")
Back to Journals » Drug Design, Development and Therapy » Volume 9
A phase I/II study of biweekly capecitabine and irinotecan plus bevacizumab as second-line chemotherapy in patients with metastatic colorectal cancer
Authors Suenaga M , Mizunuma N, Matsusaka S, Shinozaki E , Ozaka M, Ogura M , Chin K , Yamaguchi T
Received 7 January 2015
Accepted for publication 10 February 2015
Published 16 March 2015 Volume 2015:9 Pages 1653—1662
DOI https://doi.org/10.2147/DDDT.S80449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Mitsukuni Suenaga,1 Nobuyuki Mizunuma,1 Satoshi Matsusaka,1 Eiji Shinozaki,1 Masato Ozaka,1 Mariko Ogura,1 Keisho Chin,1 Toshiharu Yamaguchi2
1Department of Gastroenterology, 2Department of Gastroenterological Surgery, Cancer Institute Hospital of Japanese Foundation for Cancer Research, Ariake, Koto-ku, Tokyo, Japan
Background: Triweekly capecitabine plus irinotecan (XELIRI) is not completely regarded as a valid substitute for fluorouracil, leucovorin, and irinotecan (FOLFIRI) in metastatic colorectal cancer (mCRC) because of the potential for greater toxicity. We conducted a phase I/II study to assess the efficacy and safety of biweekly XELIRI plus bevacizumab (BV) as second-line chemotherapy for mCRC.
Methods: Patients with mCRC who had received prior chemotherapy including oxaliplatin and BV and had a UGT1A1 genotype of wild-type or heterozygous for UGT1A1*6 or *28 were eligible for this study. Treatment comprised capecitabine 1,000 mg/m2 twice daily from the evening of day 1 to the morning of day 8, intravenous irinotecan on day 1, and BV 5 mg/kg on day 1 every 2 weeks. The phase I study consisted of two steps (irinotecan 150 and 180 mg/m2), and dose-limiting toxicity was assessed during the first treatment cycle. The primary endpoint of the phase II study was progression-free survival (PFS).
Results: The recommended dose of irinotecan was determined to be 180 mg/m2 in the phase I study. Between November 2010 and August 2013, 44 patients were enrolled in phase II. The patients’ characteristics were as follows (N=44): median age, 60 years (range 32–80); male/female, 21/23; and UGT1A1 wild-type/heterozygous, 29/15. The median PFS was 6.8 months (95% confidence interval, 5.3–8.2 months), and the primary endpoint was met. Median overall survival was 18.3 months. The response rate was 22.7%. There was no significant difference in PFS or overall survival according to UGT1A1 status. Grade 3 or higher adverse events were mainly neutropenia in six patients and diarrhea in five patients. There were no other severe adverse events or treatment-related deaths.
Conclusion: In mCRC patients with wild-type or heterozygous UGT1A1*6 or *28 genotype, biweekly XELIRI + BV is effective and feasible as second-line chemotherapy. Biweekly XELIRI + BV is considered a valid substitute for FOLFIRI + BV in mCRC.
Keywords: XELIRI, bevacizumab, metastatic colorectal cancer, UGT1A1
Introduction
Colorectal cancer is the leading cause of death in women and the third leading cause of death in men in Japan.1 During the past decade, highly promising, new molecularly targeted agents have been introduced for the management of metastatic colorectal cancer (mCRC).2–5 In addition to conventional cytotoxic agents, four molecularly targeted agents have been approved in Japan: bevacizumab (BV), panitumumab, cetuximab, and regorafenib. Very recently, a novel oral nucleotide antitumor agent (TAS-102) was first approved only in Japan.6 Japanese patients with mCRC can thus receive evidence-based treatment with a range of cytotoxic and molecularly targeted agents under National Health Insurance. BV is a recombinant, humanized monoclonal antibody against vascular endothelial growth factor. In combination with chemotherapy, BV has been demonstrated to improve survival as both first- and second-line treatment in patients with mCRC.2,7–10 Chemotherapy plus BV has thus been established to be a safe and effective standard regimen for mCRC.
Capecitabine is an oral fluoropyrimidine prodrug that is converted to 5-fluorouracil (5-FU), primarily in tumor cells. In Japan, capecitabine has been mainly combined with oxaliplatin (XELOX) for the treatment of mCRC since its approval in 2009.11 Clinical trials evaluating capecitabine plus irinotecan (XELIRI) with or without BV in patients with mCRC were previously performed in the European Union and the United States,12,13 but not in Japan. Triweekly XELIRI is not completely regarded as a valid replacement for FOLFIRI in mCRC because of its potential for higher toxicity.12 Reduced-dose triweekly XELIRI was officially recommended recently.13 Biweekly XELIRI regimens have yet to be evaluated comprehensively.
UDP glucuronyl transferases (UGTs), which are the major metabolic enzymes of SN-38, the active metabolite of irinotecan, have been shown to be related to toxic effects such as neutropenia.14–18 SN-38 is metabolized to SN-38 glucuronide (SN-38G) via UGT1A1. To date, the presence of heterozygous UGT1A1*6 and *28 or homozygous UGT1A1*6 or *28 has been shown to be strongly related to severe irinotecan-related toxicity.16 Ethnic differences have been also found: the UGT1A1*28 allele is found mainly in whites, while the UGT1A1*6 allele is more common among Asians.17 A previous prospective study evaluated the maximum-tolerated dose of irinotecan in Japanese patients with gastrointestinal cancer who had UGT1A1*28 or UGT1A1*6 polymorphisms (or both). The study reported that patients who had the wild-type genotype or who were homozygous for the UGT1A1*28 (*28/*28) or *6 (*6/*6) allele, double heterozygous for UGT1A1*28 and *6 (*28/*6) allele, heterozygous for the UGT1A1*28(*28/*1) or *6(*6/*1) allele could tolerate irinotecan in a starting dose of 150 mg/m2. The area under the time–concentration curve of SN-38 from 0–24 hours (AUC0–24h) was greater in the homozygous group, resulting in a higher incidence of severe toxicity as compared with the other groups.18 In addition, careful observation and frequent blood tests are recommended when patients who are homozygous for UGT1A1 are given irinotecan in a dose of 150 mg/m2, albeit this level was confirmed to be the maximum-tolerated dose.
