")
Back to Journals » OncoTargets and Therapy » Volume 11
A peptide-CpG-DNA-liposome complex vaccine targeting TM4SF5 suppresses growth of pancreatic cancer in a mouse allograft model
Authors Park S , Kim D, Wu G, Jung H , Park JA , Kwon HJ , Lee Y
Received 5 September 2018
Accepted for publication 6 November 2018
Published 3 December 2018 Volume 2018:11 Pages 8655—8672
DOI https://doi.org/10.2147/OTT.S186606
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Sangkyu Park,1,2,* Dongbum Kim,3,* Guang Wu,3,4 Harry Jung,3 Jeong-A Park,1,2 Hyung-Joo Kwon,3,5 Younghee Lee1,2
1Department of Biochemistry, College of Natural Sciences, Chungbuk National University, Cheongju, Chungbuk 28644, Republic of Korea; 2Biotechnology Research Institute, Chungbuk National University, Cheongju, Chungbuk 28644, Republic of Korea; 3Center for Medical Science Research, College of Medicine, Hallym University, Chuncheon 24252, Republic of Korea; 4School of Laboratory Medicine and Life Sciences, Wenzhou Medical University, Wenzhou, Zhejiang 325035, China; 5Department of Microbiology, College of Medicine, Hallym University, Chuncheon 24252, Republic of Korea
*These authors contributed equally to this work
Background: Patients with pancreatic cancer have a poor prognosis and are usually diagnosed at a late stage. Because TM4SF5 is known to be overexpressed in hepatocellular carcinoma, colon cancer, and pancreatic cancer, it is considered as one of the candidate molecular targets for an anticancer strategies.
Purpose: The purpose of this study was to evaluate possible utility of TM4SF5 to treat pancreatic cancer using a mouse allograft model.
Materials and methods: We analyzed expression of TM4SF5 in pancreatic cancer tissues using immunohistochemistry. We established a mouse pancreatic cancer cell line stably expressing TM4SF5 and identified the effect of TM4SF5 expression in vitro. We used the CpG-DNA-peptide-liposome complex as a peptide vaccine and investigated antitumor effects of the vaccine in a mouse model with TM4SF5 expressing pancreatic cells. To investigate the function of produced antibody, we evaluated effects of the anti-TM4SF5 monoclonal antibody in vitro in terms of cell growth and migration properties.
Results: Immunohistochemical analysis showed that 36.4% of pancreatic cancer tissue samples expressed TM4SF5. Expression of TM4SF5 induced increased cell proliferation and motility in vitro. Injection of the TM4SF5 peptide vaccine induced the production of anti-hTM4SF5 antibodies and reduced the growth of pancreatic tumors in mice established by subcutaneous injection of the TM4SF5-expressing mouse pancreatic cancer cell line. The treatment of TM4SF5-expressing cells with the anti-hTM4SF5 monoclonal antibody reduced cell growth, modulated the expression of the epithelial–mesenchymal transition markers Vimentin and E-cadherin, and decreased cell motility in vitro.
Conclusion: Our results showed that the TM4SF5 peptide vaccine had a protective effect against pancreatic tumors expressing TM4SF5, and this effect was mediated, at least in part, by the production and suppressive function of the anti-TM4SF5 antibodies. Therefore, we suggest that targeting TM4SF5 could be a novel strategy to prevent or treat pancreatic cancer.
Keywords: transmembrane 4 superfamily member 5 protein, pancreatic cancer, peptide vaccine, antibody, targeting
Introduction
Pancreatic cancer has a poor prognosis, probably because it is diagnosed at an advanced stage in many cases. It is the eighth and ninth leading cause of death from cancer in men and women respectively throughout the world.1 Approximately 90% of pancreatic cancers are pancreatic ductal adenocarcinoma (PDAC).1 In pancreatic cancer, four genes are frequently mutated: KRAS, CDKN2A, SMAD4, and TP53.2 KRAS is mutated in ~95% of PDAC patients, and the KRAS mutation triggers the genetic progression of PDAC and drives PDAC development and maintenance.3–5 However, despite a long period of attempts, anti-KRAS therapy has yet to be proven effective.3 Recently, clinical trials based on therapeutic strategies targeting specific proteins and/or pathways have shown significant advances in many tumor types. However, in pancreatic cancer, these trials were proven not successful, and pancreatic cancer remains a lethal disease.6–8
Therapeutic strategies for cancer include surgery, chemotherapy, radiation therapy, immunotherapy, targeted therapy, etc. Peptide vaccine therapy is based on the stimulation of the immune system by immunization with peptides from defined tumor-associated antigens (TAAs) usually in combination with immune stimulators (including adjuvants).9 Since MAGE-1 was identified as the first reported human TAA gene,10 several other TAA genes have been identified and studied as targets of therapeutic peptide vaccines. These TAAs may be classified into several categories. Cancer/testis antigens are expressed in testis germ cells but not in adult somatic tissues, and differentiation antigens are specific for lineages or differentiation stage of various cell types and other tumor antigens.9,11 Many clinical trials and research studies on peptide vaccines for cancer immunotherapy are currently exploring the efficacy of peptides that act as antigens in anticancer vaccines.12–19
A liposome is a vesicle used to deliver drugs or genetic materials into a cell.20,21 Especially, cationic liposomes have advantages to efficiently bind with negatively charged DNA and cell membranes.22 Non-cationic lipids are generally used as helpers for cationic liposomes. For example, dioleoyl phosphatidylethanolamine (DOPE) increases the endosomal escape ability of DNA after endocytosis of cationic liposome/DNA complex.23,24 In a pancreatic cancer model, recent studies reported the use of a liposome complex to deliver shRNAs or anti-miRNA oligonucleotides for therapeutic purposes.25,26 Liposomes are also used for efficient antigen delivery and enhanced immune response in developing vaccines.27–29 Previously, we confirmed that natural phosphodiester bond CpG-DNA (specifically, MB-ODN 4531(O)) induced innate immune responses.30 Immune stimulating activity of CpG-DNA was enhanced by encapsulation in a liposome complex consisting of DOPE: cholesterol hemisuccinate (CHEMS) (1:1 ratio). We named CpG-DNA co-encapsulated with DOPE:CHEMS “Lipoplex(O)”.31,32 Liposome complex consisting of B cell epitope and Lipoplex(O) successfully induced antibodies specifically recognizing viral proteins and cancer-associated proteins.31,32
TM4SF5, a membrane protein with four transmembrane domains, was first identified in 1998, and high expression of TM4SF5 was reported in hepatocellular carcinoma (HCC), colon cancer, soft tissue sarcoma, gastric cancer, esophageal cancer, and pancreatic cancer.33–35 TM4SF5 expression enhances the motility and malignancy of hepatocytes by increased interaction with CD151, and TM4SF5 expression level is associated with cancer progression and poor survival of patients.35–37 TM4SF5 regulates VEGF-mediated angiogenesis through cooperation with integrin and tumorigenic proliferation.34,38 TM4SF5 reduces the RhoA activity by p27kip1 translocation and FAK phosphorylation, and RhoA inactivation induces the epithelial–mesenchymal transition (EMT) and G1/S phase progression.39–42 TM4SF5-induced FAK phosphorylation promotes immune escape in HCC.43 TM4SF5-induced growth and motility can be blocked by a synthetic compound 4′-(p-toluenesulfonyl-amide)-4-hydroxychalcone.44 In our previous studies, we reported that immunization with the TM4SF5 peptide epitope-CpG-DNA-liposome complex is effective in the prevention and therapy of HCC and colon cancer.45–47 We also confirmed that the TM4SF5-targeted monoclonal antibody inhibits the growth and metastasis of HCC and colon cancer.39,48,49
Considering that TM4SF5 is also highly expressed in pancreatic cancer tissue,33 it is likely that the TM4SF5 peptide vaccine may have preventive or therapeutic effects on pancreatic cancer. In this study, we established mouse pancreatic cancer cells expressing TM4SF5 and confirmed the preventive effects of a vaccination with the TM4SF5 peptide-CpG-DNA-liposome complex on TM4SF5-expressing mouse pancreatic tumors in a mouse allograft model.
Materials and methods
Tissue microarrays and immunohistochemistry
The formalin-fixed, paraffin-embedded AccuMax tissue array was obtained from ISUABXIS with the approval of the Institutional Review Board in Hallym University (approval number: HIRB-2014-114). The tissue array was analyzed by immunohistochemistry using the mouse anti-TM4SF5 monoclonal antibody49 (mEC2-C, 1 μg/slide) and Histostain Plus kit (Thermo Fisher Scientific, Waltham, MA, USA) as previously described.48 All images were examined using a Nikon Eclipse E-200 microscope. The percentages of cells expressing TM4SF5 were calculated as the number of TM4SF5-positive cells divided by the total number of cells in each tumor type.
