortex Shaker; Tarsons® Products Pvt. Ltd., Kolkata, India) for 1 min. Then, acetonitrile (560 μL) was added and again vortexed for 5 min. After vortexing, the supernatant solution was collected by means of cooling by centrifuging at 15,000 rpm for 15 min at 4°C. The supernatant was collected in test tubes and was subjected to nitrogen evaporation at 50°C until complete evaporation of the solvent. The residual was reconstituted in mobile phase(acetonitrile: 40 mM ammonium formate buffer with pH 2.5 [28:72%v/v]), and the concentration of the drug in plasma was estimated using developed HPLC bioanalytical method.
The plasma drug concentrations obtained from each group were fed into WinNonlin standard edition v.5.2 (Pharsight, Mountain View, CA, USA) with respect to time to calculate the pharmacokinetic parameters. The noncompartmental method was used for the determination of pharmacokinetic parameters. The peak plasma concentration (Cmax) and time to achieve peak plasma concentration (Tmax) were calculated by plotting the plasma drug concentration against time. The area under the curve from time of administration to time “t” (AUC0–t) was obtained by using trapezoidal rule. The elimination half-life (t1/2) was calculated from elimination rate constant (Ke). The clearance (Cl) of gefitinib was deduced from the product of Ke and volume of distribution (Vd). The relative bioavailability was calculated from the obtained AUC0–t of nanosuspension to AUC0–t of standard drug dispersion.36,38,39
|
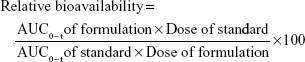
The in vivo permeability of gefitinib from the nanosuspension was also determined using the given formula:40
|
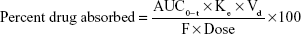
where, AUC0–t = last area under the curve, Ke = elimination rate constant, Vd = volume of distribution, and F = percent bioavailability of gefitinib from the formulation.
Stability studies
Accelerated stability studies of the optimized nanosuspension were carried out at 5°C±3°C. The nanosuspension was filled into amber-colored glass vials and sealed with rubber stopper and aluminum caps. The vials were stored at the specified temperatures for 90 days. The samples were collected at 0, 7th, 15th, 30th, and 90th day from these vials. The collected samples were analyzed with respect to particle size, PDI, and zeta potential.21,41
Statistical analysis
Two-way analysis of variance (ANOVA) followed by Bonferroni post hoc test was applied for the analysis of in vitro drug release and pharmacokinetic studies. The results were considered to be significant at probability level <5% (P<0.05). The statistical analysis was performed using GraphPad Prism® v.5 (GraphPad Software, Inc., La Jolla, CA, USA).42
Results and discussion
Preparation of gefitinib nanosuspension
Gefitinib nanosuspension was prepared using solvent evaporation technique. Eudragit® RL100, PVA, and PVP K30 were used for the preparation of the nanosuspension.
Eudragit® RL100 was chosen because of its very fine particle distribution, good physicochemical stability,7,8 high permeability profile, mucoadhesiveness, drug targeting to the gastrointestinal tract, high retention time,9–12 and no toxicity/irritation properties.8,13 High permeability, high retention, physicochemical stability, and targeting to the gastrointestinal tract are mainly due to the presence of ammonium groups in its chemical structure. Because Eudragit® RL100 is hydrophobic in nature, it is also expected that drug entrapment in the nanoparticles prepared using this polymer will be high.7,43–45
PVA was used for the stabilization of gefitinib nanoparticles.27 It is nonionic, nontoxic, noncarcinogenic, and has a high degree of swelling in aqueous solutions and good biocompatibility. However, sometimes it produces a skewed distribution pattern of nanoparticles and also increases the viscosity of suspension.46 Hence, PVP K30 was used further as a co-stabilizer to ensure spherical and small particle size. It also helps to retain low viscosity of the suspension.47 Other reasons behind the selection of PVP K30 were its hydrophilic nature, high melting point, nonionic, nontoxic nature, high physiological tolerance, and a powerful precipitation inhibitor effect. These properties maintain the supersaturation state of the drug in the gastrointestinal fluid.48–50 PVP K30 produces amorphous and/or smooth-surfaced spherical particles.50 PVP is approved by the Food and Drug Administration and is a generally regarded as a safe polymer.51 It has also been reported that the nanoparticles produced with PVA and PVP improve the correlation between the nanoparticle stability and monomeric length of the polymer chain.44 Earlier studies have also proposed that nonionic surfactants have a tendency to adsorb on hydrophobic surfaces.44,45 The adsorption aids to restrict polymer segments to the particle surface and the adsorbed chains extend into the solution, which restricts the chains and lowers the chain entropy. Finally, it enhances the stability by repulsive interaction energy. It has also been proved that PVA and PVP provide protection from oxidation and also reduce the chances of aggregation of prepared nanoparticles by providing a steric barrier.44,45,52–54
In the present study, DMSO was used to dissolve the drug. Methanol was used as an organic solvent to dissolve Eudragit® RL100, because it is compatible with DMSO. As per the USP 37/NF 32, the acceptance limit of methanol as residual solvent is high compared to other solvents of the same category, but it is placed in the safest category of ICH, class 2, with limited nongenotoxic animal carcinogens or possible causative agents of other irreversible toxicity. Hence, further magnetic stirring was applied at room temperature (3 h) for the evaporation of methanol.55 No additional process was applied for the removal of DMSO from the formulation, because DMSO is considered to be a safe solvent when compared to methanol and is placed in the safest category of ICH, class 3 solvents, with low toxicity potential. DMSO ≤50 mg per day as a residual solvent would be considered acceptable (corresponding to 0.5% under option 1).55–57 DMSO (~0.025%) was used in the preparation of gefitinib nanoparticles which was considered to be safe and within the acceptable limits of DMSO. Furthermore, DMSO is considered to be safe as is it metabolized in humans by oxidation to dimethyl sulfone (DMSO2) or by reduction to dimethyl sulfide (DMS). DMSO and DMSO2 are excreted in urine and feces. DMS is eliminated through breath and skin with a characteristic “garlic” or “oyster-like” odor.55–57
PAT – particle size analysis
Gefitinib nanosuspension was prepared successfully using Eudragit® RL100 with the applications of QbD. Particle size, PDI, and zeta potential were found to be in the range of 249.8–597 nm, 0.294–0.456, and −4.26 to −10.79 mV, respectively.