To expand irinotecan-based treatment options, we conducted a phase I/II study to assess the efficacy and safety of biweekly XELIRI (capecitabine, 1,000 mg/m2 twice daily plus irinotecan, 180 mg/m2) plus BV as second-line chemotherapy in patients with mCRC.
Patients and methods
Study design
This prospective phase I/II study was performed at a single center in Japan. We evaluated the safety and efficacy of biweekly XELIRI + BV in patients with mCRC. The study was performed in accordance with the Declaration of Helsinki and the ethical guidelines for clinical studies. The institutional review board of Cancer Institute Hospital in Tokyo, Japan approved this protocol. This study has been registered in the University hospital Medical Information Network (UMIN) Clinical Trials Registry as UMIN000003990.
The study design is shown in Figure 1. In phase I, done initially to evaluate safety, three to six patients received XELIRI + BV (irinotecan 150 mg/m2) in step 1, and six patients received XELIRI + BV (irinotecan 180 mg/m2) in step 2. If dose-limiting toxicity (DLT) developed in one patient in step 1, three additional patients were assigned to step 1. If none of the first three patients, or one or two of the six patients had DLT, step 2 began. If DLT occurred in less than or equal to two of the six patients in step 2, the phase II part of the study was started. If greater than or equal to three of the six patients in step 2 had DLT, phase II was conducted at the recommended dose of step 1. DLTs were defined as the following events occurring during the first treatment cycle: grade 3 diarrhea persisting for 8 or more days; febrile neutropenia with a body temperature of ≥38.5°C; grade 4 hematologic toxicity, clinically important grade 3 or 4 nonhematologic toxicity, or both; or toxicity requiring treatment to be delayed by more than 3 weeks.
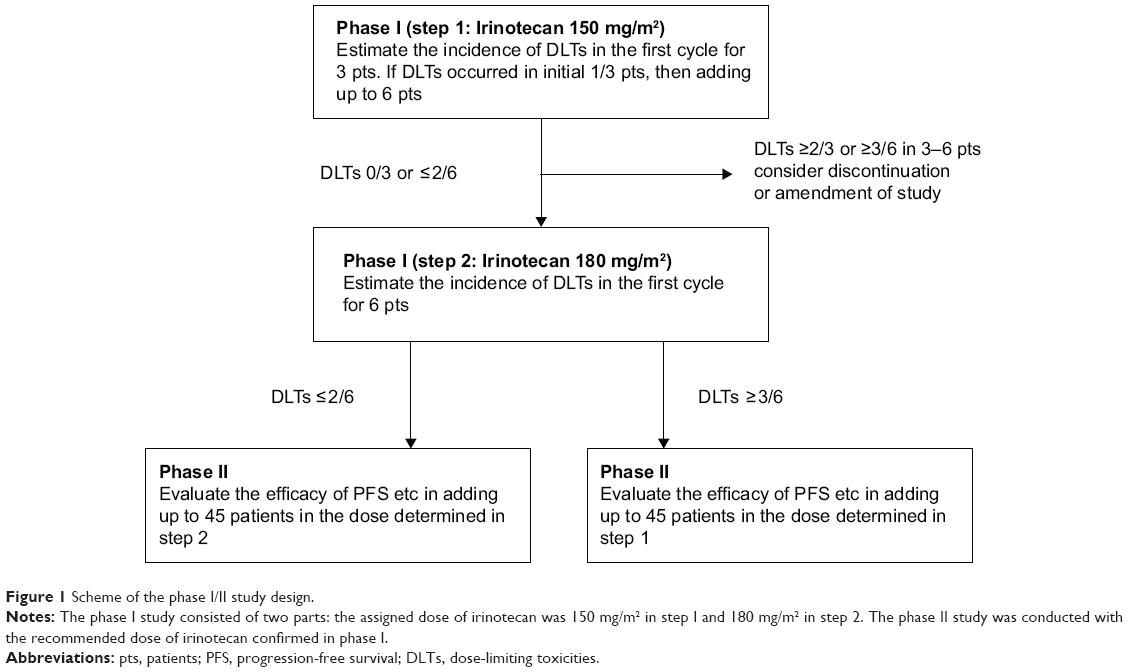 | Figure 1 Scheme of the phase I/II study design. |
Patients
Eligible patients had to have a histologically confirmed diagnosis of mCRC and had to have received oxaliplatin-based chemotherapy plus BV as first-line treatment. All patients also had to meet the following criteria: an age of 20–75 years; an Eastern Cooperative Oncology Group performance status (ECOG-PS) of 0 or 1; wild type or *28 heterozygous or *6 heterozygous UGT1A1 gene polymorphisms; neutrophil count ≥1,500/mm3; platelet count ≥100,000/mm3; hemoglobin level ≥9.0 g/dL; total bilirubin ≤1.5 times the upper limit of normal (ULN); aspartate aminotransferase and alanine aminotransferase levels ≤2.5 times the ULN (≤5.0 in patients with liver metastasis); serum creatinine ≤1.5 times the ULN; and written informed consent.