Cell culture
The mouse PDAC cell line PANC02 was provided by Professor Kyu Lim (Chungnam National University, Republic of Korea).50 The cells were maintained in DMEM (Hyclone, Logan, UT, USA) with 10% FBS (Hyclone), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C under a humidified atmosphere of 5% CO2. The use of the cell line was approved by the Institutional Animal Care and Use Committee of Hallym University (Permit Number: Hallym 2015-55).
RT-PCR
Total RNA was isolated with TRI Reagent® according to the manufacturer’s instructions (MRC, Cincinnati, OH, USA). Then, 2 μg of total RNA was reverse-transcribed in the first-strand synthesis buffer containing 6 μg/mL oligo(dT) primer, 50 U M-MLV reverse transcriptase, 2 mM dNTP, 10 mM DTT, and 40 U RNaseOUT™ recombinant ribonuclease inhibitor (Thermo Fisher Scientific). The reaction was done at 37°C for 50 minutes and heat inactivated at 70°C for 15 minutes. One microliter of synthetic cDNA was subjected to a standard PCR reaction of 25 or 30 cycles consisting of denaturation for 40 seconds at 95°C, annealing for 40 seconds at 58°C, and extension for 40 seconds at 72°C. The primer sequences used were as follows: GAPDH, 5′-TCC ACC ACC CTG TTG CTG TA-3′ (sense) and 5′-ACC ACA GTC CAT GCC ATC AC-3′ (anti-sense) (product size 452 bp); human TM4SF5, 5′-AGC TTG CAA GTC TGG CTC AT-3′ (sense) and 5′-GCT GGA TCC CAC ACA GTA CT-3′ (anti-sense) (product size 401 bp); mouse TM4SF5, 5′-CGC TTA CTT GCG AAA TGA CA-3′ (sense) and 5′-TTT CCT GCA ATC GCC ACA CA-3′ (anti-sense) (product size 174 bp).
Packaging and transduction of control and TM4SF5-encoding retroviruses
The human TM4SF5 cDNA was amplified from pcDNA3.1-hTM4SF549 by PCR using the following primer set: hTM4SF5 5′ primer, 5′-GAA TTC GCC ACC ATG GAA CAA AAA CTC ATC TCA GAA GAG GAT CTG GGT GCA ATG TGT ACG GGA AAA-3′ and hTM4SF5 3′ primer, 5′-CTC GAG TCA GTG AGG TGT GTC CTG-3′. The cDNA fragments were cloned into the expression vector pLXSN (Clontech, Mountain View, CA, USA) using the Xho I and EcoR I sites. GP2-293, a cell line derived from 293 cells, was obtained from Clontech and used as a packaging cell line for preparation of the retroviruses. GP2-293 cells were maintained in DMEM containing 10% FBS in 5% CO2 incubator at 37°C. Retroviral vectors pLXSN or pLXSN-hTM4SF5 along with pVSV-G (Clontech) encoding the pseudo-envelope protein gene were transfected into the cells by Lipofectamine 2000 (Thermo Fisher Scientific). Twelve hours later, the medium was exchanged with fresh culture medium supplemented with 10 mM sodium butyrate (Sigma-Aldrich Co., St Louis, MO, USA). After 48 hours, the supernatant of the culture medium was taken and filtered through a filter with a 0.45 μm pore size. The retrovirus supernatants were concentrated using Centricon centrifugal filters (EMD Millipore, Billerica, MA, USA) and stored at −80°C. The viral supernatant was applied to PANC02 cells along with 8 μg/mL of polybrene (Sigma-Aldrich Co.). Twenty-four hours later, G418 (Sigma-Aldrich Co.) was added at a concentration of 1 mg/mL, and the G418-resistant PANC02 cells were selected.
Western blot analysis
Harvested cells were lysed in a lysis buffer (pH 8.0, 20 mM Tris-HCl, 137 mM NaCl, 10% glycerol, 10 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40, protease inhibitor cocktail, and phosphatase inhibitor). Proteins were resolved by SDS-PAGE and electro-transferred to polyvinylidene fluoride membranes (EMD Millipore). The membranes were blocked with 5% dry milk in PBS-Tween-20 ([PBST] 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, and 0.05% Tween-20) and probed with an appropriate primary antibody. The monoclonal anti-GAPDH antibody (cat no SC-32233, 1:1,000 dilution) was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The anti-E-cadherin (cat no 3195, 1:1,000 dilution) and anti-Vimentin (cat no 5741, 1:1,000 dilution) polyclonal antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Immunoreactive proteins were visualized by horseradish peroxidase (HRP)-conjugated anti-rabbit (cat no SC-2004) or anti-mouse (cat no SC-2005) secondary antibodies (1:5,000 dilution, Santa Cruz Biotechnology Inc.) and an enhanced chemiluminescence solution (ATTO, Tokyo, Japan).
Immunoprecipitation
Whole cell lysates were lysed in a lysis buffer. After the humanized anti-hTM4SF5 monoclonal antibody, hEC2-C-2,49 was conjugated to protein A-agarose beads (Hoffman-La Roche Ltd., Basel, Switzerland), the whole cell lysates were immunoprecipitated with the antibody-conjugated agarose beads. Immunoprecipitated proteins were washed with PBS and processed for a standard Western blot analysis using the mouse anti-hTM4SF5 monoclonal antibody (mEC2-CF, 1 μg/mL), which we previously reported,51 and anti-Myc antibody (Cell Signaling Technology, cat no 2276, 1:1,000 dilution) which recognizes Myc-tag in the recombinant hTM4SF5.
Confocal microscopy
Cells were cultured on glass cover slips in 4-well plates (Thermo Fisher Scientific). The cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 (Sigma-Aldrich Co.), and stained with humanized anti-hTM4SF5 monoclonal antibody (1 μg/mL) for 1 hour.49 After extensive washing with PBS, the samples were incubated with Alexa Fluor 488-conjugated goat anti-human IgG (2 μg/mL, Thermo Fisher Scientific, cat no A11013) for 1 hour. The nuclei were stained with Hoechst 33258 (Sigma-Aldrich Co.), and the mounted samples were scanned with an LSM 710 (Carl Zeiss Meditec AG, Jena, Germany).
Cell proliferation assay
The cell proliferation ELISA, BrdU colorimetric kit (Hoffman-La Roche Ltd.), was used to measure the cell proliferation according to the manufacturer’s instructions. Cells were treated with normal IgG or humanized anti-hTM4SF5 monoclonal antibody (10 μg/mL) for 3 days. The BrdU solution was added to each well, and then, the plates were incubated for 4 hours at 37°C. After fixation of the cells, anti-BrdU antibody conjugated with peroxidase was added to each well for 90 minutes at room temperature. A colorimetric assay was developed with a substrate solution, and the absorbance at 370 nm with a reference wavelength of 492 nm was measured using a microplate reader (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Cell cycle analysis
Cell cycle status was determined by staining the cellular DNA with propidium iodide (PI). Cells were treated with normal IgG or the humanized anti-hTM4SF5 monoclonal antibody (10 μg/mL) for 3 days. For cycle detection, cells were treated with PI/RNase solution (BD Biosciences, San Jose, CA, USA) for 30 minutes after fixation overnight in 70% ethanol. The fluorescent signal was detected by flow cytometry using a FACSCalibur (BD Biosciences) and the cell cycle status was analyzed using FCS express program (De Novo Software, Glendale, CA, USA).
In vitro wound-healing assays
Cells were placed in a 6-well plate, cultured overnight to confluence in medium containing serum, and the monolayer was wounded with a pipette tip. Normal IgG or the humanized anti-hTM4SF5 monoclonal antibody (10 μg/mL) was added to the medium for the indicated periods. The cells were fixed with 4% paraformaldehyde (Biosesang, Seongnam, Republic of Korea) for 20 minutes and stained with 0.01% crystal violet (Sigma-Aldrich Co.) for 20 minutes. The wound-healing activity of cells was calculated by the following formula; the cells migrating into wound (%) = [(the wounded area at 0 day – cell-free space in the wounded area)/wounded area at 0-day ×100]. The percent ratio of migrated area to wounded area was measured in three wells per experimental treatment and three wounds per well under a microscope (Nikon Corporation, Tokyo, Japan).
