")
Back to Journals » OncoTargets and Therapy » Volume 14
A Phase I Trial of the MET/ALK/ROS1 Inhibitor Crizotinib Combined with the VEGF Inhibitor Pazopanib in Patients with Advanced Solid Malignancies
Authors Piha-Paul SA , Dumbrava EE, Nair BC, Xiong W, Xu L, Mostorino R, Subbiah V, Tannir N, Fu S, Naing A, Janku F, Karp DD, Patel S, Daw NC , Hong D, Meric-Bernstam F, Zinner R
Received 5 December 2020
Accepted for publication 2 April 2021
Published 7 May 2021 Volume 2021:14 Pages 3037—3049
DOI https://doi.org/10.2147/OTT.S291801
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Sarina A Piha-Paul,1 Ecaterina E Dumbrava,1 Binoj C Nair,1 Wendy Xiong,1 Li Xu,1 Rosa Mostorino,1 Vivek Subbiah,1 Nizar Tannir,2 Siqing Fu,1 Aung Naing,1 Filip Janku,1 Daniel D Karp,1 Shreyaskumar Patel,3 Najat C Daw,4 David Hong,1 Funda Meric-Bernstam,1,5,6 Ralph Zinner7
1Department of Investigational Cancer Therapeutics (A Phase I Clinical Trials Program), University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Department of Genitourinary Medical Oncology, University of Texas MD Anderson Cancer Center, Houston, TX, USA; 3Department of Sarcoma Medical Oncology, University of Texas MD Anderson Cancer Center, Houston, TX, USA; 4Department of Pediatrics, University of Texas MD Anderson Cancer Center, Houston, TX, USA; 5Department of Breast Surgical Oncology, University of Texas, MD Anderson Cancer Center, Houston, TX, USA; 6The Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy, University of Texas MD Anderson Cancer Center, Houston, TX, USA; 7Department of Medical Oncology, Thomas Jefferson University Hospital, Philadelphia, PA, USA
Correspondence: Sarina A Piha-Paul
Investigational Cancer Therapeutics, MD Anderson Cancer Center, 1515 Holcombe Blvd. Unit 455, Houston, TX, 77030, USA
Tel +1 713-563-1055
Fax +1 713-792-3535
Email [email protected]
Background: Crizotinib inhibits ALK, MET and ROS1 tyrosine kinases but the development of resistance to monotherapy is an issue. The anti-angiogenic properties of pazopanib could overcome crizotinib drug resistance. Additionally, the anti-angiogenic properties of crizotinib could augment the clinical efficacy of pazopanib.
Methods: We evaluated the safety and responses in patients with advanced solid tumors treated with crizotinib and pazopanib.
Results: Eighty-two patients (median age 53 years, range 18– 78 years) were enrolled. The median number of prior systemic therapies was 3 (range, 0– 8). We were able to dose escalate to dose level 8 (crizotinib 250 mg twice daily and pazopanib 800 mg daily) with no MTD identified. Grade 3 or 4 toxicities were seen in 32% of patients with the highest prevalence being fatigue (n=9, 11%), diarrhea (n=6, 7%), vomiting (n=3, 4%), anemia (n=2, 2%) and ALT increased (n=2, 2%). Of the 82 patients, 61 (74%) had measurable disease by RECISTv1.1 and reached first restaging (6 weeks). Partial response (PR) was observed in 6/61 (10%) patients, and stable disease (SD) lasting ≥ 6 months was observed in 10/61 patients (16%) (total = 16/61 (26%) of patients with SD ≥ 6 months/PR).
Conclusion: Dose level 6 (crizotinib 200 mg twice daily and pazopanib 600 mg daily) was the most tolerable dosing of the combination and can be used in future studies. We also observed moderate clinical activity in patients with advanced solid tumors that had received numerous prior therapies.
Keywords: crizotinib, pazopanib, VEGF, ALK/ROS1, MET
Introduction
Crizotinib is a potent first-generation inhibitor of the anaplastic lymphoma kinase (ALK), MET (c-MET), and c-ROS oncogene 1 (ROS1) receptor tyrosine kinases and is approved for the treatment of patients with non-small cell lung cancer (NSCLC), who have tested positive for either ALK or ROS1 rearrangement.1–4 Crizotinib approval as a first-line therapy for patients with previously untreated ALK-positive NSCLC was based on its superior objective response rate and improved progression free survival compared to standard first-line doublet chemotherapy.5 Additionally, in preclinical studies, crizotinib (previously known as PF-02341066) was found to inhibit tumor cell growth in other solid tumors cell lines and xenograft models, including prostate, squamous head and neck cancer, osteosarcoma, ovarian cancer and renal cancer.6–10
Studies have shown that the majority of patients treated with crizotinib initially have tumors that show a robust response, but eventually and almost invariably, develop drug-resistance.11,12 To address this, next-generation ALK inhibitors have been developed and are approved for first, second, and third line ALK positive NSCLC. However, ultimate recurrence through this sequence of ALK inhibitors occurs over time. Thus, there is an interest in developing combination therapies for ALK inhibitors with other therapeutic agents to increase the duration of response and sustain clinical benefit. Multiple bypass molecular pathways are ascribed to ALK resistance.13 The angiogenesis pathway, for example, is considered as one of the essential requirements for disease progression in various solid tumors.14 Blocking the angiogenesis pathway has been shown to be effective in multiple solid tumors inclusive of thyroid, lung, ovarian and other cancers.15 The combination of existing chemotherapy with anti-angiogenesis drugs has been tested in multiple cancer types;16,17 for example, bevacizumab combined with pemetrexed18,19 and sunitinib in combination with pemetrexed.20 Dual inhibition of cMET and VEGF has also been shown to be more effective than single pathway inhibition alone in pre-clinical models.21 Interestingly, crizotinib is also shown to have some anti-angiogenic activity in addition to its anti-proliferative effects.22
Pazopanib is a multi-kinase, angiogenesis inhibitor known to block VEGFR, PDGFRα, PDGFRβ and C-KIT and is approved for the treatment of patients with renal cell cancer23 and advanced soft tissue sarcoma (STS) who have received prior chemotherapy.24 We hypothesized that the combination of crizotinib with pazopanib would augment the clinical efficacy of pazopanib through dual inhibition of angiogenesis pathways in patients and additionally that in patients whose tumors harbored ALK, MET or ROS1 deleterious aberrations, pazopanib would augment crizotinib therapy and help overcome drug resistance. Here we report our experience treating patients with advanced malignancies with this combination therapy.
Patients and Methodology
Study Design and Dosing
This is a single institution, open-label, phase I dose-escalation study in patients with advanced malignancies (NCT 01548144). This trial was open to all patients with advanced or metastatic cancer refractory to standard therapy, relapsed after standard therapy, or who had no standard therapy available that could improve survival by at least three months.
Treatment was administered on an outpatient basis at the University of Texas, MD Anderson Cancer Center. The cycle of therapy was 21 days. No investigational, commercial agents or therapies other than those described here could be administered with the intent to treat the patient’s malignancy. Pazopanib was given orally once daily, while the crizotinib schedule varied according to dose levels. This single institution study protocol was approved by the Institutional Review Board at the University of Texas, MD Anderson Cancer Center and was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and all local and federal regulatory guidelines. All patients signed informed consent prior to enrolling onto the study.