Patients with any of the following conditions were excluded: the last dose of S-1 (an oral fixed combination of tegafur, gimeracil, and oteracil potassium) was received within 1 week before enrollment; clinically detectable ascites; paralytic, or mechanical bowel obstruction; brain tumors or brain metastases; major surgery, open biopsy, or clinically significant traumatic injury within 4 weeks before enrollment or fine-needle aspiration biopsy within 1 week before enrollment; a bleeding diathesis or coagulopathy; active gastrointestinal ulcer; a current or a previous (within the last year) history of gastrointestinal perforation; international normalized ratio (INR) ≤1.5 within 2 weeks before enrollment; non-healing bone fracture; urinary protein ≥1+ within 2 weeks before enrollment; uncontrolled hypertension; uncontrolled diabetes; clinically significant cardiovascular disease; long-term treatment with high-dose aspirin (≥325 mg/day) or non-steroidal anti-inflammatory medications; a current or previous history of serious hypersensitivity to 5-FU or BV; interstitial lung disease or pulmonary fibrosis; or a previous history of treatment with irinotecan.
Chemotherapy
The XELIRI plus BV regimen was given every 2 weeks. On day 1, irinotecan 180 mg/m2 was given as a 90-minute intravenous infusion, and BV 5 mg/kg was given as an intravenous infusion over the course of 30 minutes. Capecitabine 1,000 mg/m2 was given twice daily from the evening of day 1 to the morning of day 8.
The study treatment was delayed if any of the following criteria were not met on the scheduled day of administration or the previous day: neutrophil count ≥1,500/mm3; platelet count ≥75,000/mm3; hemoglobin ≥8.0 g/dL; total bilirubin ≤1.5 times the ULN; aspartate aminotransferase and alanine aminotransferase levels ≤2.5 times the ULN (≤5.0 times, if liver metastasis was present); serum creatinine ≤1.5 times the ULN, urinary protein ≤1+; hand–foot skin reaction less than or equal to grade 1; or any other nonhematologic toxicities less than or equal to grade 1, excluding hypertension, alopecia, and body weight loss.
The dose of irinotecan was reduced to 150, 125, and 100 mg/m2 if grade 4 neutropenia, grade 3 or higher thrombocytopenia, grade 3 or higher febrile neutropenia, or any nonhematologic toxicity except for hypertension and body weight loss occurred. The dose of capecitabine was also reduced to 750 and 500 mg/m2 in accordance with the criteria described above in this section.
No dose modifications were allowed for BV. The study was terminated if BV was permanently discontinued because of grade 3 or higher thrombosis, any grade hemoptysis or gastrointestinal perforation, refractory grade 2 or higher proteinuria or uncontrolled hypertension, or refractory grade 2 or grade 3 or higher bleeding or reversible posterior leukoencephalopathy syndrome (RPLS). The study was also terminated in patients who required more than 4 weeks to recover from any adverse effect.
Evaluation of safety and efficacy
Patients’ data, including the results of imaging studies, were recorded in electronic clinical records. A multidisciplinary colorectal cancer team at Cancer Institute Hospital, Tokyo, Japan, confirmed patient eligibility. Adverse effects were graded according to the Common Terminology Criteria for Adverse Events (CTCAE), version 4.0, in all patients every 2 weeks or before each treatment cycle. Treatment was continued until disease progression, unmanageable toxic effects, patient refusal, or transfer of the patient to another hospital. Baseline tumor response was assessed within 4 weeks before enrollment in the study, and tumor response was then prospectively assessed every 6 weeks on computed tomography according to the Response Evaluation Criteria for Solid Tumors (RECIST), version 1.1. An Independent Data and Safety Monitoring Committee of BIXER study group, Tokyo, Japan, confirmed all safety and efficacy data derived from this study.
Statistical analysis
The objective of the phase I study was to estimate the incidence of DLTs in the first treatment cycle. The primary objective of the phase II study was to assess progression-free survival (PFS). Secondary objectives were the evaluation of safety and overall response rate (ORR). The ORR was based on the number of patients who had a complete or partial response to treatment. PFS was defined as the time between the date of starting treatment and the date of confirming disease progression or death. Data on patients without disease progression were censored on the date at which the patient was last confirmed to be alive. Overall survival (OS) was calculated from the date of the first day of treatment until the date of death from any cause. In patients who were lost to follow-up, data were censored on the date on which the patient was last confirmed to be alive. PFS and OS were estimated by the Kaplan–Meier method and compared using the log-rank test; predictive or prognostic factors were identified by univariate analysis. Multivariate analysis of the factors was conducted based on the Cox proportional hazards model to identify factors associated with PFS and OS. All analyses were carried out with the use of SPSS software, version 22.0 (IBM Corporation, Armonk, NY, USA). P-values of <0.05 were considered to indicate statistical significance.
On the basis of previous clinical trials of irinotecan-based chemotherapy, the null hypothesis that the expected median PFS would be 4 months was tested against a two-sided alternative.19,20 Forty patients were estimated to be required to make the width of the 95% confidence interval (CI) of the median PFS about 3.2 months. The target number of patients was therefore set at 45 to take into account those who would be excluded from the analysis set.
Results
Phase I study
In the phase I study, three patients were enrolled in step 1 and received irinotecan in a dose of 150 mg/m2. Six other patients were enrolled in step 2 and received irinotecan in a dose of 180 mg/m2. None of the first nine patients had DLT, and the safety of treatment was initially confirmed. The following adverse events occurred in steps 1 and 2, respectively: neutropenia in two patients and one patient; anorexia in one and one patient; diarrhea in one and one patient; stomatitis in one and one patient; and increased alanine or aspartate aminotransferase levels in one and three patients. There were no grade 3 or higher adverse events. On the basis of the phase I study, the recommended dose of irinotecan for the phase II study was determined to be 180 mg/m2.