RhoA activity assay
The RhoA Pull-down Activation Assay Biochem Kit (bead pull-down format) (Cytoskeleton, Denver, CO, USA) was used to detect the active form of RhoA according to the manufacturer’s instructions. Cells were treated with normal IgG or the anti-hTM4SF5 monoclonal antibody for 6 hours. After the cells were lysed, the cell lysates were incubated with the beads conjugated with the RhoA binding domain of Rhotekin for 1 hour at 4°C. After extensive washing with PBS-T, the samples were processed for a standard Western blot analysis using RhoA-specific antibody.
In vitro cell migration and invasion assays
Transwell chambers with 8 μm porosity (Corning Incorporated, Corning, NY, USA) were used for these assays. For the migration assays, the lower side of the transwell chamber membranes was coated with gelatin (10 μg/well, Sigma-Aldrich Co.). For the invasion assays, a Matrigel invasion chamber (Corning Incorporated) was used. Cells were suspended in serum-free medium with normal IgG or the humanized anti-hTM4SF5 monoclonal antibody (10 μg/mL) and placed on the top of the transwell chamber. DMEM containing 10% FBS was placed in the lower chamber. After incubation for 48 hours, the cells that invaded to the lower surface of the filters were fixed, stained with crystal violet, and counted under a microscope (Nikon Corporation).
CpG-DNA and peptide synthesis
CpG-DNA (MB-ODN 4531(O), AGCAGCGTTCGTGTCGGCCT) was provided by Samchully Pharm (Seoul, Republic of Korea) as described previously.30 The B cell epitope peptide of human TM4SF5 (hTM4SF5EC2, 131TACAYLLNRTLWDRCEAPPRVVPWNCT157) was selected, and the cyclic form of the peptide (hTM4SF5EC2-C) was synthesized by Peptron (Daejeon, Republic of Korea) as described previously.49
Preparation of the B cell epitope peptide and CpG-DNA co-encapsulated in DOPE:CHEMS complexes
The liposomes DOPE and CHEMS were obtained from Sigma-Aldrich Co. Liposome complexes consisting of peptide (50 μg) and CpG-DNA (50 μg) co-encapsulated with DOPE:CHEMS (at a 1:1 ratio) were formulated as reported previously.31 Briefly, DOPE and CHEMS were mixed in 10% ethanol at a molar ratio of 1:1, evaporated with nitrogen gas to produce a solvent-free lipid film, and resuspended in a mixture containing equal volumes of water-soluble MB-ODN 4531(O) (50 μg) and peptide (50 μg), followed by vigorous stirring at room temperature for 30 minutes. After adjusting the pH to 7.0, the peptide and CpG-DNA co-encapsulated with the DOPE:CHEMS [Lipoplex(O)] complex were sonicated lightly for 30 seconds with a sonicator (Sonifier 450; Branson Ultrasonics, Danbury, CT, USA). The complex was filtered with a 0.22 μm filter and freeze-thawed three times with liquid nitrogen.
Mice and immunization
Four-week old female C57BL/6 mice (n=24; weight, 19–20 g) were obtained from Nara Biotech, Inc. (Seoul, Republic of Korea). The mice were maintained under specific pathogen-free conditions (20°C–25°C, 40%–45% humidity, 12-hour light/dark cycle; food and water access, ad libitum). All procedures for the animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals of the National Veterinary Research and Quarantine Service of Korea with the approval of the Institutional Animal Care and Use Committee of Hallym University (Permit Number: Hallym 2015-55). Mice were anesthetized under isoflurane (2%–3%) inhalation with a RC2-Rodent Circuit Controller (Lab Etc Inc., Clayton, MO, USA), and all efforts were made to minimize suffering. After letting them adapt for 1 week, the mice (weight, 20–21 g) were injected intraperitoneally with PBS or a complex of peptide and Lipoplex(O) three times at 10-day intervals. Ten days after the third immunization, the mice were injected subcutaneously in the dorsal right flank with PBS or 1×106 of PANC02-hTM4SF5 cells. The body weight and size of the tumors were measured every 7 days. Tumor volumes were evaluated as length × width2/2. The mice were sacrificed 100 days after the first immunization, and the tumors were surgically excised and weighed. Mice were euthanized by CO2 inhalation (all efforts were made to minimize suffering) when the mice lost 20% of their adult body weight, tumor size reached 2,000 mm3, observed evidence of debilitation, pain or distress such as a hunched posture, rough coat, reduced food consumption, emaciation, inactivity, difficulty ambulating, respiratory problems, and solid tumor growth. The CO2 inhalation was performed with 100% CO2 at a fill rate of 10%–30% of the chamber volume per minute.
ELISA
Mouse sera were obtained by retro-orbital bleeding under isoflurane (2%–3%) inhalation anesthesia with a RC2-Rodent Circuit Controller 10 days after the final injection. Ninety-six-well immunoplates (Nalgene Nunc International, Rochester, NY, USA) were coated with 5 μg/mL of the cyclic peptide hTM4SF5EC2-C and then blocked with PBST containing 1% BSA. Total IgG levels were measured as previously described.31 Briefly, the sera were diluted to 1:50 with PBST and added to the wells of each plate. To measure the titers of total IgG, the sera were added to the top row of each plate, and serial 1:3 dilutions in PBST were then placed into the subsequent rows. The plates were incubated for 2 hours at room temperature and washed with PBST. Then, HRP-conjugated goat anti-mouse IgG (1:5,000, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA, cat no 115-035-003) was added to the wells and incubated for 1 hour. A colorimetric assay was developed with a tetramethylbenzidine substrate solution (Kirkegaard and Perry Laboratories, Gaithersburg, MD, USA), and we used a Spectra Max 250 microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA) to measure the absorbance at 405 nm.
Statistics
The results are shown as the mean±standard error of the mean (SEM) from at least three independent experiments. Statistical significance of the differences between two samples was evaluated using Student’s t-test. P<0.05 was considered statistically significant.
Results
Expression of TM4SF5 in pancreatic cancer tissues
To confirm expression of TM4SF5 in pancreatic cancer, we analyzed human pancreatic cancer tissues by immunohistochemistry. Analysis of 33 pancreatic cancer specimens showed that 36.4% of pancreatic cancer tissue samples expressed TM4SF5 with staining in >11% of tumor cells (Figure 1 and Table 1). Therefore, we decided to investigate the effects of vaccination targeting TM4SF5 using a mouse allograft model.
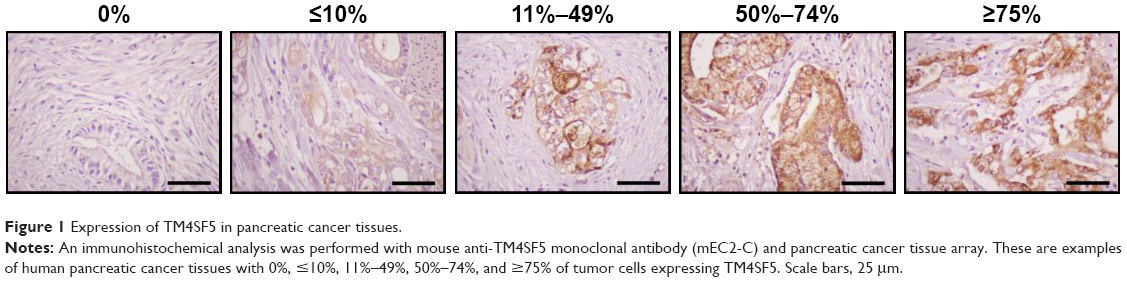 | Figure 1 Expression of TM4SF5 in pancreatic cancer tissues. |
 | Table 1 Immunohistochemical analysis of TM4SF5 expression in pancreatic cancer tissues |
Establishment of the TM4SF5-expressing mouse pancreatic cancer cells
The mouse PDAC cell line PANC02 is frequently used to form tumors in a mouse allograft model.52–56 We first checked the expression of TM4SF5 in PANC02 cells, however expression of TM4SF5 was not detected in the PANC02 cells. Therefore, we then established a mouse PDAC cell line stably expressing TM4SF5. We cloned the human TM4SF5 gene into the retroviral vector pLXSN and prepared retrovirus particles using the packaging protocol with pVSV-G in GP2-293 cells. Then, we established control PANC02 cells harboring the pLXSN vector (PANC02-mock) and PANC02 cells expressing TM4SF5 (PANC02-hTM4SF5) by transduction. Expression of TM4SF5 at the mRNA level was detected in the PANC02-hTM4SF5 cells by RT-PCR (Figure 2A, right); however, there was no detected expression of TM4SF5 in the PANC02-mock cells. We also confirmed the expression of TM4SF5 at protein level by immunoprecipitation followed by Western blotting (Figure 2B). TM4SF5 protein was detected in the PANC02-hTM4SF5 cells but not in the PANC02-mock cells. Using confocal microscopy, TM4SF5 protein was detected in the PANC02-hTM4SF5 cells and hardly detected in the PANC02-mock cells (Figure 2C).