Risk analysis, optimization, and verification
Full factorial design was implemented for the optimization of the final formulation. The main and interactive effects of three autonomous variables (such as CMAs and CPPs) on three responses (CQAs) were analyzed. The effect of autonomous variables on CQAs was assessed by using polynomial equation and 3D response surface plot constructed by the software as shown in Figure 2.
 | Figure 2 3D response surface plot illustrating the effect of autonomous variables on CQAs. |
The polynomial equations for particle size, PDI, and zeta potential are as follows:
Particle size = 389.280 − 26.880X1 − 48.750X2 − | (1) |
PDI = 0.150 + 0.170X1 + 0.049X2 + 0.013X3 − | (2) |
Zeta potential = −7.55 + 0.50X1 + 2.69X2 | (3) |
where X1, X2, and X3 are concentrations of PVP and PVA and sonication time, respectively.
A significant effect was assessed for the determination of percentage variabilities in CQAs, and a probability value of P<0.05 was considered as a significant level.
Model selection for particle size
For particle size, model R2FI with an F value of 26.58 was found to be significant (P=0.0366). The regression coefficient with a P-value <0.05 indicates that the model terms are significant, whereas values >0.1000 indicate insignificant model terms. Similarly, the significant effect of main and interactive variables on the response particle size was also assessed. The P-value of concentration of PVA was found to be 0.0463 whereas the P-value for sonication time was found to be 0.0260. The concentration of PVP and PVA as interactive variables also showed a significant effect on particle size with P-value 0.0154. The adequate precision value for R2FI model was found to be 13.475. This is a measure of signal to noise ratio. For a precise model, the minimum adequate precision value should be 4.
The R2 value is a measure of total variability that has been explained by the model, and it was found to be 0.9852 which means 98.52% variations have been explained by the present model.58–60 The actual R2 value was more than the predicted R2 value (0.7628), which also reflects the suitability of the present R2FI model.
The coefficient estimate values of concentration of PVP, concentration of PVA, and sonication time as individual (or main) variables were found to be negative. Similarly, the coefficient estimate value of concentration of PVP and sonication time as interactive (complex) variables was also found to be negative. However, the coefficient estimate value of concentration of PVP and concentration of PVA as interactive variables was positive. A positive value of coefficient estimate indicates that the particle size increases with respect to an increase in the value of autonomous variable, whereas a negative value of coefficient estimate indicates that the particle size decreases with respect to the increase in the value of autonomous variable. It is very interesting to note that PVA as an individual variable behaves as a droplet stabilizer whereas as an interactive variable it increases the size of nanoparticles. The stabilizer property of the droplets prevents the coalescence of droplets by localizing at the surface of the interface between the dispersed phase and continuous phase. This observation coincides with earlier findings in this area regarding the effect of PVA concentration on particle size.61,62
Furthermore, the interactive effect of variables was also determined with the help of a 3D response surface plot. As depicted in Figure 2A, the particle size decreased as the sonication time and concentration of PVP increased. It shows the perfect combination of these two variables. Among them, one (sonication time) helps to reduce the particle size and the other (concentration of PVP) helps to reduce the steric charge generated on the surface of the particles during size reduction. As shown in Figure 2B, the particle size increased as the concentrations of PVP and PVA increased. This might have happened because the polymer chain begins to entangle together causing increased viscosity of PVA in the medium due to the increase in concentration; thus more polymer molecules may have been coated on the surface of the nanoparticles.44
Model selection for PDI
For PDI, model R2FI with F value of 476.32 was found to be significant (P=0.0021). P<0.05 was considered as a significant level. Furthermore, the significant effect of main and interactive variables on PDI was also assessed. The P-value of all autonomous variables as main and interactive was found to be between 0.0014 and 0.0290, which indicates that the variables have a significant effect (individual/complex) on PDI. The adequate precision value for R2FI model was found to be 57.388, which is a strong measure of signal to noise ratio.
The R2 value for this model was found to be 0.9992 which means 99.92% variations have been explained by the present model.59–61 This actual R2 value (0.9992) was better than the predicted R2 value (0.9866). It strongly supports the suitability of the present R2FI model.
The coefficient estimate values of individual variables such as concentration of PVP, concentration of PVA, and sonication time were found to be positive, whereas the coefficient estimate values of sonication time, concentration of PVP, and concentration of PVA as complexed variables were found to be negative.