Patient baseline characteristics
In phase II, 40 patients were newly enrolled, but two were excluded because they did not meet the eligibility criteria. Finally, 44 patients (including six patients from step 2 of phase I study) were included in the analysis of the safety and efficacy of irinotecan at the recommended dose of 180 mg/m2.
The characteristics of the patients (N=44) were as follows: median age, 60 years (range, 32–74 years); ECOG-PS 0, 86.4%; UGT1A1 genotype wild/*6 heterozygous/*28 heterozygous, 66%/16%/18%; and first-line chemotherapy regimen fluorouracil, leucovorin, and oxaliplatin (FOLFOX)/XELOX, 41%/59%. The ORR of first-line chemotherapy was 54.5%, and the median duration of treatment was 263 days (range, 63–1,100 days). The detailed characteristics are shown in Table 1.
 | Table 1 Baseline characteristics of patients (N=44) |
Treatment
The median number of treatment cycles was 10.5 (range, 1–39 treatment cycles). The median cumulative doses of each agent were 207,900 mg/patient (range, 21,000–691,800 mg/patient) for capecitabine, 2,725.8 mg/m2 (range, 179.5–5,951.1 mg/m2) for irinotecan, and 52.5 mg/kg (range, 5–181.0 mg/kg) for BV. The median relative dose intensities of each agent were 0.83 (range, 0.34–1.08) for capecitabine, 0.82 (range, 0.41–1.07) for irinotecan, and 0.84 (range, 0.54–1.08) for BV. Treatment was delayed in 36 patients (81.8%) a median of three times (range, 1–10 times) for each drug. The main reasons for treatment delay were neutropenia in 22 patients (50%), followed by diarrhea in five and liver dysfunction in five patients. In addition, 20 patients (45.5%) required at least one dose reduction, including dose reductions of irinotecan in 19 patients and capecitabine in 14 patients. The main cause of dose modification was diarrhea followed by neutropenia for both capecitabine and irinotecan. BV was interrupted in five patients (11.4%): four of these patients had proteinuria and two had uncontrolled hypertension (one patient had both events). There were no severe BV-related adverse events during any treatment course. Overall, 37 patients (84.1%) could continue treatment until disease progression. The other reasons for discontinuance of treatment were adverse events in four patients, refusal to continue treatment in one patient, and palliative resection in one patient. One patient was still receiving treatment after about 1 year.
Efficacy
Tumor response was assessed in 44 patients (Table 2). The ORR was 22.7% (95% CI, 9.80%–35.6%), and disease control was achieved in 86.4% (95% CI, 75.8%–96.9%) of the patients. One patient had a best overall response of complete response. A waterfall-plot analysis of the best tumor response is shown in Figure 2. Tumor response was not assessable in one patient because adverse events developed soon after treatment began. The median follow-up was 364 days (range, 84–1,337 days). Median PFS was 206 days (95% CI, 153–259 days), while median OS was 608 days (95% CI, 469–747 days) (Figure 3).