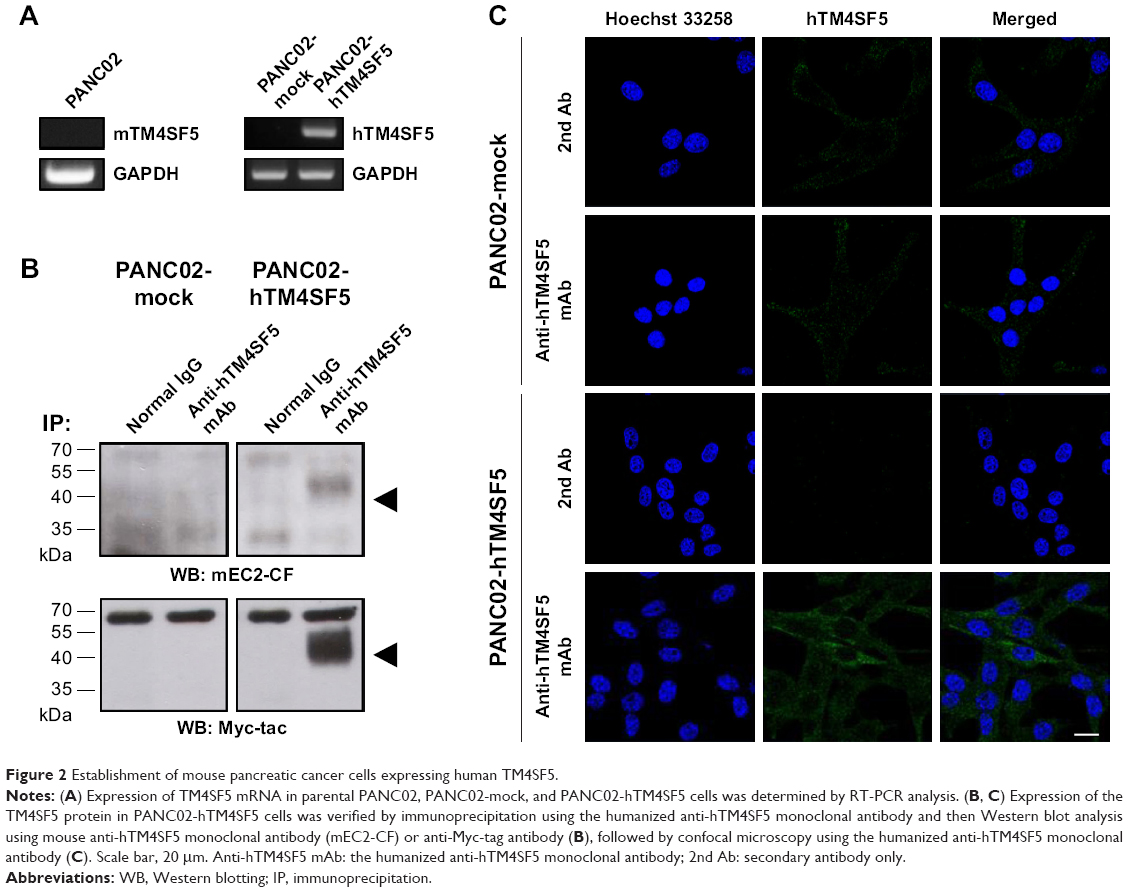 | Figure 2 Establishment of mouse pancreatic cancer cells expressing human TM4SF5. |
Effects of TM4SF5 expression in mouse pancreatic cancer cells
The tetraspanin superfamily, which includes TM4SF5, promotes tumor growth and metastasis by mediating activation of integrin signaling and/or modulating the immune response in other cancers.39,48,49,57–59 To analyze the cellular consequences of TM4SF5 expression in a pancreatic cancer model, we checked the growth and motility of the PANC02-mock and PANC02-hTM4SF5 cells. First, we measured the change in cell proliferation using the BrdU incorporation assay. Expression of TM4SF5 increased the cell proliferation (Figure 3A). In addition, G1 phase was decreased and S phase was increased in PANC02-hTM4SF5 cells compared to PANC02-mock cells (Figure 3B and C). RhoA activity negatively regulates cell cycle progression through control of cyclin gene expression, and TM4SF5 promotes progression of G1/S phase by inhibition of RhoA activity in HCC.42 In Figure 3D, the level of active RhoA (RhoA-GTP) was decreased by hTM4SF5 expression in PANC02 cells. Next, we investigated the effect of TM4SF5 on cell migration using the wound healing assay and found that the migration of PANC02-hTM4SF5 cells into the wounded area was increased compared to the PANC02-mock cells (Figure 3E).
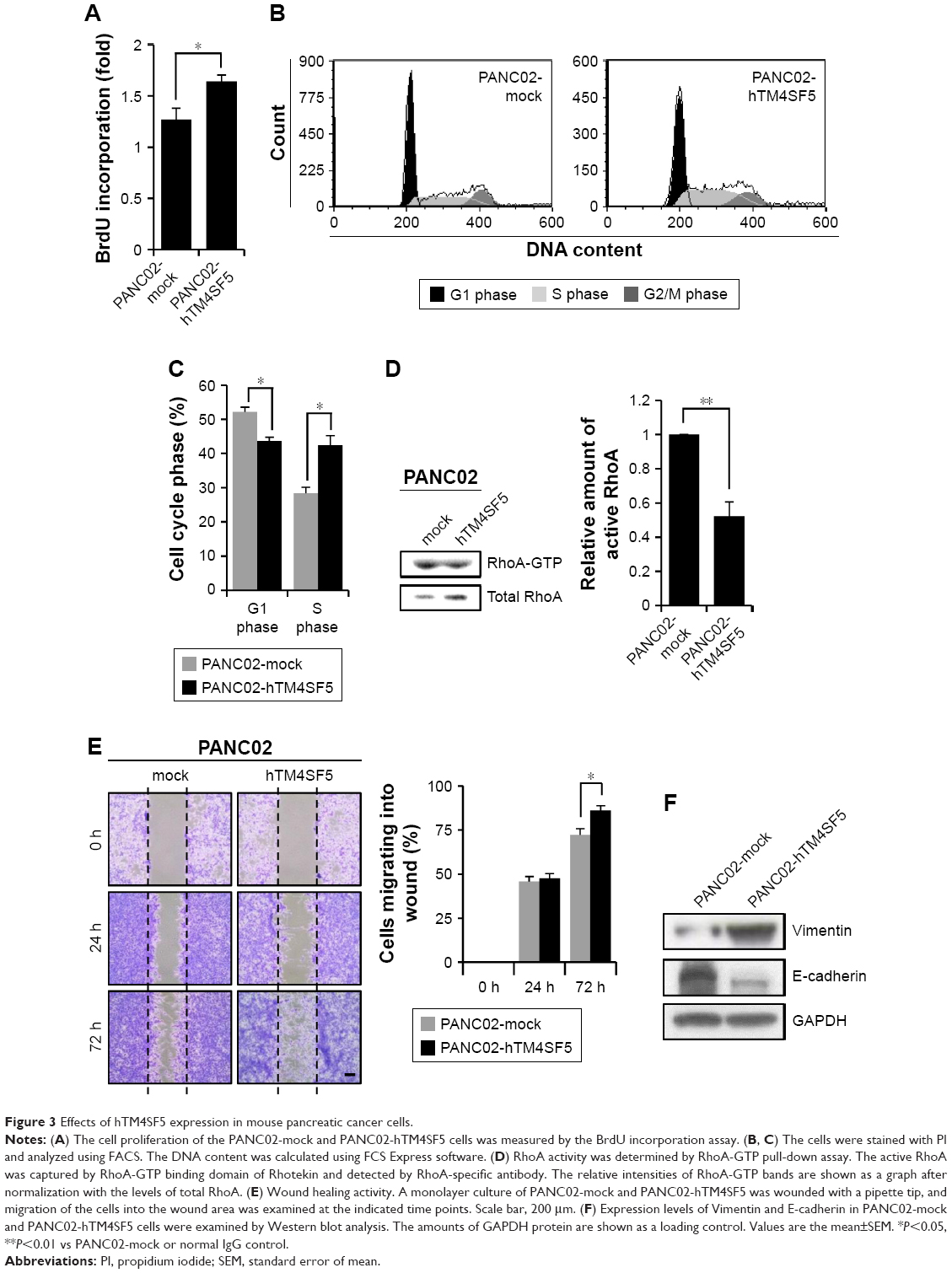 | Figure 3 Effects of hTM4SF5 expression in mouse pancreatic cancer cells. |
Vimentin, one of the mesenchymal markers, contributes to tumorigenesis and the EMT mediated by the organization of cytoskeletons and loss of an epithelial junction protein E-cadherin.60 The expression level of E-cadherin is reduced during the EMT in tumor progression and inversely correlated with tumor malignancy.61 Because the TM4SF5 expression increased the cell motility, we further checked the expression levels of Vimentin and E-cadherin. As shown in Figure 3F, the expression levels of Vimentin and E-cadherin were increased and decreased, respectively, in the PANC02-hTM4SF5 cells compared to the PANC02-mock cells. Therefore, we confirmed that TM4SF5 expression induced cell growth and reinforced mesenchymal properties in mouse PDAC cells.