Furthermore, the interactive effect of variables was also determined with the help of the 3D response surface plot. As shown in Figure 2C, PDI decreased as the sonication time and concentration of PVP increased. It also confirms the suitability of these two autonomous variables as complexed. As shown in Figure 2D, PDI decreased as the concentration of PVP and concentration of PVA increased, which means the nanoparticles have a monodisperse nature.42 This also reflects that PVA produces the uniform coating on the surface of nanoparticles, which contributes to enhance the physical stability of colloidal formulation.44
Model selection for zeta potential
For zeta potential, linear model with F value of 719.54 was found to be very significant (P<0.0001). Furthermore, the significant effect of concentration of PVP and concentration of PVA was also assessed. The P-values of concentration of PVP and concentration of PVA were found to be 0.0010 and <0.0001, respectively. This indicates that the variables have a significant effect on zeta potential. The adequate precision value for this model was found to be 51.021. It can strongly measure the signal to noise ratio.
The R2 value for this model was found to be 0.9965 which means 99.65% variations have been explained by the present model.58–60 The actual R2 value (0.9965) was found to be almost similar to the predicted R2 value (0.9911). Hence, this is also a supportive evidence for the selected model. The coefficient estimate values of concentration of PVP and concentration of PVA were found to be positive, which clearly defines that the zeta potential values increased with respect to the increase in concentration of each variable.
Further, the effect has been demonstrated with a 3D response surface plot. As shown in Figure 2E, the zeta potential values increased with increase of concentration of PVP and concentration of PVA, which also confirms that PVP and PVA help to improve the physical stability of colloidal formulation.44
Optimization of CMAs and CPPs with verification of CQAs
The targeted criteria were fed into the software to achieve the predicted composition (software suggestions). On the basis of desirability value, a software-suggested solution was selected as a region of interest and was practically used for its verification. The desirability value of the selected software suggestion was found to be 0.986, which provides an assurance of 98.60% possibilities to achieve the target with optimized CMAs and CPPs. Higher the value of desirability, more the possibility to achieve the target.63 A formulation was prepared with optimized CMAs and CPPs, and its CQAs were analyzed. The actual obtained results and predicted results of CQAs were further used to calculate the residual values to ensure the achievement of design space. The calculation of residual values is also a verification/validation of the model and CQAs. The residual values were calculated as percent residual using the following formula:63
|
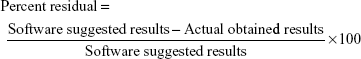
The optimized CMAs and CPPs with residual values of CQAs are summarized in Table 2. The residual values were found to be between the range of −3.49 and 1.01, and they were found to be very low, which shows that the actual obtained results have very strong correlation with software-predicted results. Lower residual value is also an indicator of less variation and more reproducibility of CQAs with the optimized CMAs and CPPs.
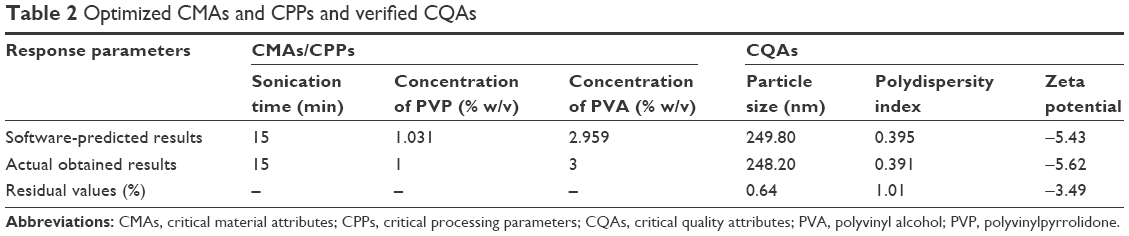 | Table 2 Optimized CMAs and CPPs and verified CQAs |
The optimized formulation showed the particle size to be <250 nm which indicates that the cellular uptake of the prepared formulation may be good, as cellular uptake depends upon the particle size.64,65 PDI value <0.4 confirms uniform and narrow PDI which also emphasizes that the obtained particles are of same size. The zeta potential values were found to be very low which indicates that formulation may be less stable. As the zeta potential values were found to be negative, which may not much support cellular uptake, it is expected to be safe as negatively charged nanoparticles decrease the chances of cytotoxicity. Because of the negative charge on the surface of nanoparticles, they may undergo nonspecific adsorption to the cellular membrane.
Characterization of optimized nanosuspension
Drug content
The optimized nanosuspension was prepared thrice and analyzed using HPLC. The percent drug content of the optimized nanosuspension was found to be 87.74%±1.19%. A slight decrease in the drug content was found because gefitinib is light and heat sensitive.66 In the present study, the temperature was under control during the preparation of nanoparticles. Hence, light may be the main degradation pathway which was responsible for the degradation of gefitinib in nanosuspension.
AFM
AFM is a powerful tool for the determination of surface morphology.28 A thin film of nanoparticles was subjected to AFM, which was set in tapping mode. The reason for selection of the tapping mode was to avoid damage and removal of weakly bound and defective layers of PVP and PVA (as adsorbate) on the surface of Eudragit® nanoparticles (as adsorbent). In the contact mode, the hard surfaces may destroy the adsorbed morphology.28,67 The results for the particle size obtained with AFM were closer to that obtained with the Zetasizer. The AFM 2D and 3D images of gefitinib nanoparticles (recorded at 20×20 μm magnification) are shown in Figure 3A and B. The gefitinib nanoparticles (GNPs) were spherical in shape with smooth surface. These results are similar to those nanoparticles prepared with Eudragit® L100 and PLGA using PVA as a stabilizer.68 It also proves that PVA helps to impart the spherical and smooth surface on nanoparticles.