The protocol followed a standard 3+3 design.25 If one patient in a cohort experienced a dose-limiting toxicity (DLT) during the first cycle, three additional patients were enrolled and treated at that dose level. If at any time more than 33% of patients in a cohort experienced a DLT, that cohort was closed to additional patients. Adverse events (AE) were graded, based on the Common Terminology Criteria for Adverse Events, version 4.0 (CTCAEv4.0). DLTs were defined as any grade 3 or 4 non-hematologic toxicity related to any of the study drugs (except nausea and vomiting responsive to appropriate medical interventions, correctable electrolyte imbalances or alopecia); any grade 4 hematologic toxicity lasting 3 weeks or longer despite supportive care; any grade 4 nausea or vomiting > 5 days despite maximum anti-nausea regimens; any other grade 3 non-hematologic toxicity including symptoms/signs of vascular leak or cytokine release syndrome; any severe or life-threatening complication/abnormality not defined in the CTCAEv4.0 that was attributable to the therapy. The maximum tolerated dose (MTD) was defined by DLTs that occurred in the first cycle (three weeks). If a response was observed in a particular tumor type with the study drug combination, expanded enrollment was permitted for up to a total of 14 patients with that tumor type at the highest dose level deemed safe at the time of patient enrollment. Furthermore, expansion group also included patients with activating abnormalities for which there is evidence that crizotinib has antitumor activity including ALK translocations, amplification and mutations, MET amplification and mutations and ROS1 rearrangements (e.g. FIG1-ROS1 translocation).
All enrolled participants, including patients in the expansion group, were considered in the DLT analysis. For the purpose of dose expansion, a tumor response was defined as one or more of the following: 1) stable disease for more than or equal to four months (SD ≥ 4 months), or 2) decrease in the sum of target lesions by more than or equal to 20% by Response Evaluation Criteria in Solid Tumors (RECIST criteria v1.1).
Eligibility Criteria
Key inclusion criteria included patients with any advanced cancer, either refractory to standard therapy or for which no effective standard therapy exists; evaluable or measurable disease by RECISTv1.1 criterion; Eastern Cooperative Oncology Group (ECOG) status ≤2; adequate organ functions with absolute neutrophils > 1000 cells/uL, platelets ≥ 75,000/uL, total bilirubin ≤ 2 x ULN (upper limit of normal), alanine transaminase (ALT) ≤ 2.5 x ULN (≤ 5 x ULN if liver metastases present) and serum creatinine < 2 x ULN; and women of child-bearing potential and men must agree to use adequate contraception. Key exclusion criteria were patient receiving any concurrent chemotherapy other than study drugs; any uncontrolled inter-current illness including, but not limited to, ongoing or active infection requiring intravenous antibiotics; any symptomatic congestive heart failure; any history of stroke or transient ischemic attack within 6 months prior to study enrollment; any history of abdominal fistula, gastrointestinal perforation, or intra-abdominal abscess within 6 months prior to study enrollment and patients with proteinuria ≥ 2+ by urine test.
Assessment of Tumor Response
Tumor measurements were performed on patients with measurable disease at baseline and every two cycles (6 weeks) thereafter. Measurable target lesions were evaluated for response using RECIST v1.1.26,27 RECIST v1.1 defines partial response (PR) as at least 30% reduction in the sum of target lesions compared to baseline. Progressive disease (PD) is defined as an increase in disease of at least 20% compared to the smallest sum recorded (nadir) with an increase of at least 5 mm in absolute value. Stable disease (SD) is defined as neither PR, nor PD. For the purpose of this report, prolonged stable disease (SD) was defined as lasting ≥ 6 months.
Results
Patient Characteristics
Eighty-two patients with advanced or metastatic solid malignancies were enrolled between April 2012 and December 2017. Out of 82 patients, 49 patients participated in dose-escalation and the remaining 33 patients participated in dose-expansion. Demographics, clinical characteristics and patient distribution are summarized in Table 1. The median age of patients was 53 years (range, 18–78 years). The median number of prior systemic therapies was 3 (range, 0–8). Before enrollment onto the trial, one NSCLC patient with an undocumented/unknown history of an ALK translocation had received prior crizotinib for 9 months while 16 patients including sarcoma (6) and renal cancer (10) had received prior pazopanib. The median duration of prior pazopanib therapy was 9.3 months (range, 1.4 to 38.5 months). Out of 82 patients, 34 patients had other prior anti-angiogenic therapy, either as single agent or in combination. These anti-angiogenic therapies mainly consisted of bevacizumab (23), sunitinib (12), and/or axitinib (6). The most common cancer type was renal cell carcinoma (n=16) followed by ovarian cancer (n=10). All patients had experienced disease progression during their prior therapy. The median number of cycles completed for all patients was 4 (range, 1–69). Fifty patients (61%) received more than 2 cycles. For patients with SD ≥ 6 months or better, the median number of cycles completed was 10 (range, 8–69). A total of 33 patients were recruited into the expansion cohort designed to further evaluate toxicity and antitumor activity in select tumor types. The following tumor types were enrolled: renal cancer (n=11), ovarian cancer (n=7), colorectal cancer (n=3), soft tissue sarcomas (n=5), salivary gland tumors (n=2), breast cancer (n=1) and additional tumors with either ALK or MET aberrations (n=4).
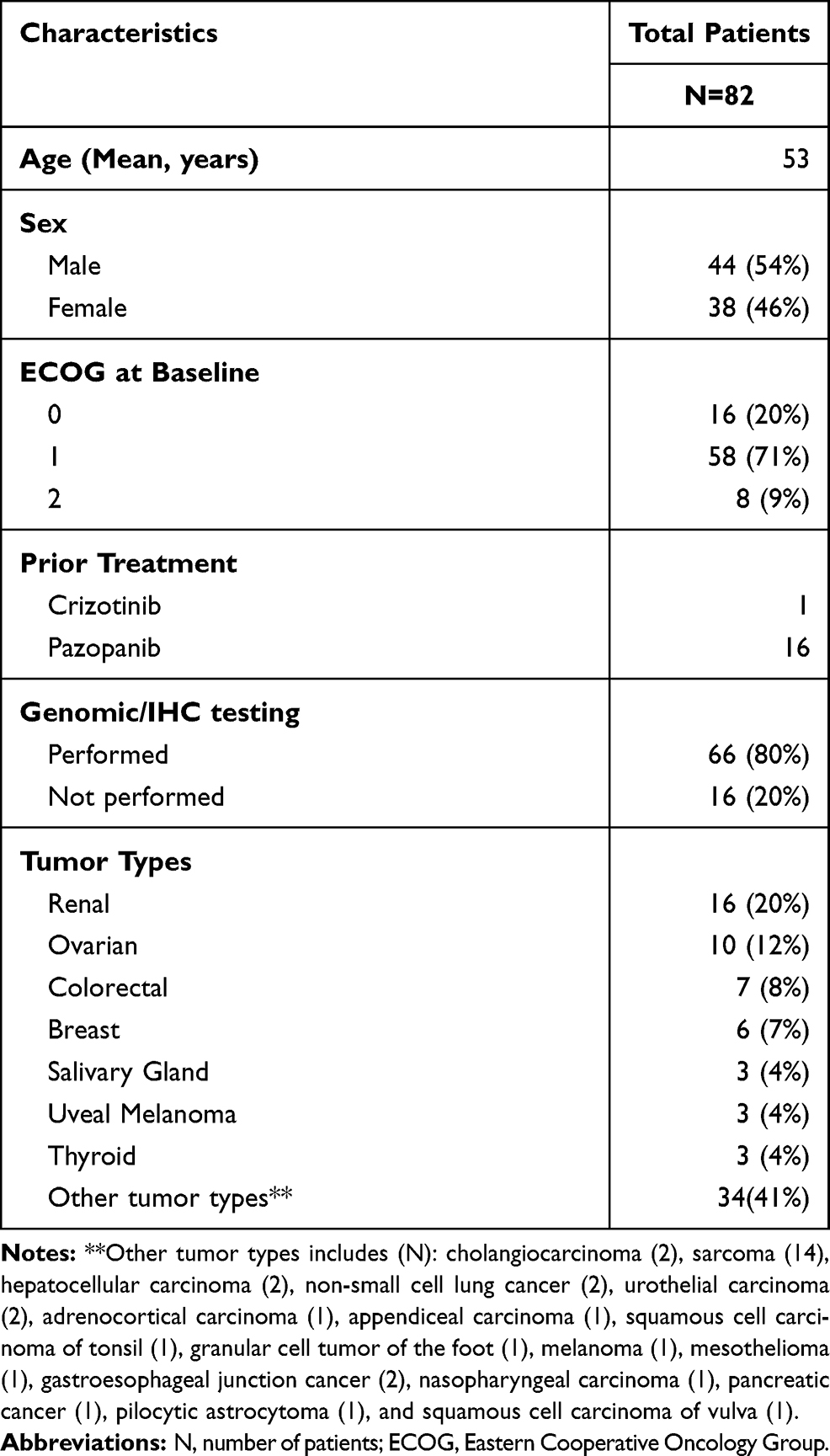 |
Table 1 Baseline Demographics and Clinical Characteristics |
Toxicity Assessment
Patients were enrolled in accordance with the planned 3+3 study design until dose level 4 (Table 2, Table 3), at which point an expansion cohort for response (as described in the Methodology section) was initiated. Dose escalation for the remaining four levels continued in accordance with the original dose escalation plan. Dose level 8 (crizotinib 250 mg twice daily and pazopanib 800 mg daily) was reached and no MTD was obtained as we were able to reach the highest FDA-approved doses of both drugs.