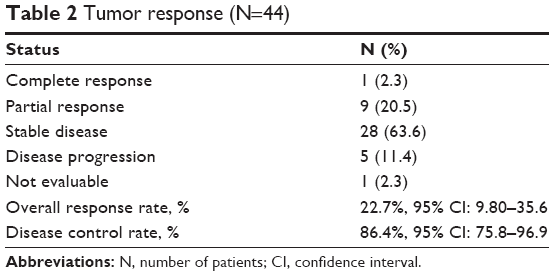 | Table 2 Tumor response (N=44) |
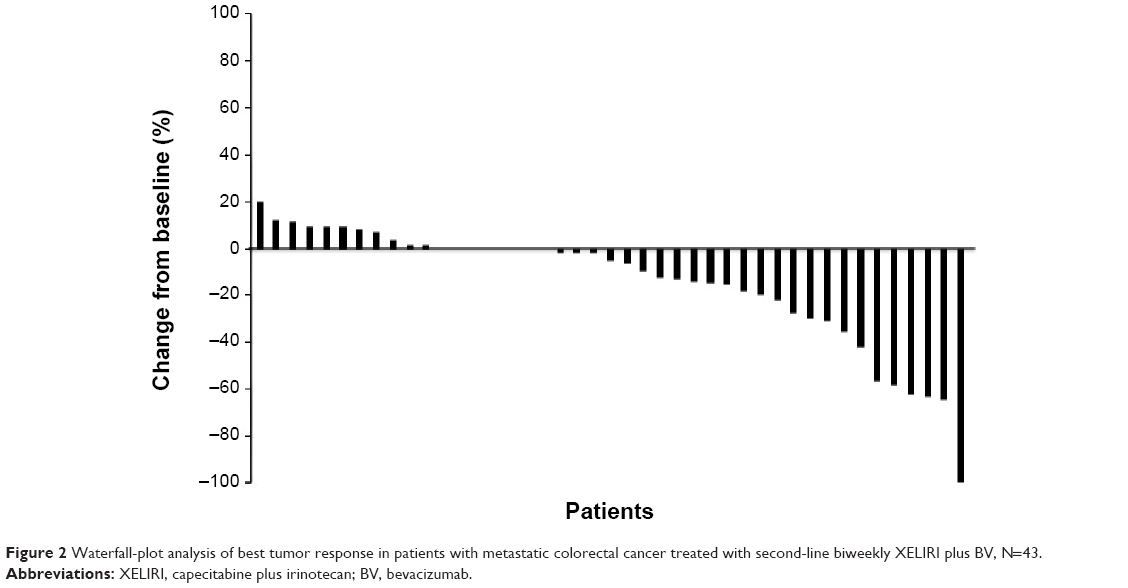 | Figure 2 Waterfall-plot analysis of best tumor response in patients with metastatic colorectal cancer treated with second-line biweekly XELIRI plus BV, N=43. |
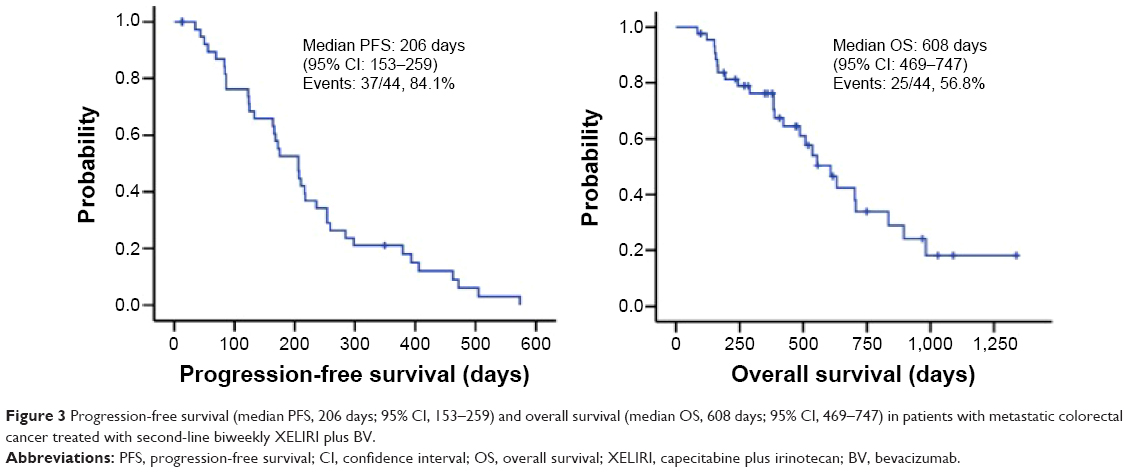 | Figure 3 Progression-free survival (median PFS, 206 days; 95% CI, 153–259) and overall survival (median OS, 608 days; 95% CI, 469–747) in patients with metastatic colorectal cancer treated with second-line biweekly XELIRI plus BV. |
On univariate analysis, significantly longer PFS was associated with ECOG-PS 0 (log-rank, P=0.018), a tumor response to XELIRI + BV (log-rank, P=0.001), and a duration of first-line treatment of 260 days or longer (log-rank, P=0.002). There was also a trend toward better PFS in patients with rectal tumors (log-rank, P=0.066).
Longer OS was significantly associated with better ECOG-PS (log-rank, P=0.002), a good tumor response (log-rank, P=0.006), and use of anti-epidermal growth factor receptor (anti-EGFR) agents for third-line chemotherapy (log-rank, P=0.034). In addition, KRAS wild type (log-rank, P=0.065) was associated with trends toward better OS. Neither PFS (log-rank, P=0.705) nor OS (log-rank, P=0.119) differed significantly among the UGT1A1 genotypes. In addition, PFS (log-rank, P=0.360) and OS (log-rank, P=0.623) were independent of whether the first-line regimen was XELOX or FOLFOX. Multivariate analysis was performed for independent factors as follows: age, ECOG-PS, primary site, adjuvant treatment, duration of the first-line treatment and tumor response for PFS, and ECOG-PS, KRAS genotype, UGT1A1 genotype, tumor response, and exposure to EGFR inhibitor for OS. Multivariate analysis of the 44 patients showed that better ECOG-PS (hazard ratio [HR], 0.26; 95% CI, 0.09–0.77, P=0.015), primary site in rectum (HR, 0.46; 95% CI, 0.22–0.94, P=0.034), a longer duration of first-line treatment (HR, 0.29; 95% CI, 0.13–0.66, P=0.003), and a tumor response to XELIRI + BV (HR, 0.37; 95% CI, 0.15–0.90, P=0.028) were independently related to longer PFS. As for OS, better ECOG-PS (HR, 0.30; 95% CI, 0.11–0.84, P=0.022) and a tumor response to XELIRI + BV (HR, 0.12; 95% CI, 0.02–0.87, P=0.036) were shown to be positive prognostic factors for OS (Table 3).
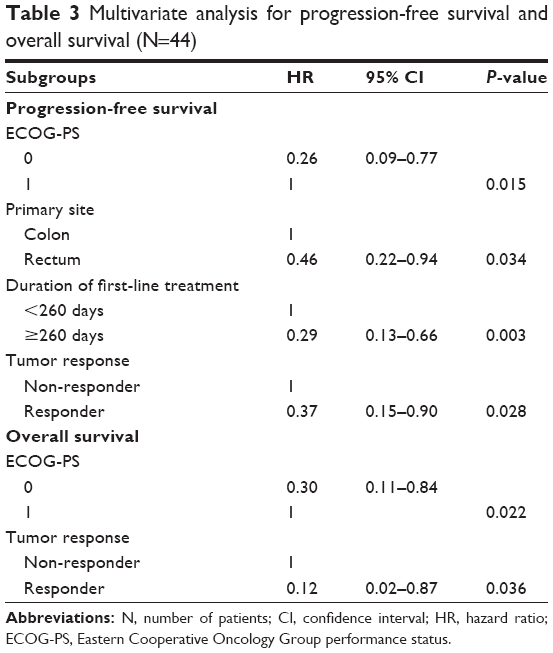 | Table 3 Multivariate analysis for progression-free survival and overall survival (N=44) |
Safety
Adverse events are summarized in Table 4. The most common grade 3 or 4 adverse events were neutropenia (13.6%), diarrhea (11.4%), and nausea (9.1%). One patient discontinued the protocol treatment because of grade 3 interstitial pneumonia considered to be related to irinotecan. There was no marked tendency of aggravation of BV-related adverse events, including hypertension, proteinuria, and cardiovascular events. There were no other treatment-related severe adverse events or deaths during treatment.