Suppression of tumor growth by vaccination with a complex of TM4SF5 peptide and Lipoplex(O) in a mouse pancreatic tumor model established with the TM4SF5-expressing PDAC cells
To evaluate the prophylactic efficacy of the TM4SF5-targeting vaccine with regards to tumor growth, we immunized C57BL/6 mice with the peptide hTM4SF5EC2-C in combination with Lipoplex(O),45 and then, PANC02-hTM4SF5 cells were implanted into the mice (Figure 4A). The formula consisting of the cyclic peptide hTM4SF5EC2-C, corresponding to the amino acids 131–157 in the extracellular domain 2 of human TM4SF5, and Lipoplex(O) were previously proven to successfully induce immune responses targeting cell membrane TM4SF5 in mice.49 After the third vaccination, we checked the titers of TM4SF5-specific IgG in the mouse sera using ELISA plates coated with the cyclic peptide hTM4SF5EC2-C and confirmed that the immunized mice had a large amount of anti-hTM4SF5 antibodies in their sera (Figure 4B). In contrast, the control mice injected with PBS had no production of TM4SF5-reactive antibodies. The PANC02-hTM4SF5 cells implanted into C57BL6 mice grew continuously and formed a tumor mass, whereas tumor development was significantly inhibited by the vaccination with the TM4SF5 cyclic peptide with Lipoplex(O) (Figure 4C–E); tumor volumes (Figure 4C and D); and tumor weight (Figure 4E) were reduced in the vaccinated mice compared to the control mice. The vaccination is not likely to induce significant side effects because there was no difference during the experiment in the body weights of the immunized mice compared to the control group (Figure 4F). On the other hand, when PANC02 cells were implanted into C57BL/6 mice, the volumes and weights of the tumors were not reduced by vaccination with Lipoplex(O) alone or a complex of the TM4SF5 cyclic peptide in combination with Lipoplex(O) (Figure S1). Therefore, we conclude that the TM4SF5 cyclic peptide and Lipoplex(O) complex induced a protective immune response and inhibited the growth of TM4SF5-expressing tumor in vivo.
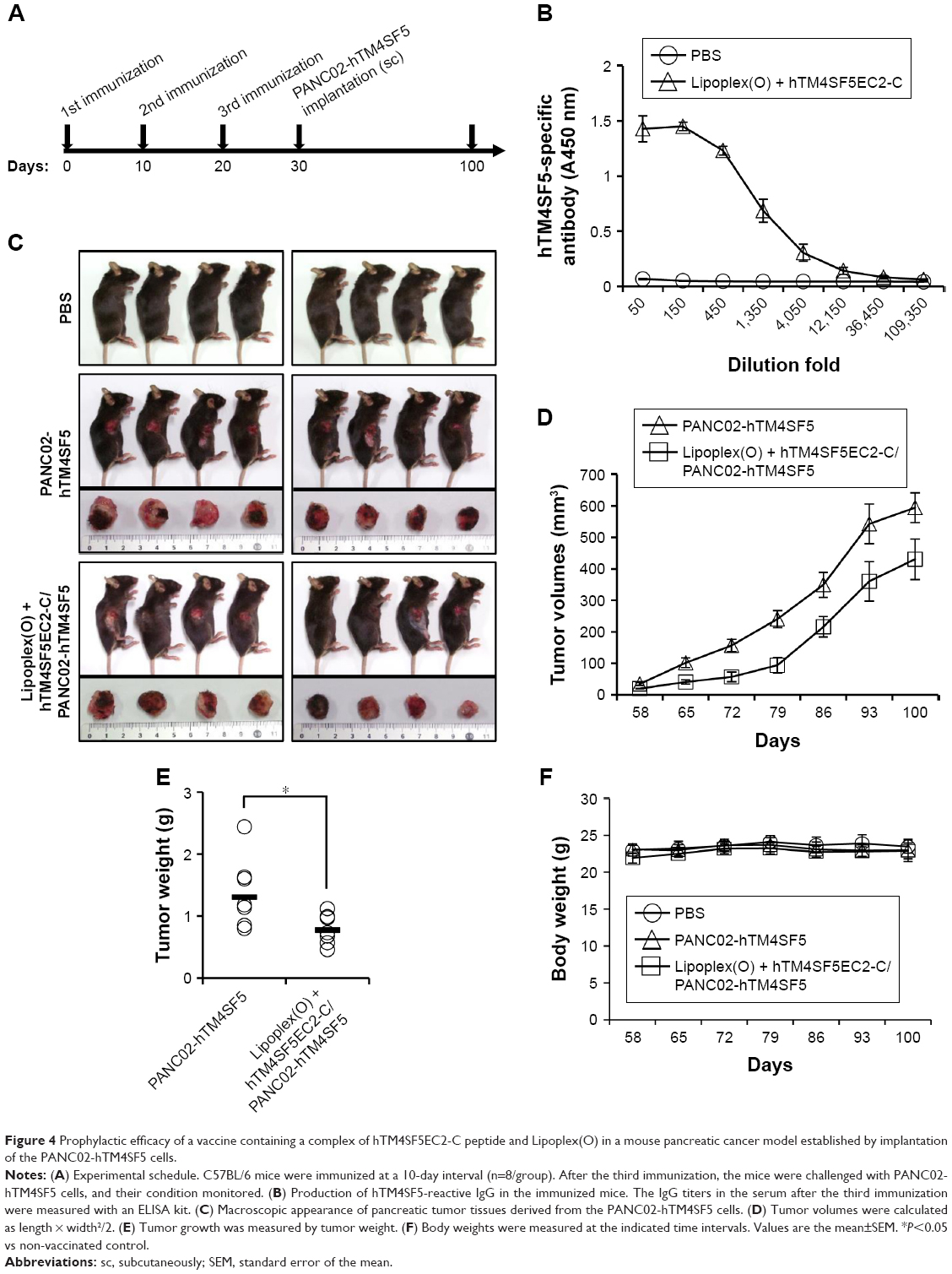 | Figure 4 Prophylactic efficacy of a vaccine containing a complex of hTM4SF5EC2-C peptide and Lipoplex(O) in a mouse pancreatic cancer model established by implantation of the PANC02-hTM4SF5 cells. |
Suppression of tumor cell growth by the humanized anti-hTM4SF5 antibody in vitro
Because we confirmed that the TM4SF5 peptide vaccination induced TM4SF5-specific antibody production and suppressed the growth of TM4SF5-expressing pancreatic tumors in vivo (Figure 4), we checked the effects of the anti-hTM4SF5 antibody on the growth of control and TM4SF5-expressing pancreatic cancer cells in vitro. Here, we used the humanized anti-hTM4SF5 monoclonal antibody (hEC2-C-2) that we established with the cyclic peptide hTM4SF5EC2-C as an antigen.49 When we measured the proliferation rate of the PANC02-mock and PANC02-hTM4SF5 cells using the BrdU incorporation assay, the proliferative activity of the PANC02-hTM4SF5 cells was significantly decreased by the humanized anti-hTM4SF5 monoclonal antibody compared to the control PANC02-mock cells in a dose-dependent manner (Figure 5A). When we checked cell cycle status after treatment with the humanized anti-hTM4SF5 monoclonal antibody, cell population in the G1 phase was increased and S phase population was slightly decreased in the PANC02-hTM4SF5 cells (Figure 5B). In addition, the level of active RhoA was increased by the humanized anti-hTM4SF5 monoclonal antibody in the PANC02-hTM4SF5 cells (Figure 5C). In the PANC02-mock cells, the cell cycle status and RhoA activity were not changed by the humanized anti-hTM4SF5 monoclonal antibody (Figure 5B and C). These data suggest that the targeting of TM4SF5 with the antibody decreased cell proliferation via regulation of cell cycle.