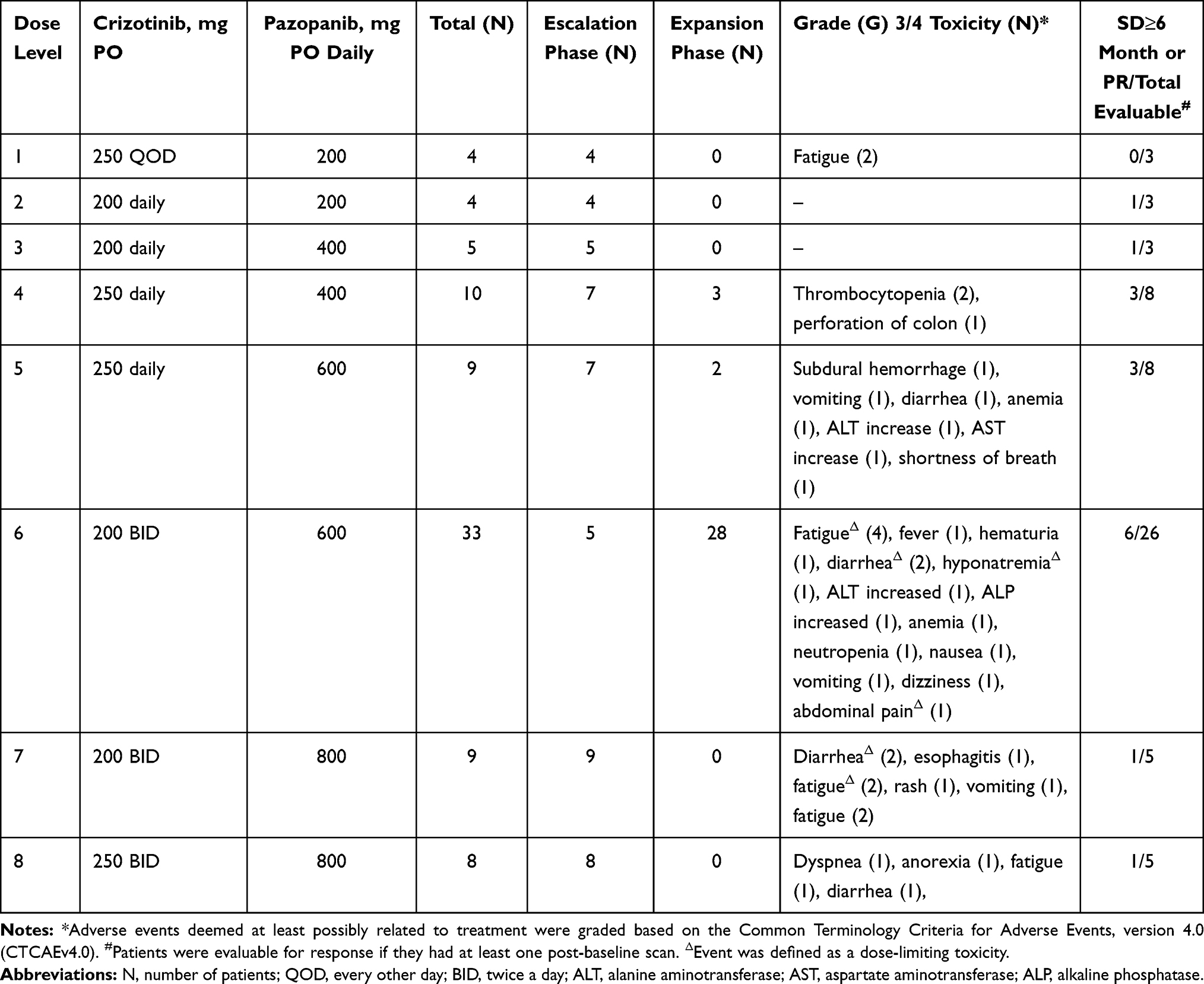 |
Table 2 Crizotinib and Pazopanib Dose-Escalation Schedule (21-Day Cycle), Grade 3/4 Toxicities and Response |
Seventy-seven patients (94%) experienced at least one adverse event that was possibly drug related. These events were mostly grade 1 or 2 and reversible. In fact, 56 patients (68%) experienced no treatment-related toxicity greater than grade 2. The most common grade 3 or 4 toxicities were as follows: fatigue (n=9, 11%), diarrhea (n=6, 7%), vomiting (n=3, 4%), anemia (n=2, 2%) and ALT increased (n=2, 2%) (Table 3). Among this subset of patients (≥ grade 3 AEs), we observed 3 DLTs (Table 2). At dose level 7, we observed 2 DLTs out of 9 patients enrolled (<33%). One patient had grade 3 fatigue while another patient had concurrent grade 3 fatigue and grade 3 diarrhea. The first patient recovered after a brief interruption in dosing and subsequent dose-reduction. He went on to complete 6 cycles of treatment before being taken off the study for disease progression. The second patient was taken off the study after only 4 days of dosing due to increased fatigue and poor tolerance to therapy. At dose level 8, one patient had grade 4 dyspnea outside the DLT period. The therapy was discontinued for this patient to remediate toxicity. Considering two occurrences of DLTs at dose level 7 and 1 patient’s discontinuation at dose level 8 due to toxicity, it was decided to continue all further expansion enrollment at dose level 6. During expansion, an additional patient at dose level 6 experienced DLT, with concurrent grade 3 hyponatremia, grade 3 fatigue and grade 3 diarrhea. In total, we observed DLT in 1 of 33 (3%) patients treated at dose level 6 including our expansion cohort patients. During this study, seven patients died while active in this trial, but none of these deaths were attributed to the study drugs. All deaths were due to disease progression except in one patient (NSCLC) at dose level 1 who died of post-obstructive pneumonia.
Overall, 16/82 (20%) patients had dose reduction of either crizotinib (n=3) or pazopanib (n=9) or both (n=4) during their course of treatment. Of these 16 patients, nine patients (56%) were dose-reduced for pazopanib at dose level 6 for various toxicities, including fatigue (5), increased transaminases (2), diarrhea (1) and nausea (1). Three patient additionally had crizotinib dose reduced from 200mg BID to 250mg daily for fatigue (1), diarrhea (1) and nausea (1). At dose level 7, two patients had pazopanib dose reduced from 800 mg daily to either 600 mg daily or 400 mg daily for diarrhea (1) or fatigue (1) while one patient had crizotinib dose reduced from 250 mg BID to 250 mg daily for grade 3 rash. Another patient had both pazopanib dose reduction to 400mg daily and crizotinib dose reduction to 250mg daily for grade 2 elevated bilirubin. At dose level 8, two patients had crizotinib dose reduced for bradycardia (1) and esophagitis (1) while one patient had pazopanib dose reduced for fatigue. Most of the dose reductions helped patients continue on the trial for at least one more cycle of treatment.