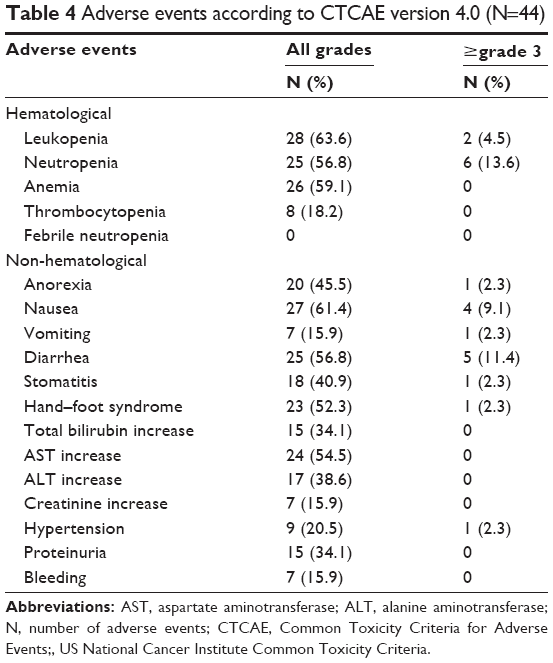 | Table 4 Adverse events according to CTCAE version 4.0 (N=44) |
Discussion
Irinotecan is one of the key drugs for the management of mCRC, along with oxaliplatin. These drugs are often combined with 5-FU plus leucovorin in regimens such as FOLFIRI or FOLFOX. Capecitabine is an oral fluoropyrimidine drug that is used instead of infusional 5-FU and combined with oxaliplatin in the XELOX regimen. As for irinotecan, the safety and effectiveness of the XELIRI regimen (capecitabine plus irinotecan) have yet to be fully evaluated in clinical trials.
Various XELIRI regimens using different dosages of irinotecan have been reported: for example, 70–150 mg/m2 on day 1 every week21,22 or 200–300 mg/m2 on day 1 every 3 weeks.23 A randomized phase II trial has compared a weekly regimen of XELIRI (capecitabine 2,000 mg/m2 on days 1–14 and 22–35, plus irinotecan 70 mg/m2 on days 1, 8, 15, 22, and 29 of a 6-week cycle) with a triweekly regimen (capecitabine 2,000 mg/m2 on days 1–14 and 22–35, plus irinotecan 300 mg/m2 on days 1 and 22 of a 6-week cycle) as first-line chemotherapy for mCRC.13 The starting dose of irinotecan was reduced from 300 to 240 mg/m2 after the safety results of another phase II trial had been reported.12 The tumor response rate as evaluated by an independent committee was 18% (95% CI: 8%–34%) in the weekly group as compared with 35% (95% CI: 20%–53%) in the triweekly group. The median PFS was 6.9 months (95% CI: 4.6–10.1 months) and 9.2 months (95% CI: 7.9–11.5 months), and the OS was 17.4 months (95% CI: 12.6–23.0 months) and 24.7 months (95% CI: 16.3–26.4 months), respectively. The results showed a trend toward better outcomes in the triweekly group, but the differences between the groups did not reach statistical significance. As for grade 3 or higher adverse events, diarrhea was marked in the weekly group, while the incidences of neutropenia and alopecia were slightly but not significantly higher in the triweekly group. The phase III BICC-C study compared three well-known irinotecan-based regimens as first-line treatment in patients with mCRC: FOLFIRI, modified IFL (irinotecan, 5-FU, and leucovorin), and triweekly XELIRI with irinotecan in a dose of 250 mg/m2. Grade 3 or greater diarrhea (47.5%) and dehydration (19.1%) were more common in the XELIRI group than in the two other groups.24 Therefore, the study was discontinued, and an additional study in which BV was added to each regimen was not conducted. Irinotecan in a dose of 240 to 300 mg/m2 was thus shown to be unsuitable for a triweekly XELIRI regimen. In 2008, the AIO 0604 study, a randomized phase II trial comparing a new modified triweekly XELIRI regimen (including 200 mg/m2 irinotecan) plus BV with XELOX plus BV, reported a reduction in severe diarrhea (grade 3: 16%, grade 4: 0%) as well as neutropenia (grade 3: 8% and grade 4: 2%) in the XELIRI + BV group. Other safety results also indicated that treatment was well tolerated.23 The objective response rate was 55%, with a median PFS of 12.1 months in the XELIRI + BV group, similar to the PFS in the XELOX + BV group. Another randomized phase II trial, the FNCLCC ACCORD 13/0503 study, compared triweekly XELIRI + BV, as used in the AIO 0604 study, with FOLFIRI + BV.25 The effectiveness of these regimens was generally similar: the response rate was 39% in the XELIRI + BV group and 43% in the FOLFIRI + BV group, and the median PFS and OS were 9 months and 23 months in both treatment groups, respectively, indicating no significant difference between the regimens. Toxicity was also similar. The results suggested that oral capecitabine could be used instead of 5-FU in combination with irinotecan, as previously demonstrated for oxaliplatin in regimens such as XELOX.2
Several studies have evaluated biweekly XELIRI regimens.26–28 A phase II trial of a biweekly XELIRI regimen (capecitabine 2,000 mg/m2 on days 2–8 plus irinotecan 175 mg/m2 on day 1, every 2 weeks) obtained an objective response rate of 32%, a median time to progression of 9 months, and a median OS of 19.2 months.27 The same regimen plus BV was evaluated in another phase II trial, which obtained a response rate of 67%, a median PFS of 12.3 months (95% CI, 6.5–18.1 months), and an OS of 23.7 months (95% CI, 16.7–30.6 months). The main grade 3 or greater toxic effects were asthenia (7%), diarrhea (7%), nausea (9%), and vomiting (7%).28 The results of these studies have indicated that a biweekly regimen of XELIRI is comparable to a triweekly regimen and has an acceptable safety profile.