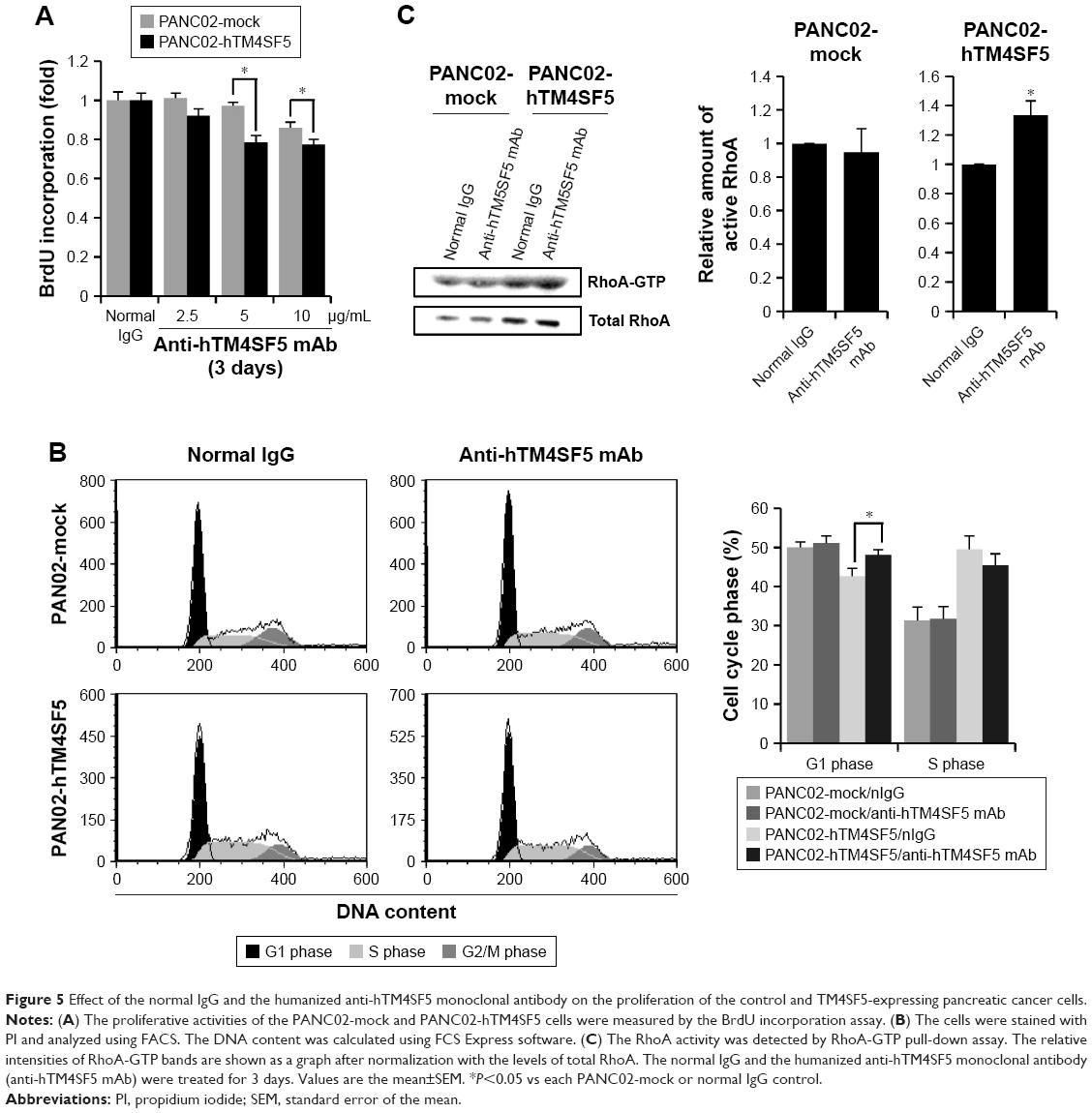 | Figure 5 Effect of the normal IgG and the humanized anti-hTM4SF5 monoclonal antibody on the proliferation of the control and TM4SF5-expressing pancreatic cancer cells. |
Suppression of tumor cell motility by the humanized anti-hTM4SF5 antibody
Because the motility of pancreatic cells was increased when TM4SF5 was expressed (Figure 3E), we investigated the effects of the humanized anti-hTM4SF5 monoclonal antibody on cell migration and invasion using the PANC02-mock and PANC02-hTM4SF5 cells. As shown by the wound-healing assay (Figure 6A), the migration of PANC02-hTM4SF5 cells into the wounded area was significantly reduced by treatment with the humanized anti-hTM4SF5 monoclonal antibody compared to the normal IgG whereas there was no effect in the control PANC02-mock cells. In addition, we measured the migration/invasion using a transwell migration/invasion chamber. The humanized anti-hTM4SF5 monoclonal antibody treatment, but not normal IgG, inhibited the migration and invasion of the PANC02-hTM4SF5 cells (Figure 6B and C). In contrast, the humanized anti-hTM4SF5 monoclonal antibody had no effect on the migration/invasion of the PANC02-mock cells. These results show that targeting of TM4SF5 with the antibody reduced the motility of TM4SF5-expressing pancreatic cancer cells in vitro.
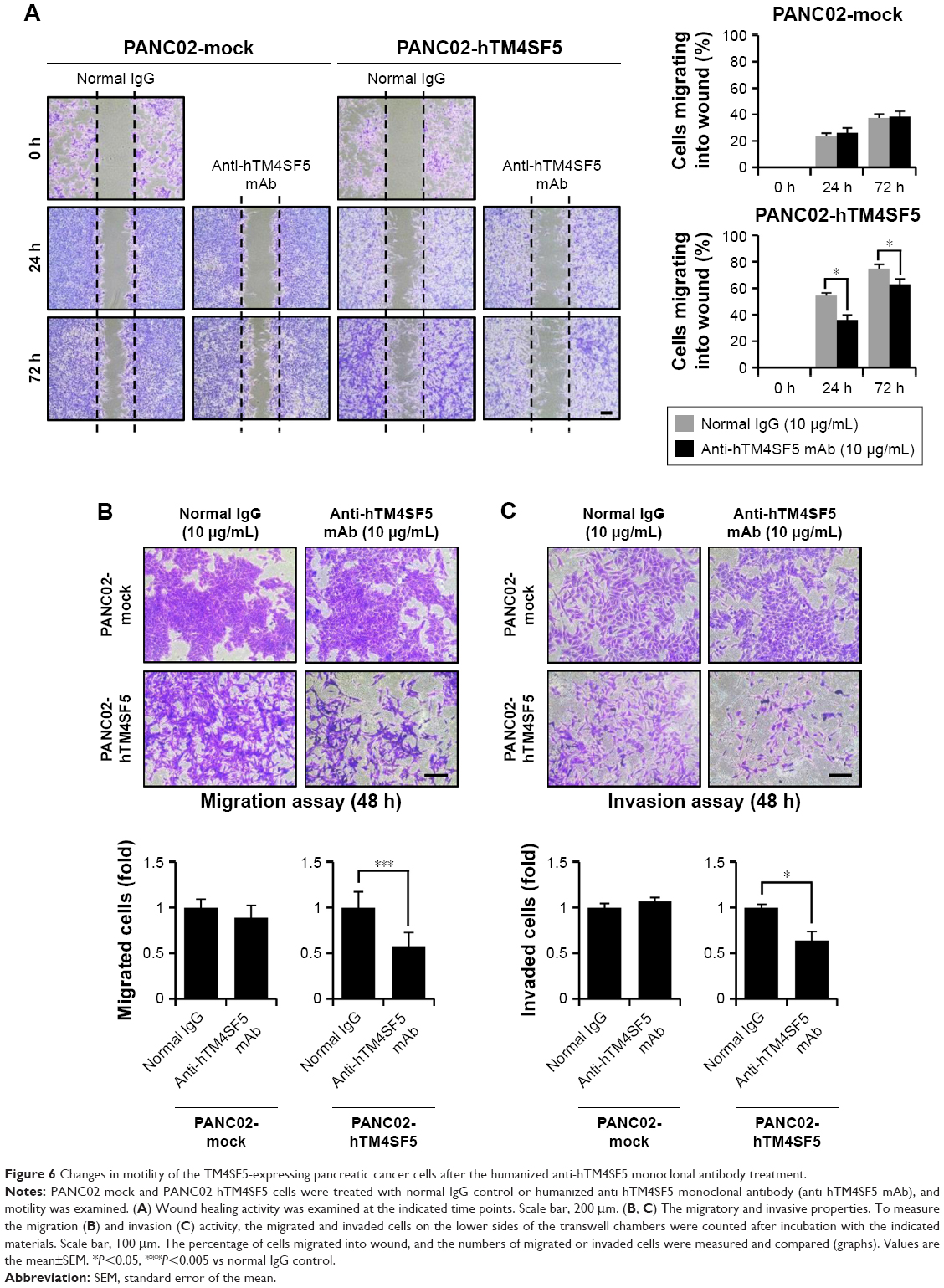 | Figure 6 Changes in motility of the TM4SF5-expressing pancreatic cancer cells after the humanized anti-hTM4SF5 monoclonal antibody treatment. |
Molecular change in the EMT markers by the humanized anti-hTM4SF5 antibody
Because we previously found that the anti-hTM4SF5 antibody (clone #2D4-18) treatment modulated EMT molecules in HCC and colon cancer,39,48 and here we confirmed the reduced cell motility of the PANC02-hTM4SF5 cells by treatment with the humanized anti-hTM4SF5 monoclonal antibody (Figure 6), we checked the expression levels of Vimentin and E-cadherin after the antibody treatment. In the PANC02-hTM4SF5 cells, the expression level of Vimentin was decreased and that of E-cadherin was increased at 3 and 5 days after the humanized anti-hTM4SF5 monoclonal antibody treatment compared to the normal IgG treatment (Figure 7B). In contrast, the expression levels of Vimentin and E-cadherin during the culture were similar when the normal IgG or the humanized anti-hTM4SF5 monoclonal antibody was treated in the PANC02-mock cells (Figure 7A). These findings suggest that the TM4SF5-targeting antibody can reduce EMT during tumor progression in TM4SF5-expressing pancreatic cancer cells.