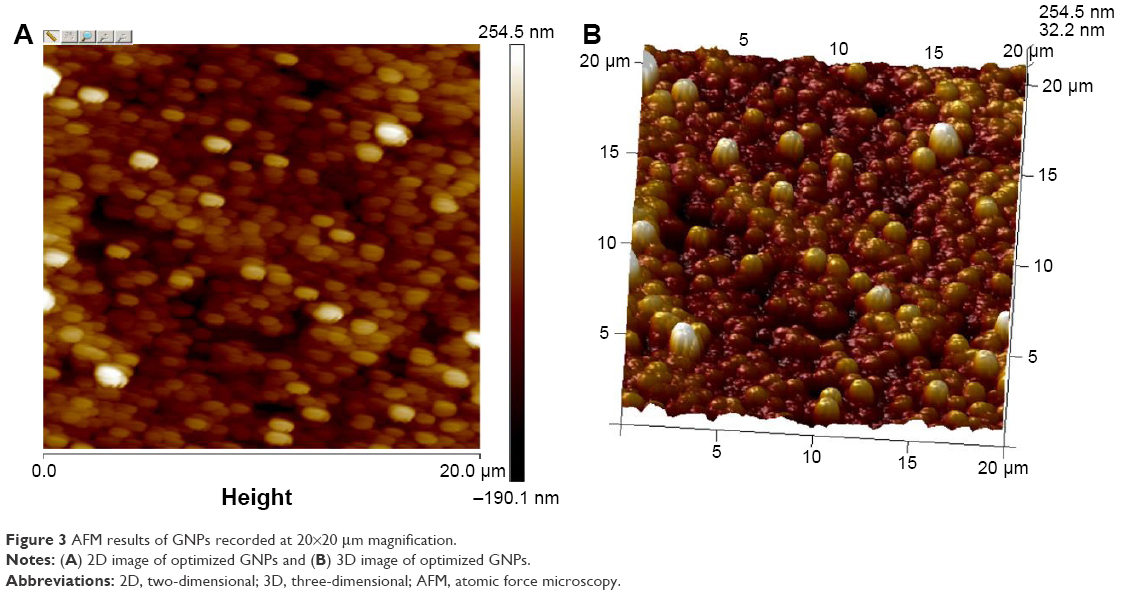 | Figure 3 AFM results of GNPs recorded at 20×20 μm magnification. |
In vitro drug release study
In vitro drug release studies were conducted using USP type II apparatus. The main reason for the selection of the dissolution method was to maintain the required temperature and stimulate biological conditions throughout the experiment, which is slightly difficult in diffusion studies of nanosuspensions. Up to 8 h, the release from gefitinib nanosuspension was very slow, that is, 5.30%±1.87%, which may be because the outer shell polymer took longer to produce the stagnant layer (or hydrated)69,70 or may be because of slow diffusion of drug from the stagnant layer formed by the outer shell polymer. It evidenced that the drug is entrapped in the core nanoparticles. These results are similar to the results reported by Lee et al.70 After producing the stagnant layer (or polymer hydration), it followed the first-order drug release pattern as a result in 84 h; drug release from the formulation was found to be 60.03%±4.09%. This type of drug release behavior observed may be due to the combination of polymers. The release from the standard drug dispersion was found to be very less, that is, 10.39%±3.37% in 84 h, in comparison with the gefitinib nanosuspension. As shown in Figure 4, in 84 h with the nanosuspension of gefitinib ~6-fold increase in solubility was found in comparison with the standard drug dispersion. This study confirmed that the decrease of particle size (as nanoparticles) helps to improve the solubility of the drug.
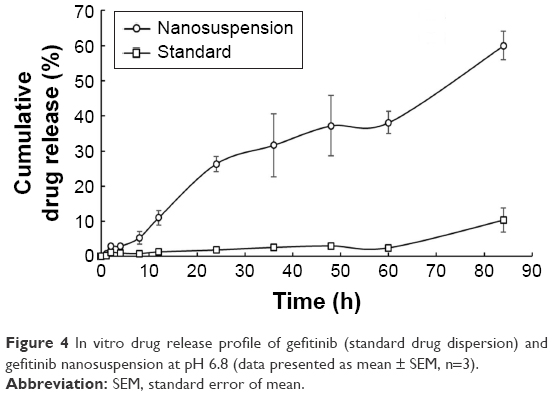 | Figure 4 In vitro drug release profile of gefitinib (standard drug dispersion) and gefitinib nanosuspension at pH 6.8 (data presented as mean ± SEM, n=3). |
In vitro drug release data were further analyzed for statistical analysis by applying two-way ANOVA followed by Bonferroni post hoc test. Significance (at P<0.001) was observed between the drug release profile of gefitinib (standard drug dispersion) and gefitinib nanosuspension.
In vitro cytotoxicity study
In vitro cytotoxicity studies, performed on Vero cells, indicate that the cell inhibition capacity of gefitinib (standard) is higher than the gefitinib nanosuspension, making the formulation safer to use. As shown in Table 3, CC50 value of viable cells is nearly 10 times in gefitinib nanosuspension wells as compared to the gefitinib wells. This phenomenon indicates that nanosuspension of gefitinib has low cytotoxicity in comparison with gefitinib.
 | Table 3 In vitro cytotoxicity assay of gefitinib and gefitinib nanosuspension against Vero cells |
Pharmacokinetic study
Plasma pharmacokinetic profile of gefitinib and gefitinib nanosuspension is illustrated in Figure 5. Pharmacokinetic parameters were calculated using WinNonlin Software v.5.2. (Pharsight Corporation) and are presented in Table 4.