Antitumor Activity
Of 82 total patients on the trial, 61/82 (74%) patients had disease that was measurable by RECISTv1.1 and reached first restaging (6 weeks); 1/82 (1%) patient had a non-measurable disease at baseline and was excluded from image analysis for tumor efficacy; and 20/82 (24%) patients were taken off the study before first restaging for various reasons including drug-related toxicities (n=2), clinical disease progression (n=11), death (n=4) or voluntary withdrawal from the clinical trial (n=3). Figure 1 is a waterfall plot depicting the best response of the 61 response-evaluable patients. Partial response (PR) was observed in 6/61 (10%) patients and stable disease (SD) lasting ≥ 6 months was observed in 10/61 patients (16%) (total = 16/61 (26%) of patients with SD ≥6 months/PR). Supplementary Figure 1 shows swimmer plots depicting best RECISTv1.1 response and duration of time during treatment.
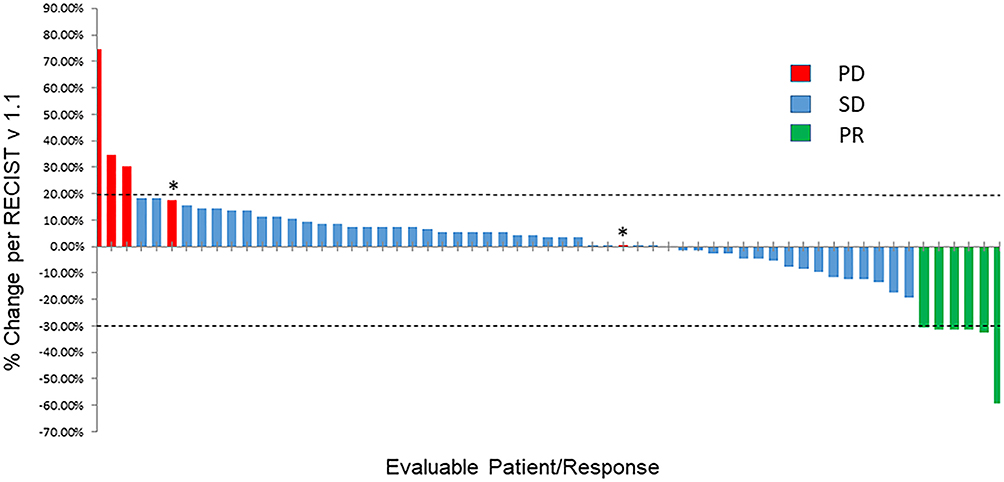 |
Figure 1 Waterfall plot depicting best RECISTv1.1 response. Individual patients are represented by vertical bars on the X-axis and best RECISTv1.1 response is depicted as percentage on the Y-axis. Sixty one of 82 patients had at least one post-baseline scan and were evaluable for response. Two patients were marked as progressive disease even though their percentage of tumor increase was less than 20% because of new lesions (*). Dotted lines show 20% increase and 30% decrease in tumor size by RECISTv1.1. |
Tumor types of patients with best response of PR include ovarian cancer (1), colorectal cancer (1), sarcoma (2), clear cell renal cancer (1) and granular cell tumor of the foot (1). In total, 3 out of 9 (33%) evaluable patients with ovarian cancer and 3 out of 14 (21%) evaluable patients with renal cancer had SD≥6 months/PR. Out of 9 patients enrolled in this trial with either MET or ALK aberrations, one patient with colon cancer and ALK p.R1209Q mutation had a best response of PR (−33%) on dose level 6 and one patient with salivary gland tumor and MET p.N375S mutation had a best response of SD ≥6 months on dose level 5. Among other patients with PR as best response to therapy, we had a clear cell renal cancer patient (−32%, PR) with a Kinase Insert Domain Receptor (KDR) p.C482R mutation previously treated with multiple lines of therapy including pazopanib monotherapy for a year (best response on monotherapy was SD). After progression on pazopanib monotherapy, she completed 36 weeks on our crizotinib and pazopanib combination study before being taken off the trial because of a new lesion in the spine.
Interestingly, patients with fusion transcripts also showed good response including patients (N=1 for each fusion) with Ewing Sarcoma Breakpoint 1 (EWSR1)-CAMP Responsive Element Binding Protein (CREB1) (−60%, PR), Dynactin Subunit 1 (DCTN1)-ALK (−32%, PR) and EWSR1-CREB3L1 (4%, SD). The patient with an EWSR1-CREB1 fusion who had clear cell sarcoma of the bowel completed 69 cycles at dose level 5 before withdrawing consent to participate in another clinical study closer to home. The patient with a DCTN1-ALK gene fusion (detected by next-generation sequencing genomic testing and confirmed by diffuse ALK expression by immunohistochemistry) who had a myxoid neoplasm of the uterus completed 41 cycles at dose level 6. Finally, the patient with EWSR1-CREB3L1 fusion who had a sclerosing epithelioid fibrosarcoma (SEF) of the abdominal wall completed 26 cycles of treatment at dose level 8.
Discussion
This is the first report of combining crizotinib with pazopanib and results demonstrate that this combination has a tolerable safety profile, with mild to moderate adverse events in patients with advanced solid tumors. No new or unexpected adverse events were observed during this study. Despite having eight dose levels, we were unable to define the MTD of crizotinib plus pazopanib in combination as the Food and Drug Administration (FDA) approved doses for both drugs were administered without any significant DLTs. The toxicity evaluation revealed that 94% (77/82) of patients had at least one adverse event that was possibly drug-related but approximately two-thirds of these patients (68%) had toxicity of grade 2 or less. The most common grade 3 and 4 toxicities included: fatigue (11%), diarrhea (7%), vomiting (4%), anemia (2%) and increased ALT (2%). The most common (occurring in more than 30% of all patients) non-hematologic adverse events, irrespective of grade, were fatigue (71%), nausea (58%), diarrhea (39%), vomiting (35%) and anorexia (30%) and for hematological toxicities, irrespective of grade, were anemia (22%) and leukopenia (13%).
Most of the AEs experienced in our study are consistent with prior reported AEs with either crizotinib and/or pazopanib monotherapy. Clinical trials with crizotinib monotherapy in patients with advanced NSCLC that was ALK-positive or ROS-positive reported visual effects, nausea, diarrhea, vomiting, constipation, edema, elevated transaminases, and fatigue as the common adverse events (in >25% of patients) irrespective of their grades.5,28–31 Visual disorders of grade 1 or 2 severity were the most frequently observed AEs with crizotinib monotherapy and were captured by using a patient-reported questionnaire. Unfortunately, our study lacked this type of assessment and may be a reason for the lack of visual changes being reported in any of our treated patients.
More serious toxicities associated with crizotinib monotherapy include hepatotoxicity, interstitial lung disease/pneumonitis, and QT-interval prolongation.32 In fact, ALT increase (17%) was the most common grade 3–4 toxicity observed in these prior studies. In our study, hepatotoxicity was also observed, albeit much milder in comparison to these previous reports as 26% of patients in our study experienced grade 1–2 increased AST and 20% patients experienced grade 1–2 increased ALT. Further, only 2% of all enrolled patients in our study experienced grade ≥ 3 increase in ALT.