A few clinical trials have evaluated not only XELIRI, but also FOLFIRI with or without biologic agents as second-line treatment for mCRC. The BEVACOLOR study evaluated BV combined with standard regimens for chemotherapy such as FOLFIRI or FOLFOX in the second-line treatment of mCRC.19 Exactly 74% of patients received irinotecan-based regimens, including FOLFIRI in 57% of patients. The overall response rate, median PFS, and OS were 33%, 7.8 months (95% CI, 6.1–8.7 months), and 21 months (95% CI, 17.3–25.7 months), respectively. Recently, the ML18147 study explored a new strategy for second-line treatment, in which the use of BV is continued beyond the first disease progression. The response rate, median PFS, and median OS in the BV-combined group including irinotecan-based regimens (34%) with XELIRI (12%) were 5.4%, 5.7 months, and 11.2 months, respectively.29 These results confirmed that second-line treatment with BV plus irinotecan-based chemotherapy was effective in patients who had previously received oxaliplatin-based chemotherapy, regardless of the history of BV usage. However, the effectiveness of second-line BV seems to be higher in patients who did not receive BV as first-line chemotherapy.
Because the approved dose of irinotecan in Japan is 150 mg/m2, few prospective studies have administered irinotecan in a dose of 180 mg/m2 in FOLFIRI or FOLFIRI regimens plus BV. The inclusion criteria of our study therefore required that patients have wild-type or heterozygous UGT1A1*6 or *28 genotypes to use irinotecan in a dose of 180 mg/m2, and patients with homozygous UGT1A1 genotypes were excluded.
In our study, the efficacy data were comparable to those of previous studies. In particular, the overall tumor response rate was better than expected, given that the study was designed to evaluate the use of BV beyond first-line progression. Trends toward longer PFS and OS were also observed in the study. The planned dose intensity of irinotecan in this study (90 mg/m2/week) was equal to or higher than that used in previously reported biweekly XELIRI (87.5 mg/m2/week) or triweekly XELIRI (66.7 mg/m2/week) regimens, although these studies treated patients in a first-line setting.23,26 As for capecitabine, the planned dose intensity was similar to that used in these previous studies. As for safety, the incidences of severe diarrhea, neutropenia, and hand–foot syndrome were acceptable despite the higher dose of irinotecan. Exclusion of patients who had homozygous UGT1A1 genotypes might have been an important reason for the good safety results, and 7-day treatment with capecitabine was considered useful for reducing not only toxicity but also mental strain during treatment, although the number of hospital visits increased. Thus, we have confirmed that a biweekly XELIRI regimen was well tolerated in the enrolled patients. On the other hand, a prospective phase II study (BIX study) evaluating a second-line triweekly XELIRI + BV regimen similar to the regimen used in the AIO 0604 trial was recently reported from Japan. Triweekly XELIRI + BV was shown to be well tolerated and effective as a second-line treatment for patients who are eligible for BV beyond disease progression or have wild-type or heterozygous UGT1A1 genotype, similar to the patients in our study.30 The efficacy and safety are similar in both XELIRI regimens, though dose intensities or medication schedule are different. A pooled analysis of these two studies is needed to ascertain which regimen is superior.
In previous studies, UGT1A1 genotype and KRAS gene status were not associated with PFS or OS on multivariate analysis.29,30 As for the results from first-line treatment in the current study, a longer duration of treatment was associated with better PFS; however, tumor response was unrelated to the results of second-line treatment. Interestingly, chemosensitivity to second-line XELIRI was not affected by the first-line regimen, ie, capecitabine or 5-FU combined with oxaliplatin. Capecitabine-related toxicity such as hand–foot syndrome was not worsened by the continuous use of capecitabine. In general, FOLFIRI is used mainly in patients who are refractory to oxaliplatin-based regimens in Japan, and our study results support the use of XELIRI as a new option for second-line chemotherapy. We now have two oral fluoropyrimidine preparations available, capecitabine and S-1, both of which have been developed as combination therapy with oxaliplatin or irinotecan. The sequential use of these drugs or their continued use beyond initial progression should be investigated in further clinical trials. In addition, we are currently planning the biomarker exploration study in our phase I/II study.
In Japanese patients with wild-type or heterozygous UGT1A1*6/*28 genotype, the recommended dose of irinotecan was determined to be 180 mg/m2. Biweekly XELIRI + BV is well balanced in terms of efficacy and safety as second-line chemotherapy and is considered comparable to the modified triweekly XELIRI regimen. Biweekly XELIRI + BV may be a valid substitute for FOLFIRI + BV in mCRC.
Acknowledgments
We thank the participating patients, their family members, and all researchers in this study. We are grateful to Drs Hiroyuki Uetake and Atsushi Sato for their support as independent review committee members and to Yuki Horike and Kaori Kobayashi for collecting the data.
Disclosure
The authors report no conflict of interest related to this work.