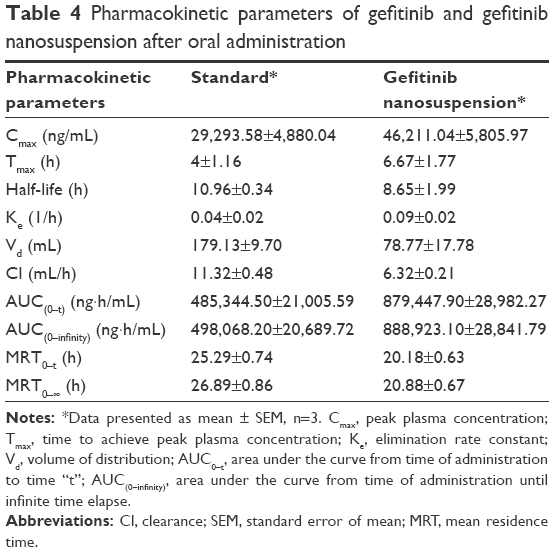 | Table 4 Pharmacokinetic parameters of gefitinib and gefitinib nanosuspension after oral administration |
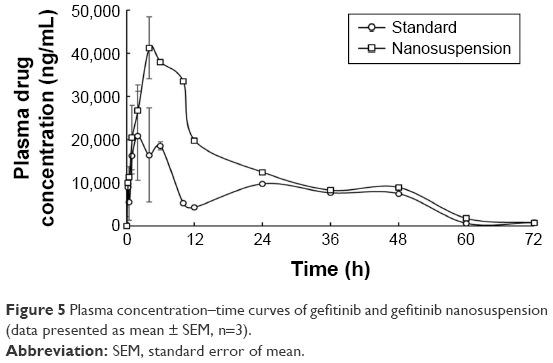 | Figure 5 Plasma concentration–time curves of gefitinib and gefitinib nanosuspension (data presented as mean ± SEM, n=3). |
Pharmacokinetic parameters of gefitinib (standard drug dispersion) were compared with gefitinib nanosuspension. In comparison with the standard, Cmax, Tmax, and AUC values increased with gefitinib nanosuspension, which indicates the fast onset of action and long absorption phase of gefitinib from nanosuspension, but t1/2, Vd, Ke, and Cl values were found to decrease with gefitinib nanosuspension. The decrease in Vd indicates that the solubility of the drug is increased after preparation of the nanosuspension. This is also supported by the in vitro drug release studies as the drug release is found to be greater than the standard. The corresponding effect of Vd can also be seen on the obtained lower values of t1/2, Ke, Cl, and MRT. This also reflects that the gefitinib nanosuspension affects the termination of action. The increase in Cmax and AUC values in the case of gefitinib nanosuspension indicates that the present formulation may be helpful to reduce the dose of gefitinib. The relative bioavailability results of gefitinib nanosuspension also confirmed the 1.812-fold increase in oral bioavailability. The bioavailability results evidenced that the present formulation may be helpful to improve therapeutic activity and to reduce the dose of gefitinib.
The percent in vivo absorption of gefitinib from standard drug dispersion and nanosuspension was found to be 38.38% and 68.82%, respectively. This is a measure of in vivo permeability of the drug and is clearly reflected in the increase of in vivo permeability of gefitinib after preparing the nanosuspension.
Pharmacokinetic data were also analyzed for statistical analysis by applying ANOVA followed by Bonferroni post hoc test. Significance (at P<0.001) was observed between the plasma drug concentrations of gefitinib (standard drug dispersion) and gefitinib nanosuspension. This significant effect was mainly observed between 2 and 10 h.
Stability studies
The first reason for the selection of this temperature for the stability study was that gefitinib is stable at this temperature in well-closed amber-colored containers. Second, it is already proven with earlier studies that liquid dosage forms are less stable at higher temperature. Hence, to avoid repetition, lower temperature was selected for stability studies, which is also a standard temperature as per the ICH guidelines. The gefitinib-loaded polymeric nanoparticles showed an increment in particle size and PDI by approximately three times and two times, respectively, over a period of 90 days after storage at 5°C±3°C. A slight reduction in zeta potential was also observed after 90 days storage at 5°C±3°C. The physical stability results reflect that the gefitinib nanosuspension is less stable; hence, there is a need to convert this liquid dosage form into a solid dosage form to improve its stability.
Conclusion
In the present study, gefitinib nanosuspension was successfully prepared using continuous homogenization and ultrasonication techniques. The QbD approach was also applied in the study to understand the effect of CMAs and CPPs on CQAs and to improve the quality and safety of formulation. To study the effect of CMAs and CPPs, 23 full factorial design was used. The particle size, PDI, and zeta potential of the optimized formulation were 248.20 nm, 0.391, and −5.62 mV, respectively. Drug content of the optimized nanoformulation was found to be 87.74%±1.19%. AFM study of the optimized formulation confirmed that the prepared nanoparticles are smooth and spherical in nature. In vitro cytotoxicity studies of the nanosuspension on Vero cell line revealed that the formulation is nontoxic. The gefitinib nanosuspension released 60.03%±4.09% drug over a period of 84 h whereas as standard drug dispersion released only 10.39%±3.37% drug in the same duration. From the pharmacokinetic studies, the half-life, Cmax, and Tmax of the drug in an optimized nanosuspension was found to be 8.65±1.99 h, 46,211.04±5,805.97 ng/mL, and 6.67±1.77 h, respectively. A 1.812-fold increase in relative bioavailability of nanosuspension was found, which confirmed that the present formulation is suitable to enhance the oral bioavailability of gefitinib.