A previous Phase III randomized study (VEG105192) with pazopanib as single agent in patients with advanced renal cell carcinoma (RCC) reported diarrhea (52%), increased AST (53%), increased ALT (53%), hypertension (40%), hair color changes (38%), nausea (26%), vomiting (21%) and fatigue (19%) as the most common AEs of any grade.23 A phase III study (COMPARZ) in RCC evaluated pazopanib versus sunitinib and reported diarrhea (63%), increased AST (61%), increased ALT (60%), fatigue (55%), hypertension (46%), nausea (45%), hair depigmentation (30%), hand-foot syndrome (29%), and vomiting (28%), irrespective of severity of grade. In our study, we observed less hepatotoxicity and diarrhea compared to the COMPARZ and VEG105192 studies but more fatigue which occurred in 71% of the patients.23,33
In terms of efficacy, in 61 response evaluable patients, we had 6 PRs (10%) and 10 patients (16%) with SD ≥ 6 months (SD ≥ 6 months/PR of 26%). Three patients (3/16; 19%) with SD ≥ 6 months or PR, had deleterious ALK or MET aberrations (Table 4). Of these 3 patients, none had received prior ALK or ROS1 inhibitors and only 1 patient had received a prior MET inhibitor. FDA accelerated approval of crizotinib for advanced NSCLC (ALK positive) was based on findings from two phase I and II trials.4,34,35 In a phase I study (PROFILE 1001) with ALK-positive NSCLC patients, Camidge et al29 reported a 60.8% objective response rate (ORR) among 143 response-evaluable patients. Of these 143 patients, 3 patients had a CR and 84 patients had a PR and the clinical benefit rate (CR+PR+SD) at weeks 8 and 16 was 82.5% and 70.6%, respectively.29 Updated results from a Phase II study with crizotinib (PROFILE 1005) demonstrated 54% ORR among 908 response-evaluable patients with central ALK-testing with 11 (1%) patients having achieved CR and 480 (53%) patients having achieved PR.28 Six patients (6/16; 38%) with SD ≥ 6 months or PR, had wild type ALK, MET and ROS1 on molecular testing (Table 4). Of these 6 patients, 3 had had prior antiangiogenics (prior bevacizumab n=2; prior axitinib n=1). A phase II study with pazopanib in patients with advanced metastatic RCC showed a response rate (CR+PR) of 35% with 1.3% CR, 33.3% PR and 44.9% SD (>8 weeks).36 Reasons for the differences in response between our study and these studies are multifactorial, including: 1) heterogeneous tumor types enrolled in our study; 2) heavily pre-treated patients with a median of 3 prior systemic therapies in our study; and, 3) lack of genomic selection for enrollment into our study.
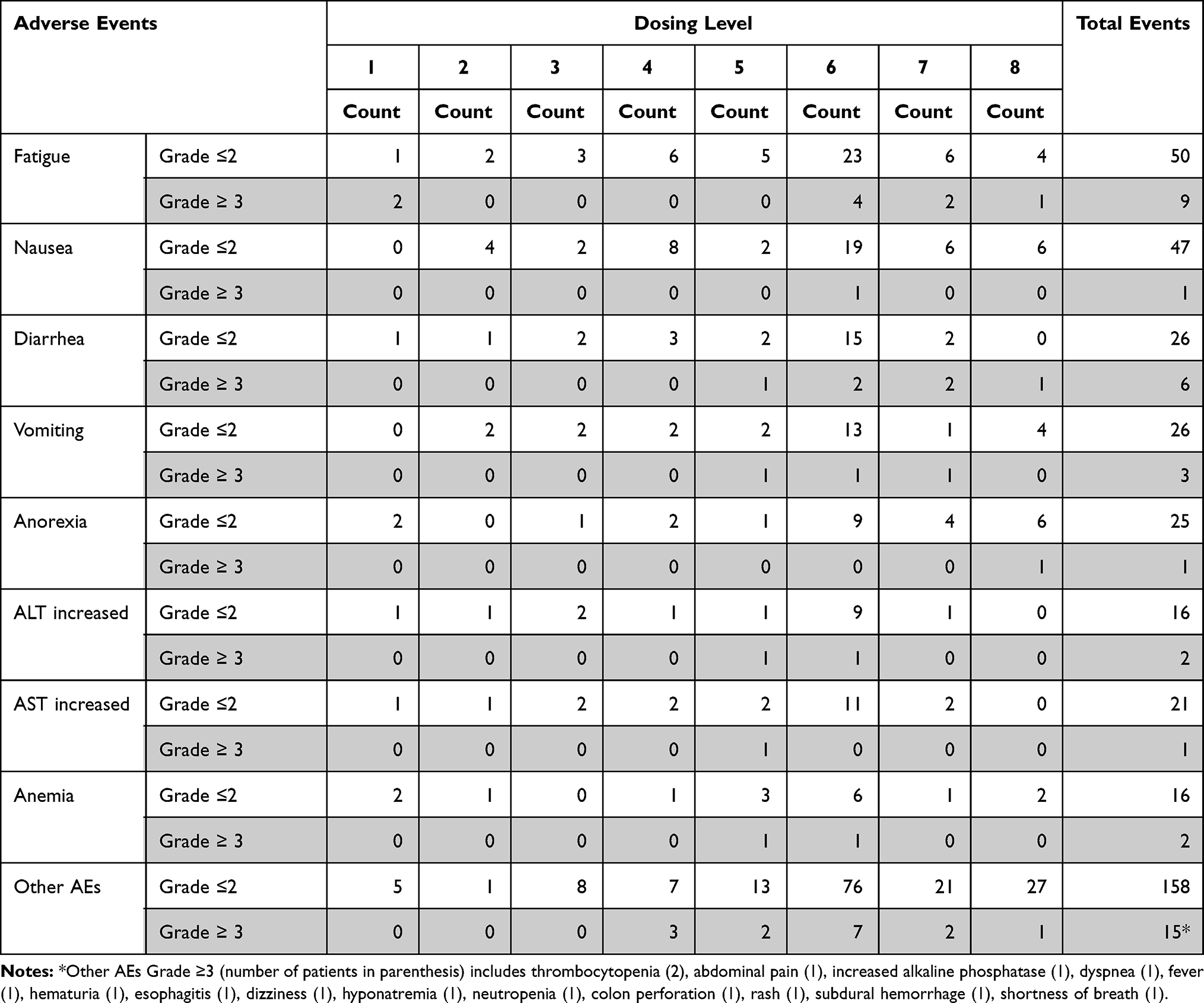 |
Table 3 Adverse Events at Any Dose Level |
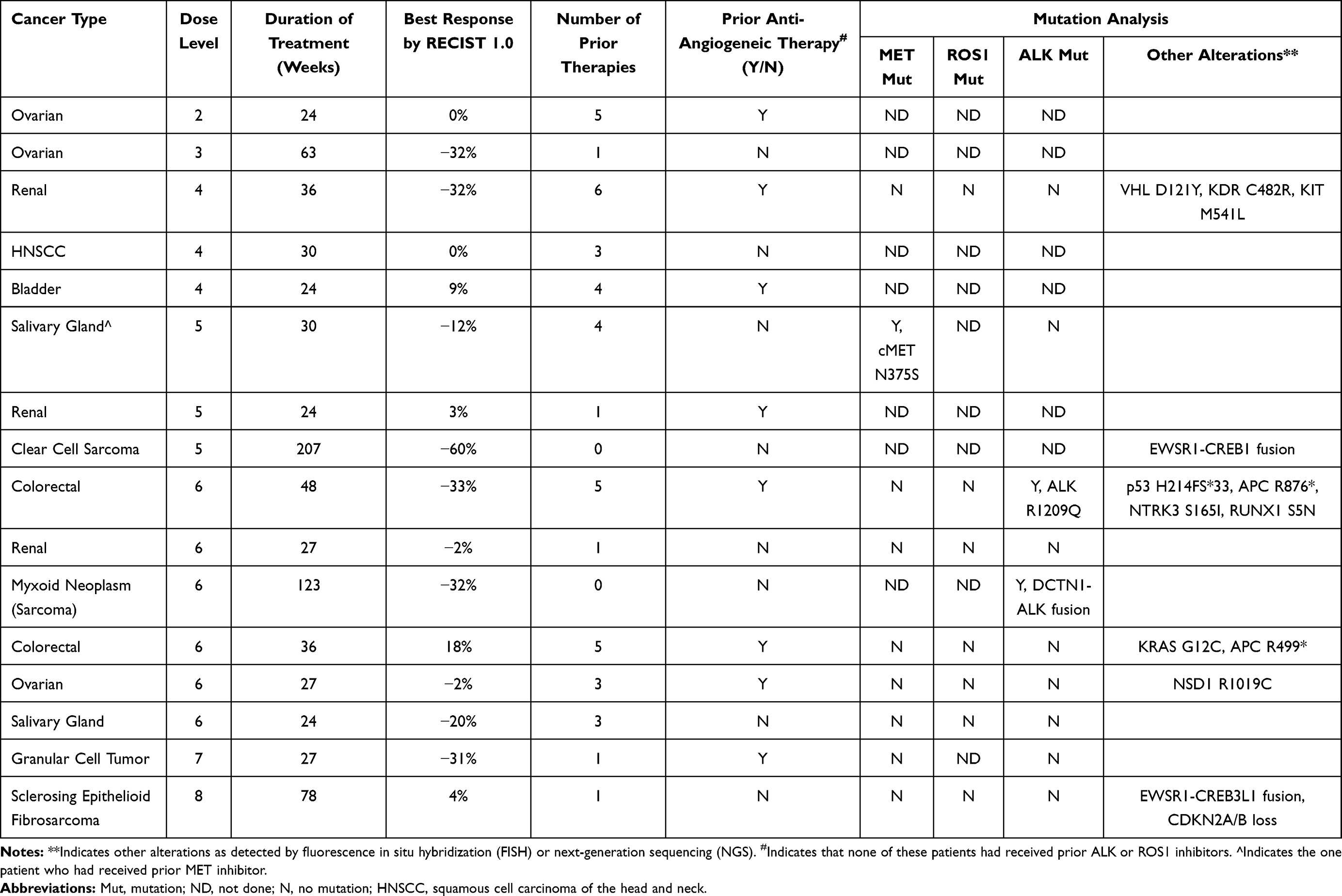 |
Table 4 Stable Disease (SD) ≥6 Months or Partial Response (PR) by RECIST and Characterization by Patient |
Interestingly, a patient with DCTN1-ALK gene fusion showed a partial response (−32%) to therapy.37 DCTN1-ALK fusions have been observed recurrently in multiple tumor types including spitz tumors, lung cancer and inflammatory myofibroblastic tumors.38–40 Shimada et al41 has characterized the DCTN1-ALK fusion protein and showed that it is a potential oncogene that can be used as a target for ALK tyrosine kinase inhibitors including crizotinib and alectinib. The gene fusion event confers constitutive activation of ALK and treatment with crizotinib inhibited constitutive phosphorylation of ALK and activation of downstream PI3K and MAPK signaling cascades.38 Recently, Michels et al42 has demonstrated that an ALK p.G1269A mutation was detected in a patient with DCTN1-ALK fusion who developed acquired resistance to crizotinib. Unfortunately, we were unable to obtain a repeat biopsy in our patient and therefore we could not confirm if any new mutations in ALK could have possibly contributed to the emergence of therapeutic resistance.
We also observed that a patient with EWSR1-CREB1 fusion favorably responded to crizotinib and pazopanib combination treatment. Our clear cell sarcoma patient was negative for EWSR1-activating transcription factor-1 (ATF1) fusion transcripts and was able to complete 69 cycles (>4 years) on dose level 5 with a best response of PR (−60%). Ultimately, the patient withdrew consent from the study in order to pursue treatment closer to home.43 Most EWSR1 fusions involve the 5ʹ portion of EWSR1 which acts as an activator of the DNA binding region of its fusion partner targeting the fusion of specific promoters. CREB1 binds cAMP response elements within target genes to upregulate gene expression. EWSR1-ATF1 fusion protein is known to activate the melanocyte master transcription factor (MITF) which further activates the MET gene.44 It is however not known if all EWSR1-fusion proteins are capable of activating cMET expression. Nevertheless, it is hypothesized that these fusions sensitize tumor cells to cMET and/or ALK inhibitors.
Similarly, another patient with sclerosing epithelioid fibrosarcoma (SEF) and EWSR1-CREB3L1 fusion completed 26 cycles of treatment with a best response of SD. SEFs are considered rare but aggressive tumors arising from deep tissues and characterized by EWSR1-CREB3L1 translocation.45 Unfortunately, there are no standardized treatment regimens for SEFs and these tumors are quite challenging to manage. The unusual response of SEFs with EWSR1-CREB3L1 fusion to combination therapy with crizotinib and pazopanib has not been reported. Intriguingly, a recent phase II trial (90,101 “CREATE”) evaluated the efficacy and safety of crizotinib in patients with advanced clear cell sarcoma (CCSA) with EWSR1/ATF1 fusions and showed an ORR of only 3.8%,46 and adds to speculation that fusion partner genes of EWSR1 may be an important determinant of clinical efficacy to crizotinib. This however needs further testing.