References
Center for Cancer Control and Information Services National Cancer Centre. Cancer Statistics in Japan; 2013. Available from: http://ganjoho.jp/data/professional/statistics/backnumber/2013/cancer_statistics_2013.pdf. Accessed Jan, 2015. | ||
Saltz LB, Clarke S, Díaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2007;26:2013–2019. | ||
Van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. | ||
Douillard JY, Siena S, Cassidy J, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol. 2014;25:1346–1355. | ||
Grothey A, Van Cutsem E, Sobrero A, et al; CORRECT Study Group. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–312. | ||
Yoshino T, Mizunuma N, Yamazaki K, et al. TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2012;13:993–1001. | ||
Kabbinavar F, Hurwitz HI, Fehrenbacher L, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21:60–65. | ||
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. | ||
Kabbinavar FF, Schulz J, McCleod M, et al. Addition of bevacizumab to bolus fluorouracil and leucovorin in first-line metastatic colorectal cancer: results of a randomized phase II trial. J Clin Oncol. 2005;23:3706–3712. | ||
Giantonio BJ, Catalano PJ, Meropol NJ, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2006;25:1539–1544. | ||
Doi T, Boku N, Kato K, et al. Phase I/II study of capecitabine plus oxaliplatin (XELOX) plus bevacizumab as first-line therapy in Japanese patients with metastatic colorectal cancer. Jpn J Clin Oncol. 2010;40:913–920. | ||
Bajetta E, Di Bartolomeo M, Mariani L, et al; Italian Trials in Medical Oncology (I.T.M.O.) Group. Randomized multicenter Phase II trial of two different schedules of irinotecan combined with capecitabine as first-line treatment in metastatic colorectal carcinoma. Cancer. 2004;100:279–287. | ||
Borner MM, Bernhard J, Dietrich D, et al; Swiss Group for Clinical Cancer Research (SAKK), Berne, Switzerland. A randomized phase II trial of capecitabine and two different schedules of irinotecan in first-line treatment of metastatic colorectal cancer: efficacy, quality-of-life and toxicity. Ann Oncol. 2005;16:282–288. | ||
Innocenti F, Undevia SD, Iyer L, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–1388. | ||
Toffoli G, Cecchin E, Corona G, et al. The role of UGT1A1*28 polymorphisms in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol. 2006;24:3061–3068. | ||
Ando Y, Saka H, Ando M, et al. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res. 2000;60:6921–6926. | ||
Han JY, Lim HS, Shin ES, et al. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol. 2006;24:2237–2244. | ||
Satoh T, Ura T, Yamada Y, et al. Genotype-directed, dose-finding study of irinotecan in cancer patients with UGT1A1*28 and/or UGT1A1*6 polymorphisms. Cancer Sci. 2011;102:1868–1873. | ||
Bennouna J, Borg C, Delord JP, et al. Bevacizumab combined with chemotherapy in the second-line treatment of metastatic colorectal cancer: results from the phase II BEVACOLOR study. Clin Colorectal Cancer. 2012;11:38–44. | ||
Tournigand C, Andre T, Achille E, Lledo G, Flesh M, Mery-Mignard D, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22(2):229–237. | ||
Tewes M, Schleucher N, Achterrath W, et al. Capecitabine and irinotecan as first-line chemotherapy in patients with metastatic colorectal cancer: results of an extended phase I study. Ann Oncol. 2003;14:1442–1448. | ||
Jordan K, Kellner O, Kegel T, Schmoll HJ, Grothey A. Phase II trial of capecitabine/irinotecan and capecitabine/oxaliplatin in advanced gastrointestinal cancers. Clin Colorectal Cancer. 2004;4:46–50. | ||
Schmiegel W, Reinacher-Schick A, Arnold D, et al. Capecitabine/irinotecan or capecitabine/oxaliplatin in combination with bevacizumab is effective and safe as first-line therapy for metastatic colorectal cancer: a randomized phase II study of the AIO colorectal study group. Ann Oncol. 2013;24:1580–1587. | ||
Fuchs CS, Marshall J, Mitchell E, et al. Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: results from the BICC-C Study. J Clin Oncol. 2007;25:4779–4786. | ||
Ducreux M, Adenis A, Pignon JP, et al. Efficacy and safety of bevacizumab-based combination regimens in patients with previously untreated metastatic colorectal cancer: final results from a randomised phase II study of bevacizumab plus 5-fluorouracil, leucovorin plus irinotecan versus bevacizumab plus capecitabine plus irinotecan (FNCLCC ACCORD 13/0503 study). Eur J Cancer. 2013;49:1236–1245. | ||
García Alfonso P, Muñoz Martin A, Alvarez Suarez S, et al. Bevacizumab in combination with capecitabine plus irinotecan as first-line therapy in metastatic colorectal cancer: a pooled analysis of 2 phase II trials. Onkologie. 2013;36:363–367. | ||
Garcia-Alfonso P, Muñoz-Martin A, Mendez-Ureña M, et al. Capecitabine in combination with irinotecan (XELIRI), administered as a 2-weekly schedule, as first-line chemotherapy for patients with metastatic colorectal cancer: a phase II study of the Spanish GOTI group. Br J Cancer. 2009;101:1039–1043. | ||
García-Alfonso P, Muñoz-Martin AJ, Alvarez-Suarez S, et al. Bevacizumab in combination with biweekly capecitabine and irinotecan, as first-line treatment for patients with metastatic colorectal cancer. Br J Cancer. 2010;103:1524–1528. | ||
Bennouna J, Sastre J, Arnold D, et al; ML18147 Study Investigators. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol. 2013;14:29–37. | ||
Hamamoto Y, Yamaguchi T, Nishina T, et al. A phase I/II study of XELIRI plus bevacizumab as second-line chemotherapy for Japanese patients with metastatic colorectal cancer (BIX study). Oncologist. 2014;19:1131–1142. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.