Furthermore, related to the applications of QbD in this study, few experimental runs used for the optimization are the evidence for reduction of manufacturing cost. The least values of residual error obtained are the evidence for reduction of manufacturing variability. Small particle size, least PDI values, and smooth and spherical particles confirm the desired quality of gefitinib nanosuspension. Similarly, the Vero cell line studies of gefitinib nanosuspension confirmed the achievement of safety by the nanoformulation. On the basis of the obtained results as evidences, it can be concluded that QbD is a helpful tool in nanoparticulate drug delivery systems to reduce the manufacturing cost, reduce the manufacturing variability, and improve the quality and safety, which is a primary requirement by USDFA.
Acknowledgments
The authors acknowledge the support of Neon Laboratories Ltd., Mumbai, India and Dr Reddy’s Laboratories Ltd., Hyderabad, India for providing drug and polymer as gift samples. They also acknowledge the support of Manipal University, Manipal, Karnataka, India in providing infrastructural facilities.
Disclosure
The authors report no conflicts of interest in this work.
References
Arora EM, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315:971–979. | ||
Schaeybroeck SV, Karaiskou-McCaul A, Kelly D, et al. Epidermal growth factor receptor activity determines responses of colorectal cancer cells to gefitinib alone and in combination with chemotherapy. Clin Cancer Res. 2005;11:7480–7489. | ||
Zhang G, Xie X, Liu T, Yang J, Jiao S. Effects of pemetrexed, gefitinib and their combination on human colorectal cancer cells. Cancer Chemother Pharmacol. 2013;72:767–775. | ||
McKillop D, McCormick AS, Miles GS, et al. In vitro metabolism of gefitinib in human liver microsomes. Xenobiotica. 2004;34:983–1000. | ||
Budha NR, Frymoyer A, Smelick GS, et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther. 2012;92:203–213. | ||
EGFR Tyrosine Kinase Inhibitor, 2016. Product monograph – IRESSA®. Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor. Available from: https://www.drugs.com/cdi/gefitinib.html. Accessed September 4, 2016. | ||
Pignatello R, Bucolo A, Puglisi G. Ocular tolerability of Eudragit® RS100 and Eudragit® RL100 nanosuspensions as carriers for ophthalmic controlled drug delivery. J Pharm Sci. 2002;91:2636–2641. | ||
Garad S, Wang J, Joshi Y, et al. Preclinical development for suspensions. In: Kulshreshtha AK, Singh ON, Wall GM, editors. Pharmaceutical Suspensions – From Formulation Development to Manufacturing. London: AAPS Press, Springer; 2010:127–176. | ||
Venkateshwaramurthy N, Sambathkumar R, Vijayabaskaran M, et al. Clarithromycin mucoadhesive microspheres for anti-Helicobacter pylori therapy: formulation and in vitro evaluation. Int J Curr Pharm Res. 2010;2:24–27. | ||
Venkateshwaramurthy N, Sambathkumar R, Vijayabaskaran M, Perumal P. Formulation and in vitro evaluation of furazolidone mucoadhesive microspheres. Int J Curr Pharm Res. 2010;2:104–106. | ||
Lopes D, Nunes C, Martins MCL, Sarmento B, Reis S. Eradication of Helicobacter pylori: past, present and future. J Control Release. 2014;189:169–186. | ||
Sambathkumar R, Venkateshwaramurthy N, Vijayabaskaran M, Perumal P. Formulation of clarithromycin loaded mucoadhesive microspheres by emulsification internal gelation technique for anti-Helicobacter pylori therapy. Int J Pharm Pharm Sci. 2011;3:171–177. | ||
Lamprecht A, Yamamoto H, Takeuchi H, et al. pH-sensitive microsphere delivery increases oral bioavailability of calcitonin. J Control Release. 2004;98:1–9. | ||
Xu X, Khan MA, Burgess DJ. A quality by design (QbD) case study on liposomes containing hydrophilic API: I. Formulation, processing design and risk assessment. Int J Pharm. 2011;419:52–59. | ||
Lionberger RA, Lee SL, Lee L, et al. Quality by design: concepts for ANDAs. AAPS J. 2008;10:268–276. | ||
Mora-Huertas CE, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. Int J Pharm. 2010;385:113–142. | ||
ICH Q8(R2), 2009. Guidance for industry – Pharmaceutical development Q8(R2), 2009. Available from: http://www.ich.org. Accessed June 30, 2016. | ||
Pund S, Shete Y, Jagadale S. Multivariate analysis of physicochemical characteristics of lipid based nanoemulsifying cilostazol – quality by design. Colloids Surf B Biointerfaces. 2004;115:29–36. | ||
Shah B, Khunt D, Bhatt H, et al. Application of quality by design approach for intranasal delivery of rivastigmine loaded solid lipid nanoparticles: effect on formulation and characterization parameters. Eur J Pharm Sci. 2015;78:54–66. | ||
Amasya G, Badilli U, Aksu B, et al. Quality by design case study 1: design of 5 – fluorouracil loaded lipid nanoparticles by the W/O/W double emulsion – solvent evaporation method. Eur J Pharm Sci. 2016;84:92–102. | ||