Apart from the interesting gene-fusion patients discussed, we observed that a patient with KDR p.C482R mutation achieved PR with our combination therapy. KDR is also known as vascular endothelial growth factor receptor-2 (VEGFR-2). The p.C482R mutation is a gain-of-function mutation leading to constitutive dimerization and activation of VEGFR-2 and this variant of KDR has been shown to highly correlate with serum-soluble VEGFR-2 level which is recognized as a pharmacodynamic response marker for pazopanib.47,48 Our study lends support to the use of pazopanib either as a single agent or in combination with crizotinib for patients with KDR p.C482R mutation.
In conclusion, we determined that dose level 6 (crizotinib 200 mg twice daily and pazopanib 600 mg daily) was the most tolerable dosing of this drug combination and could be used for future studies. We also demonstrated moderate clinical activity in patients with advanced solid tumors that had received numerous prior therapies.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.
Acknowledgments
We thank the patients who participated in this study and their families for supporting them.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Pfizer Pharmaceutical Company.
Disclosure
Dr Sarina A. Piha-Paul reports Clinical Trial Research Support (Received through the institution) from AbbVie, Inc., ABM Therapeutics, Inc., Acepodia, Inc., Alkermes, Aminex Therapeutics, Amphivena Therapeutics, Inc., BioMarin Pharmaceutical, Inc., Boehringer Ingelheim, Bristol Myers Squibb, Cerulean Pharma, Inc., Chugai Pharmaceutical Co., Ltd., Curis, Inc., Daiichi Sankyo, Inc., Eli Lilly, ENB Therapeutics, Five Prime Therapeutics, Gene Quantum, Genmab A/S, Glaxo SmithKline, Helix BioPharma Corp., Incyte Corp., Jacobio Pharmaceuticals Co., Ltd., MedImmune, LLC., Medivation, Inc., Merck Sharp and Dohme Corp., Novartis Pharmaceuticals, Pieris Pharmaceuticals, Inc., Pfizer, Principia Biopharma, Inc., Puma Biotechnology, Inc., Rapt Therapeutics, Inc., Seattle Genetics, Silverback Therapeutics, Taiho Oncology, Tesaro, Inc., TransThera Bio, Core Grant (CCSG Shared Resources Clinical Trial Research Support (Received through the institution)) from NCI/NIH P30CA016672, outside the submitted work. Dr Vivek Subbiah reports Research funding/Grant support for clinical trials (to institution): Novartis, Bayer, Berghealth, Incyte, Fujifilm, Pharmamar, D3, Pfizer, Multivir, Amgen, AbbVie, Alfa-sigma, Agensys, Boston Biomedical, Idera Pharma, Inhibrx, Exelixis, Blueprint Medicines, Loxo Oncology, MedImmune, Altum, Dragonfly Therapeutics, Takeda, and National Comprehensive Cancer Network, NCI-CTEP and UT MD Anderson Cancer Center, Turning point therapeutics, Boston Pharmaceuticals. Travel funding: Novartis, Pharmamar, ASCO, ESMO, Helsinn, Incyte, Ad Hoc Advisory Board: Helsinn, LOXO Oncology/Eli Lilly, R-Pharma US, INCYTE, QED pharma, MedImmune, Novartis, Signant Health. Dr Aung Naing reports grants from National Cancer Institute, grants from EMD Serono, grants from MedImmune, grants, non-financial support from ARMO BioSciences, grants from Karyopharm Therapeutics, grants from Incyte, grants, personal fees from Novartis, grants from Regeneron, grants from Merck, grants from Bristol-Myers Squibb, grants from Pfizer, grants, personal fees from CytomX Therapeutics, grants from Neon Therapeutics, grants from Calithera Biosciences, grants from TopAlliance Biosciences, grants from Eli Lilly, grants, personal fees from Kymab, grants from PsiOxus, grants from Arcus Biosciences, grants from NeoImmuneTech, grants from ImmuneOncia, grants from Surface Oncology, grants, personal fees from Genome & Company, grants from OncoSec KEYNOTE-695, grants from STCube Pharmaceuticals, outside the submitted work; and Spouse • Research funding: Immune Deficiency Foundation, Jeffrey Modell Foundation and chao physician-scientist, and Baxalta. • Advisory board: Takeda, CSL, Behring, Horizon, and Pharming. Dr Filip Janku reports grants from Agios, grants, personal fees from Asana, grants from Astellas, grants from Astex, grants from Bayer, grants from Bicara, grants from BioMed Valley Discoveries, grants from Bioxcel, grants from Bristol-Myers Squibb, grants, personal fees from Deciphera, grants from FujiFilm Pharma, grants from Genentech, personal fees from Ideaya, grants from JS Innopharm, grants from Lilly, grants from Merck, grants, personal fees from Novartis, grants from Novellus, grants from Plexxikon, grants from Proximagen, grants from Sanofi, grants, personal fees from Sotio, grants from SpringBank Pharmaceuticals, grants from SQZ Biotechnologies, grants, personal fees from Synlogic, grants from Synthorx, grants from Symphogen, personal fees from Bausch Health, personal fees, Ownership Interests from Cardiff Oncology, personal fees from Guardant Health, personal fees from IFM Therapeutics, personal fees from Immunomet, personal fees from Illumina, personal fees from Jazz Pharmaceuticals, personal fees from PureTech Health, Bio-Rad, Biocartis, outside the submitted work. Dr Shreyaskumar Patel reports personal fees from DAiichi SAnkyo, personal fees from Deciphera, consultant fees from Epizyme, Dova and Bayer, grants from Blueprint Medicines, Clinical Trial fees from Hutchison Medipharma, outside the submitted work. Dr David Hong reports grants from AbbVie, grants, Grant/Research Support & Consulting/Advisory Role from Adaptimmune, grants from Aldi-Norte, grants, Grant/Research Support & Consulting/Advisory Role from Amgen, grants from Astra-Zeneca, grants, Grant/Research Support, Consulting/Advisory Role, Travel, Accommodations, Expenses from Bayer, grants from BMS, grants from Daiichi-Sankyo, grants from Eisai, grants from Fate Therapeutics, grants, Grant/Research Support & Consulting/Advisory Role from Genentech, grants from Genmab, grants from Ignyta, grants, Grant/Research Support & Consulting/Advisory Role from Infinity, grants from Kite, grants from Kyowa, grants from Lily, grants, Grant/Research Support & Travel, Accommodations, Expenses from LOXO, grants from Merck, grants from MedImmune, grants