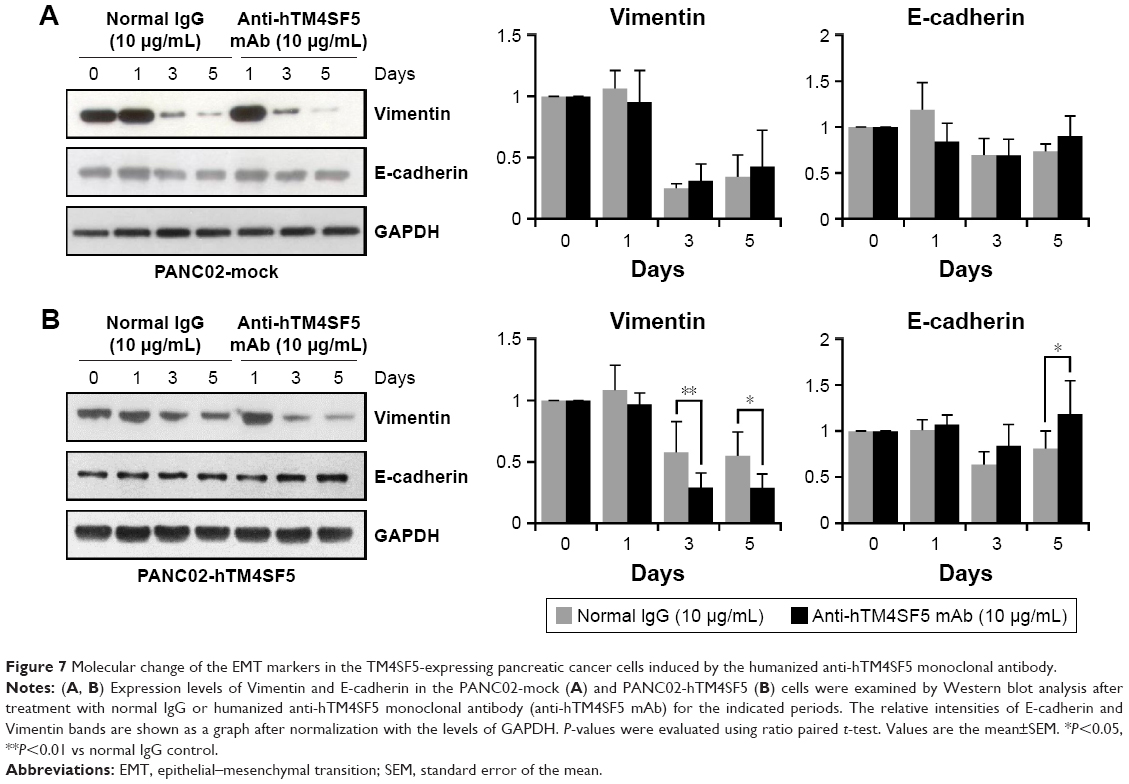 | Figure 7 Molecular change of the EMT markers in the TM4SF5-expressing pancreatic cancer cells induced by the humanized anti-hTM4SF5 monoclonal antibody. |
Discussion
High expression of TM4SF5 has been reported in several cancers including pancreatic cancer.33–35 Therefore, TM4SF5 has gained much attention as a target for anticancer strategies against those cancers. Previously, we found that immunization with a TM4SF5 peptide-CpG-DNA-liposome complex has preventive and therapeutic effects on tumor growth in a mouse HCC and colon cancer model.31,45–49 In this study, we showed that the peptide vaccine targeting TM4SF5 can induce the production of anti-hTM4SF5 antibodies and suppress the growth of TM4SF5-expressing pancreatic cancer in a mouse allograft model.
Almost all pancreatic cancers are PDAC.1 When we examined expression of TM4SF5 in the pancreatic tissues prepared from cancer patients, TM4SF5 was expressed in 36% of the samples. Even though the percentage is relatively low, 58% of the positive samples do have expression of TM4SF5 in 50% or more of tumor cells. Therefore, to investigate whether TM4SF5 could be a potential target for an anticancer strategy in pancreatic cancer, we searched for a TM4SF5-expressing mouse PDAC cell line as a model. However, we could not find a commercially or privately available one. Therefore, we established TM4SF5-expressing mouse PDAC cells (PANC02-hTM4SF5) and control cells (PANC02-mock) using a retroviral system. The PANC02-hTM4SF5 cells showed higher cancerous properties in the context of growth, migration, and mesenchymal marker expression compared to the control cells, supporting that TM4SF5 can act as an oncogenic factor. On the other hand, the proliferative activity of the PANC02-hTM4SF5 cells was significantly reduced by the anti-hTM4SF5 antibody compared to the normal IgG. Migration and EMT marker expression were also changed by the anti-hTM4SF5 antibody in PANC02-hTM4SF5. Therefore, we believe that suppression of TM4SF5 could be a strategy to treat pancreatic cancer expressing TM4SF5.
Generally, cyclic peptides have high biological activity compared to linear peptides due to the conformational structure.62,63 The rigidity of cyclic peptides reduces the entropy, therefore enabling increased binding or receptor selectivity. In addition, a cyclic structure has resistance to hydrolysis by exopeptidase.64 Cyclic peptides have been investigated for their utility as an antitumor drug. Integrins-recognition sequence Arg-Gly-Asp (RGD) inhibits tumor cell growth and metastasis, and a cyclic peptide with the RGD sequence is more potent than a linear peptide with the RGD sequence.65–67 The use of cyclic RGD peptides in drug delivery systems has also been studied.68 Antibodies produced by cyclic peptide immunization bind more strongly to the recombinant whole protein than antibodies produced by linear peptide immunization.69 Here, we found that immunization with the cyclic peptide epitope in combination with the CpG-DNA-liposome complex induced the successful production of anti-hTM4SF5 antibodies in mice. Furthermore, the growth of the tumor was suppressed in the vaccinated mice. Therefore, we confirmed that the peptide vaccine targeting TM4SF5 had a preventive effect against pancreatic tumor expressing TM4SF5 in an allograft model. Considering that the cancerous properties of PANC02-hTM4SF5 were suppressed by the treatment with the humanized anti-hTM4SF5 monoclonal antibody in vitro, antibodies produced by the vaccination clearly contributed to the suppressive effect of the peptide vaccine targeting TM4SF5 in vivo. Given the adjuvant effect of CpG-DNA inducing various immunostimulatory effects, other humoral and cellular effects in concert with the anti-hTM4SF5 antibodies could surely contribute to the defense against the pancreatic tumors in the vaccinated mice.