ICH Q1A(R2). ICH harmonised tripartite guideline. Stability testing of new drug substances and products Q1A(R2). Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1A_R2/Step4/Q1A_R2__Guideline.pdf. Accessed June 30, 2016. | ||
Marto J, Gouveria LF, Gonçalves LM, et al. A quality by design (QbD) approach on starch-based nanocapsules: a promising platform for topical drug delivery. Colloids Surf B Biointerfaces. 2016;143:177–185. | ||
Vitorino C, Carvalho FA, Almeida AJ, Sousa JJ, Pais AA. The size of solid lipid nanoparticles: an interpretation from experiment design. Colloids Surf B Biointerfaces. 2011;84:117–130. | ||
Kan S, Lu J, Liu J, Wang J, Zhao Y. A quality by design (QbD) case study on enteric coated pellets: screening of critical variables and establishment of design space at laboratory scale. Asian J Pharm Sci. 2014;9:268–278. | ||
ICH Q9, Guidance for Industry – Quality risk management, 2006. Available from: http://www.fda.gov/downloads/Drugs/.../Guidances/ucm073511.pdf. Accessed June 30, 2016. | ||
How ICH Q8, Q9, Q10 guidelines are working together throughout the product life cycle? 2010. Available from: https://www.ispe.org/index.php/ci_id/6286/la_id/1.htm. Accessed June 30, 2016. | ||
Elsayed I, Abdelbery AA, Elshafeey AH. Nanosizing of a poorly soluble drug: technique optimization, factorial analysis and pharmacokinetic study in healthy human volunteers. Int J Nanomedicine. 2004;9:2943–2953. | ||
Jiang P, Xie SS, Pang SJ, et al. The combining analysis of height and phase images in tapping-mode atomic force microscopy: a new route for the characterization of thiol-coated gold nanoparticle film on solid substrate. Appl Surf Sci. 2002;191:240–246. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. | ||
Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival – modifications to the tetrazolium dye produce giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–277. | ||
Wong YH, Tan WY, Tan CP, Long K, Nyam KL. Cytotoxic activity of kenaf (Hibiscus cannabinus L.) seed extract and oil against human cancer cell lines. Asian Pac J Trop Biomedicine. 2014;4:S510–S515. | ||
Revathi R, Perumal RV, Pai KSR, Arunkumar G, Sriram D, Kini SG. Design, development and drug-likeness and molecular docking studies of novel piperidin-4-imine derivatives as antitubercular agents. Drug Des Devel Ther. 2015;9:3779–3787. | ||
Thomas AC, Sidhartha AK, Bairy I, Bhat GV, Shenoy VP, Shenoy GG. Design, synthesis and evaluation of antitubercular activity of triclosan analogues. Arab J Chem. In press 2015. | ||
Singh M, Sasi P, Rai G, Gupta VH, Amarapurkar D, Wangikar PP. Studies on toxicity of antitubercular drugs namely isoniazid, rifampicin and pyrazinamide in an in vitro model of HepG2 cell line. Med Chem Res. 2011;20:1611–1615. | ||
Kumar VV, Chandrasekar D, Ramakrishna S, Kishan V, Rao YM, Diwan PV. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: influence of wax and glyceride lipids on plasma pharmacokinetics. Int J Pharm. 2007;335:167–175. | ||
Chatterjee A, Kumar L, Bhowmik BB, Gupta A. Microparticulated anti-HIV vaginal gel: in vitro – in vivo drug release and vaginal irritation study. Pharm Dev Technol. 2011;16:466–473. | ||
Yilmaz B, Arslan S, Asci A. HPLC method for determination of atenolol in human plasma and application to a pharmacokinetic study in turkey. J Chromatogr Sci. 2012;50:914–919. | ||
Chen CC, Tsai TH, Huang ZR, Fang JY. Effects of lipophilic emulsifiers on the oral administration of lovastatin from nanostructured lipid carriers: physicochemical characterization and pharmacokinetics. Eur J Pharm Biopharm. 2010;74:474–482. | ||
Tsai MJ, Wu PC, Hunag YB, et al. Baicalein loaded in tocol nanostructured lipid carriers (tool NLCs) for enhanced stability and brain targeting. Int J Pharm. 2012;423:461–470. | ||
Kondamudi PK, Tirumalasetty PP, Malayandi R, Mutalik S, Pillai R. Lidocaine transdermal patch: pharmacokinetic modelling and in vitro – in vivo correlation (IVIVC). AAPS PharmSciTech. 2016;17:588–596. | ||
Das S, Ng WK, Kanaujia P, Kim S, Tan RB. Formulation design, preparation and physicochemical characterizations of solid lipid nanoparticles containing a hydrophobic drug: effects of process variables. Colloids Surf B Biointerfaces. 2011;88:483–489. | ||
Kumar L, Reddy MS, Verma R, Koteshwara KB. Selection of cryoprotective agent for freeze drying of valsartan solid lipid nanoparticles. Latin Am J Pharm. 2016;35:284–290. | ||
Dillen K, Vandervoort J, Van den Mooter G, Ludwig A. Evaluation of ciprofloxacin loaded Eudragit® RS 100 or RL 100/PLGA nanoparticles. Int J Pharm. 2006;314:72–82. | ||
Singh R, Soni RK. Laser synthesis of aluminium nanoparticles in biocompatible polymer solutions. Appl Phys A. 2014;116:689–701. | ||
Tiberg F, Brinck J, Grant L. Adsorption and surface-induced self-assembly of surfactants at the solid-aqueous interface. Curr Opin Colloid Interface Sci. 1999;4:411–419. | ||