from Mirati, grants, Grant/Research Support & Travel, Accommodations, Expenses from miRNA, grants from Molecular Templates, grants from Mologen, grants from NCI-CTEP, grants from Novartis, grants, Grant/Research Support & Consulting/Advisory Role from Numab, grants, Grant/Research Support & Consulting/Advisory Role from Pfizer, grants, Grant/Research Support & Consulting/Advisory Role from Seattle Genetics, Grant/Research Support & Consulting/Advisory Role from Takeda, grants from Turning Point Therapeutics, Verstatem, Consulting/Advisory Role from Alpha Insights, Acuta, Axion, Baxter, Boxer Capital, COG, ECOR1, Expert Connect, GLG, Group H, Guidepoint, H.C. Wainwright, Janssen, Merrimack, Medscape, NTRK Connect, Prime Oncology, Slingshot, Trieza Therapeutics, WebMD, Travel, Accommodations, Expenses from ASCO, AACR, SITC, Advisor from Molecular Match, Founder from OncoResponse, Advisor from Presagia, outside the submitted work. Dr Funda Meric-Bernstam reports personal fees from Aduro BioTech Inc., Alkermes, AstraZeneca, DebioPharm, eFFECTOR Therapeutics, F. Hoffman-La Roche Ltd., Genentech Inc., IBM Watson, Jackson Laboratory, Kolon Life Science, OrigiMed, PACT Pharma, Parexel International, Pfizer Inc., Samsung Bioepis, Seattle Genetics Inc., Tyra Biosciences, Xencor, Zymeworks, Immunomedics, Advisory Committee from Inflection Biosciences, Mersana Therapeutics, Puma Biotechnology Inc., Silverback Therapeutics, Spectrum Pharmaceuticals Zentalis, grants from Aileron Therapeutics, Sponsored Research from AstraZeneca, Bayer Healthcare Pharmaceutical, Calithera Biosciences Inc., Curis Inc., CytomX Therapeutics Inc., Daiichi Sankyo Co. Ltd., Debiopharm International, eFFECTOR Therapeutics, Genentech Inc., Guardant Health Inc., Millennium Pharmaceuticals, Novartis, Puma Biotechnology Inc., Taiho Pharmaceutical Co., personal fees from Chugai Biopharmaceuticals, Mayo Clinic, Rutgers Cancer Institute of New Jersey, Beth Israel Deaconess Medical Center, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Shaw AT, Ou S-HI, Bang Y-J, et al. Crizotinib in ROS1 rearranged non–small-cell lung cancer. N Engl J Med. 2014;371(21):1963–1971. doi:10.1056/NEJMoa1406766
2. Bergethon K, Shaw AT, Ignatius Ou S-H, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863–870. doi:10.1200/JCO.2011.35.6345
3. Crino L, Kim D, Riely G. Initial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): PROFILE 1005. J Clin Oncol. 2011;29(15_suppl):7514. doi:10.1200/jco.2011.29.15_suppl.7514
4. Kwak EL, Bang Y-J, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. doi:10.1056/NEJMoa1006448
5. Solomon BJ, Mok T, Kim D-W, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. doi:10.1056/NEJMoa1408440
6. Giubellino A, Linehan WM, Bottaro DP. Targeting the met signaling pathway in renal cancer. Expert Rev Anticancer Ther. 2009;9(6):785–793. doi:10.1586/era.09.43
7. Tu W, Zhu C, Clark C, et al. Efficacy of c-Met inhibitor for advanced prostate cancer. BMC Cancer. 2010;10(1):556. doi:10.1186/1471-2407-10-556
8. Seiwert TY, Jagadeeswaran R, Faoro L, et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009;69(7):3021–3031. doi:10.1158/0008-5472.CAN-08-2881
9. Sampson ER, Martin BA, Morris AE, et al. The orally bioavailable met inhibitor PF-2341066 inhibits osteosarcoma growth and osteolysis/matrix production in a xenograft model. J Bone Miner Res. 2011;26(6):1283–1294. doi:10.1002/jbmr.336
10. Zillhardt M, Christensen JG, Lengyel E. An orally available small-molecule inhibitor of c-Met, PF-2341066, reduces tumor burden and metastasis in a preclinical model of ovarian cancer metastasis. Neoplasia. 2010;12(1):1–10. doi:10.1593/neo.09948
11. Wu J, Savooji J, Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol. 2016;9(1):19. doi:10.1186/s13045-016-0251-8
12. Rolfo C, Passiglia F, Castiglia M, et al. ALK and crizotinib: after the honeymoon what else? Resistance mechanisms and new therapies to overcome it. Transl Lung Cancer Res. 2014;3(4):250–261. doi:10.3978/j.issn.2218-6751.2014.03.01
13. Dagogo-Jack I, Shaw AT. Crizotinib resistance: implications for therapeutic strategies. Ann Oncol. 2016;27(Suppl 3):iii42–iii50. doi:10.1093/annonc/mdw305
14. Mitchell DC, Bryan BA. Anti-angiogenic therapy: adapting strategies to overcome resistant tumors. J Cell Biochem. 2010;111(3):543–553. doi:10.1002/jcb.22764
15. Schutz FAB, Choueiri TK, Sternberg CN. Pazopanib: clinical development of a potent anti-angiogenic drug. Crit Rev Oncol Hematol. 2011;77(3):163–171. doi:10.1016/j.critrevonc.2010.02.012
16. Sandler A, Gray R, Perry MC, et al. Paclitaxel–carboplatin alone or with bevacizumab for non–small-cell lung cancer. N Engl J Med. 2006;355(24):2542–2550. doi:10.1056/NEJMoa061884
17. Herbst RS, Takeuchi H, Teicher BA. Paclitaxel/carboplatin administration along with antiangiogenic therapy in non-small-cell lung and breast carcinoma models. Cancer Chemother Pharmacol. 1998;41(6):497–504. doi:10.1007/s002800050773
18. Adjei AA, Mandrekar SJ, Dy GK, et al. Phase II trial of pemetrexed plus bevacizumab for second-line therapy of patients with advanced non–small-cell lung cancer: NCCTG and SWOG Study N0426. J Clin Oncol. 2010;28(4):614–619. doi:10.1200/JCO.2009.23.6406
19. Patel JD, Bonomi P, Socinski MA, et al. Treatment rationale and study design for the pointbreak study: a randomized, open-label phase III study of pemetrexed/carboplatin/bevacizumab followed by maintenance pemetrexed/bevacizumab versus paclitaxel/carboplatin/bevacizumab followed by maintenance bevacizumab in patients with stage IIIB or IV nonsquamous non-small-cell lung cancer. Clin Lung Cancer. 2009;10(4):252–256. doi:10.3816/CLC.2009.n.035
20. Chow L, Blais N, Jonker DJ, et al. A phase I dose-escalation and pharmacokinetic study of sunitinib in combination with pemetrexed in patients with advanced solid malignancies, with an expanded cohort in non-small cell lung cancer. Cancer Chemother Pharmacol. 2012;69(3):709.
21. You W-K, Sennino B, Williamson CW, et al. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011;71(14):4758–4768. doi:10.1158/0008-5472.CAN-10-2527
22. Zou HY, Li Q, Lee JH, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67(9):4408–4417. doi:10.1158/0008-5472.CAN-06-4443
23. Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28(6):1061. doi:10.1200/JCO.2009.23.9764
24. van der Graaf WT, Blay J-Y, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled Phase 3 trial. Lancet. 2012;379(9829):1879–1886. doi:10.1016/S0140-6736(12)60651-5
25. Storer BE. Design and analysis of phase I clinical trials. Biometrics. 1989;45(3):925–937. doi:10.2307/2531693
26. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. doi:10.1016/j.ejca.2008.10.026
27. Nishino M, Jagannathan JP, Ramaiya NH, et al. Revised RECIST guideline version 1.1: what oncologists want to know and what radiologists need to know. AJR Am J Roentgenol. 2010;195(2):281–289. doi:10.2214/AJR.09.4110
28. Blackhall F, Ross camidge D, Shaw AT, et al. Final results of the large-scale multinational trial PROFILE 1005: efficacy and safety of crizotinib in previously treated patients with advanced/metastatic ALK-positive non-small-cell lung cancer. ESMO Open. 2017;2(3):e000219. doi:10.1136/esmoopen-2017-000219
29. Camidge DR, Bang Y-J, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a Phase 1 study. Lancet Oncol. 2012;13(10):1011–1019. doi:10.1016/S1470-2045(12)70344-3
30. Shaw AT, Kim D-W, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. doi:10.1056/NEJMoa1214886
31. Shaw A, Riley GJ, Bang Y-J, et al. Crizotinib in advanced ROS1-rearranged non-small cell lung cancer (NSCLC): updated results from PROFILE 1001. Ann Oncol. 2016;27(suppl_6):1206PD–1206PD. doi:10.1093/annonc/mdw383.07
32. Kazandjian D, Blumenthal GM, Chen H-Y, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist. 2014;19(10):e5–e11. doi:10.1634/theoncologist.2014-0241
33. Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013;369(8):722–731. doi:10.1056/NEJMoa1303989
34. Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non–small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20(8):2029–2034. doi:10.1158/1078-0432.CCR-13-3077
35. Kim D-W, Ahn M-J, Shi Y, et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). J Clin Oncol. 2012;30(15_suppl):7533. doi:10.1200/jco.2012.30.15_suppl.7533
36. Hutson TE, Davis ID, Machiels J-PH, et al. Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2010;28(3):475–480. doi:10.1200/JCO.2008.21.6994
37. Subbiah V, McMahon C, Patel S, et al. STUMP un“stumped”: anti-tumor response to anaplastic lymphoma kinase (ALK) inhibitor based targeted therapy in uterine inflammatory myofibroblastic tumor with myxoid features harboring DCTN1-ALK fusion. J Hematol Oncol. 2015;8(1):66. doi:10.1186/s13045-015-0160-2
38. Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in spitz tumours and spitzoid melanomas. Nat Commun. 2014;5(1):3116. doi:10.1038/ncomms4116
39. Iyevleva AG, Raskin GA, Tiurin VI, et al. Novel ALK fusion partners in lung cancer. Cancer Lett. 2015;362(1):116–121. doi:10.1016/j.canlet.2015.03.028
40. Wang X, Krishnan C, Nguyen EP, et al. Fusion of dynactin 1 to anaplastic lymphoma kinase in inflammatory myofibroblastic tumor. Hum Pathol. 2012;43(11):2047–2052. doi:10.1016/j.humpath.2012.02.014
41. Shimada Y, Kohno T, Ueno H, et al. An oncogenic ALK fusion and an RRAS mutation in KRAS mutation-negative pancreatic ductal adenocarcinoma. Oncologist. 2017;22(2):158–164. doi:10.1634/theoncologist.2016-0194
42. Michels SYF, Scheel AH, Wündisch T, et al. ALK G1269A mutation as a potential mechanism of acquired resistance to crizotinib in an ALK-rearranged inflammatory myofibroblastic tumor. NPJ Precis Oncol. 2017;1(1):4. doi:10.1038/s41698-017-0004-3
43. Subbiah V, Holmes O, Gowen K, et al. Activity of c-Met/ALK inhibitor crizotinib and multi-kinase VEGF inhibitor pazopanib in metastatic gastrointestinal neuroectodermal tumor harboring EWSR1-CREB1 fusion. Oncology. 2016;91(6):348–353. doi:10.1159/000449204
44. Davis IJ, McFadden AW, Zhang Y, et al. Identification of the receptor tyrosine kinase c-Met and its ligand, hepatocyte growth factor, as therapeutic targets in clear cell sarcoma. Cancer Res. 2010;70(2):639–645. doi:10.1158/0008-5472.CAN-09-1121
45. Stockman DL, Ali SM, He J, et al. Sclerosing epithelioid fibrosarcoma presenting as intraabdominal sarcomatosis with a novel EWSR1-CREB3L1 gene fusion. Hum Pathol. 2014;45(10):2173–2178. doi:10.1016/j.humpath.2014.05.006
46. Schoffski P, Wozniak A, Stacchiotti S, et al. Activity and safety of crizotinib in patients with advanced clear-cell sarcoma with MET alterations: European Organization for Research and Treatment of Cancer phase II trial 90101 ‘CREATE’. Ann Oncol. 2017;28(12):3000–3008. doi:10.1093/annonc/mdx527
47. Maitland ML, Xu C-F, Cheng Y-C, et al. Identification of a variant in KDR associated with serum VEGFR2 and pharmacodynamics of pazopanib. Clin Cancer Res. 2015;21(2):365–372. doi:10.1158/1078-0432.CCR-14-1683
48. Sarabipour S, Ballmer-Hofer K, Hristova K. VEGFR-2 conformational switch in response to ligand binding. eLife. 2016;5:e13876. doi:10.7554/eLife.13876
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.