Li JK, Wang N, Wu XS. Poly(vinyl alcohol) nanoparticles prepared by freezing-thawing process for protein/peptide drug delivery. J Control Release. 1998;56:117–126. | ||
Nakach M, Authelin JR, Tadros T, Galet L, Chamayou A. Engineering of nano-crystalline drug suspensions: employing a physic-chemistry based stabilizer selection methodology or approach. Int J Pharm. 2014;476:277–288. | ||
Martínez-Ohárriz MC, Rodríguez-Espinosa C, Martín C, et al. Solid dispersions of diflunisal-PVP: polymeric and amorphous states of the drug. Drug Dev Ind Pharm. 2002;28:717–725. | ||
Chauhan H, Hui-Gu C, Ate E. Correlating the behaviour of polymers in solution as precipitation inhibitor to its amorphous stabilization ability in solid dispersions. J Pharm Sci. 2013;102:1924–1935. | ||
Yang M, He S, Fan Y, et al. Microenvironmental pH-modified solid dispersion to enhance the dissolution and bioavailability of poorly water-soluble weakly-soluble weakly basic GT0918, a developing anti-prostate cancer drug: preparation, characterization and evaluation in vivo. Int J Pharm. 2014;475:97–109. | ||
Lopez FL, Shearman GC, Gaisford S, Williams GR. Amorphous formulations of indomethacin and griseofulvin prepared by electrospinning. Mol Pharm. 2014;11:4327–4338. | ||
Dutta N, Egorov S, Green D. Quantification of nanoparticle interactions in pure solvents and a concentrated PDMS solution as a function of solvent quality. Langmuir. 2013;29:9991–10000. | ||
Dutta N, Green D. Nanoparticle stability in semi dilute and concentrated polymer solutions. Langmuir. 2008;24:5260–5269. | ||
Xia X, Yang M, Wang Y, et al. Quantifying the coverage density of poly(ethylene glycol) chains on the surface of gold nanostructures. ACS Nano. 2012;6:512–522. | ||
Residual solvents, Chemicals Tests/(467) residual solvents 1. USP 37/NF 32, 2014. Available from: https://hmc.usp.org/sites/default/files/documents/HMC/GCs-Pdfs/c467.pdf. Accessed Nov 1, 2016. | ||
McKim AS, Strub R. Dimethyl sulfoxide USP, PhEur in approved pharmaceutical products and medical devices. Pharmaceutical Technology®; 2008. Available from: https://lifechoice.net/wp-content/uploads/2015/03/DMSO-USP-in-Approved-Pharmaceutical-products-and-Medical-Devices-Gaylord-Chemical-Company.pdf. Accessed Nov 1, 2016. | ||
Dimethyl sulphoxide – health and safety information. Gaylord Chemical Company, L.L.C. Bulletin 106, June 2014. Available from: http://www.gaylordchemical.com/wp-content/uploads/2015/07/GC-Literature-106.pdf. Accessed Nov 1, 2016. | ||
Turk CTS, Oz UC, Serim TM, Hascicek C. Formulation and optimization of non-ionic surfactant emulsified nimesulide-loaded PLGA-based nanoparticles by design of experiments. AAPS PharmSciTech. 2014;15:161–176. | ||
Yadav KS, Jacob S, Sachdev G, Chuttani K, Mishra AK, Sawant KK. Long circulating PEGylated PLGA nanoparticles of cytarabine for targeting leukemia. J Microencapsul. 2011;28:729–742. | ||
Yadav KS, Sawant KK. Modified nanoprecipitation method for preparation of cytarabine-loaded PLGA nanoparticles. AAPS PharmSciTech. 2010;11:1456–1465. | ||
Mastiholimath VS, Dandagi PM, Gadad AP, Mathews R, Kulkarni AR. In vitro and in vivo evaluation of ranitidine hydrochloride ethyl cellulose floating microparticles. J Microencapsul. 2008;25:307–314. | ||
Jiang Y, Wang F, Xu H, Liu H, Meng Q, Liu W. Development of andrographolide loaded PLGA microspheres: optimization, characterization and in vitro – in vivo correlation. Int J Pharm. 2014;475:475–484. | ||
Kumar L, Reddy MS, Managuli RS, Pai KG. Full factorial design for optimization, development and validation of HPLC method to determine valsartan in nanoparticles. Saudi Pharm J. 2015;23:549–555. | ||
Desai MP, Labhasetwar V, Walter E, Levy RJ, Amidon GL. The mechanism of uptake of biodegradable microparticles in Caco-2 cells is size dependent. Pharm Res. 1997;14:1568–1573. | ||
Yan F, Zhang C, Zheng Y, et al. The effect of poloxamer 188 on nanoparticle morphology, size, cancer cell uptake and cytotoxicity. Nanomedicine. 2010;6:170–178. | ||
Gefitinib. Available from: https://www.drugs.com/cdi/gefitinib.html. Accessed August 24, 2016. | ||
Paria S, Khilar KC. A review on experimental studies of surfactant adsorption at the hydrophilic solid-water interface. Adv Colloid Interface Sci. 2004;110:75–95. | ||
Cetin M, Atila A, Kadioglu Y. Formulation and in vitro characterization of Eudragit® L100 and Eudragit® L100-PLGA nanoparticles containing diclofenac sodium. AAPS PharmSciTech. 2010;11:1250–1256. | ||
Mehta RC, Jeyanthi R, Calis S, Thanoo BC, Burton K, Deluca PP. Biodegradable microspheres as depot system for parenteral delivery of peptide drugs. J Control Release. 1994;29:375–384. | ||
Lee YS, Johnson PJ, Robbins PT, Bridson RH. Production of nanoparticles-in-microparticles by a double emulsion method: a comprehensive study. Eur J Pharm Biopharm. 2013;83:168–173. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.