For the treatment of pancreatic cancer, investigators have tried various therapeutic strategies.70 Treatment with gemcitabine (dFdC: 2′,2′-difluorodeoxycytidine) has been recommended as a first-line therapy for advanced pancreatic cancer patients since 1997; however, gemcitabine treatment provides only a slight improvement in survival compared to no treatment. The overall survival of gemcitabine-treated patients is approximately 6 months.71 Pancreatic cancer cells are more sensitive to gemcitabine than other anticancer drugs; however, most patients get resistance within weeks after gemcitabine treatment.72 To improve the survival rate of gemcitabine monotherapy, combination therapy has been attempted with other cytotoxic agents such as capecitabine, oxaliplatin, and erlotinib.73–77 However, the improvement was very weak and limited. Therefore, many researchers have investigated better therapeutic strategies including targeted therapy.78,79 Because EGFR is overexpressed in 40%–60% of pancreatic cancers, lapatinib, an EGFR and HER-2 inhibitor, in combination with capecitabine, which is converted to 5-flurouracil, was tested in clinical trials to treat pancreatic cancer.80 The combination of gemcitabine with nimotuzumab, an anti-EGFR monoclonal antibody, was also tried.80,81 For targeting the angiogenesis pathway, aflibercept, a VEGF antagonist, or sorafenib, a VEGF-R2/3 kinase repressor, was combined with gemcitabine in pancreatic cancer patients. However, these combination trials did not significantly improve the therapeutic effects compared to gemcitabine monotherapy.82,83 PI3K/Akt is important in cell growth, survival, and apoptosis of various tumors, such as PDAC.84 Akt is activated in approximately 45% of PDAC patients, and the level of activated Akt is related with the survival rate after surgery.85 The alkylphospholipid perifosine, a suppressor of PI3K and Akt phosphorylation, in combination with gemcitabine showed a synergetic effect in pancreatic cancer cells expressing a high level of activated Akt.86,87 Despite these efforts, there has been no significant clinical outcome yet in pancreatic cancer, and it remains the most lethal among solid tumors. Therefore, we believe that our trial, to verify the effect of targeting and suppressing TM4SF5, is meaningful to pursue. Because our results suggest that targeting TM4SF5 could be a novel strategy to prevent or treat pancreatic cancer, further study on the effects of the humanized anti-hTM4SF5 monoclonal antibody in combination with other anticancer reagents may provide more useful information.
Conclusion
This study suggests that the targeting of TM4SF5 could be a novel strategy to prevent or treat pancreatic cancer. Vaccination targeting TM4SF5 induced production of anti-TM4SF5 antibodies and suppressed growth of TM4SF5-expressing pancreatic cancer in a mouse model. The cell growth and motility were decreased by treatment with the anti-hTM4SF5 monoclonal antibody in TM4SF5-expressing mouse pancreatic cancer cells in vitro. However, a strategy to strengthen the therapeutic effect of TM4SF5-targeting has to be investigated in the future.
Acknowledgments
This research was supported by grants from the National Research Foundation (NRF-2015R1A2A2A01007209, NRF-2018R1A2B6002504) funded by the Ministry of Science and ICT in the Republic of Korea. We would like to thank Professor Kyu Lim at Chungnam National University for kindly providing the pancreatic cancer cell line PANC02. This research was also partly supported by a research grant from Chungbuk National University in 2015.
Disclosure
The authors report no conflicts of interest in this work.
References
Moyer MT, Gaffney RR. Pancreatic adenocarcinoma. N Engl J Med. 2014;371(22):2140. | ||
Oldfield LE, Connor AA, Gallinger S. Molecular events in the natural history of pancreatic cancer. Trends Cancer. 2017;3(5):336–346. | ||
Zeitouni D, Pylayeva-Gupta Y, Der C, Bryant K. KRAS mutant pancreatic cancer: no lone path to an effective treatment. Cancers (Basel). 2016;8(4):45. | ||
Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. | ||
Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122(2):639–653. | ||
Baig S, Seevasant I, Mohamad J, Mukheem A, Huri HZ, Kamarul T. Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand? Cell Death Dis. 2016;7:e2058. | ||
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–4276. | ||
Oberstein PE, Olive KP. Pancreatic cancer: why is it so hard to treat? Therap Adv Gastroenterol. 2013;6(4):321–337. | ||
Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 2013;119:421–475. | ||
van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–1647. | ||
Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32. | ||
Melero I, Gaudernack G, Gerritsen W, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11(9):509–524. | ||
Brunsvig PF, Aamdal S, Gjertsen MK, et al. Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol Immunother. 2006;55(12):1553–1564. | ||
Bernhardt SL, Gjertsen MK, Trachsel S, et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer. 2006;95(11):1474–1482. | ||
Kyte JA, Trachsel S, Risberg B, Thor Straten P, Lislerud K, Gaudernack G. Unconventional cytokine profiles and development of T cell memory in long-term survivors after cancer vaccination. Cancer Immunol Immunother. 2009;58(10):1609–1626. | ||
Kyte JA, Gaudernack G, Dueland S, Trachsel S, Julsrud L, Aamdal S. Telomerase peptide vaccination combined with temozolomide: a clinical trial in stage IV melanoma patients. Clin Cancer Res. 2011;17(13):4568–4580. | ||
Greten TF, Forner A, Korangy F, et al. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer. 2010;10:209. | ||
Yoshitake Y, Fukuma D, Yuno A, et al. Phase II clinical trial of multiple peptide vaccination for advanced head and neck cancer patients revealed induction of immune responses and improved OS. Clin Cancer Res. 2015;21(2):312–321. | ||
Fenstermaker RA, Ciesielski MJ, Qiu J, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother. 2016;65(11):1339–1352. | ||
Akbarzadeh A, Rezaei-Sadabady R, Davaran S, et al. Liposome: classification, preparation, and applications. Nanoscale Res Lett. 2013;8(1):102. | ||
Singh S, Asal R, Bhagat S. Multifunctional antioxidant nanoliposome-mediated delivery of PTEN plasmids restore the expression of tumor suppressor protein and induce apoptosis in prostate cancer cells. J Biomed Mater Res A. Epub 2018 Sep 7. | ||
Simões S, Filipe A, Faneca H, et al. Cationic liposomes for gene delivery. Expert Opin Drug Deliv. 2005;2(2):237–254. | ||
Shim G, Kim MG, Park JY, Oh Y-K. Application of cationic liposomes for delivery of nucleic acids. Asian J Pharm Sci. 2013;8(2):72–80. | ||
Chen J, Yu Z, Chen H, Gao J, Liang W. Transfection efficiency and intracellular fate of polycation liposomes combined with protamine. Biomaterials. 2011;32(5):1412–1418. | ||
Rao DD, Luo X, Wang Z, et al. KRAS mutant allele-specific expression knockdown in pancreatic cancer model with systemically delivered bi-shRNA KRAS lipoplex. PLoS One. 2018;13(5):e0193644. | ||
Passadouro M, Faneca H. Combination of anti-miRNAs oligonucleotides with low amounts of chemotherapeutic agents for pancreatic cancer therapy. Methods Mol Biol. 2018;1699:135–154. | ||
Phillips NC, Emili A. Enhanced antibody response to liposome-associated protein antigens: preferential stimulation of IgG2a/b production. Vaccine. 1992;10(3):151–158. | ||
Chikh G, Schutze-Redelmeier MP. Liposomal delivery of CTL epitopes to dendritic cells. Biosci Rep. 2002;22(2):339–353. | ||
Chang JS, Choi MJ, Cheong HS, Kim K. Development of Th1-mediated CD8+ effector T cells by vaccination with epitope peptides encapsulated in pH-sensitive liposomes. Vaccine. 2001;19(27):3608–3614. | ||
Lee KW, Jung J, Lee Y, et al. Immunostimulatory oligodeoxynucleotide isolated from genome wide screening of Mycobacterium bovis chromosomal DNA. Mol Immunol. 2006;43(13):2107–2118. | ||
Kim D, Kwon S, Rhee JW, et al. Production of antibodies with peptide-CpG-DNA-liposome complex without carriers. BMC Immunol. 2011;12:29. | ||
Lee Y, Lee YS, Cho SY, Kwon HJ. Perspective of peptide vaccine composed of epitope peptide, CpG-DNA, and liposome complex without carriers. Adv Protein Chem Struct Biol. 2015;99:75–97. | ||
Müller-Pillasch F, Wallrapp C, Lacher U, et al. Identification of a new tumour-associated antigen TM4SF5 and its expression in human cancer. Gene. 1998;208(1):25–30. | ||
Lee SA, Lee SY, Cho IH, et al. Tetraspanin TM4SF5 mediates loss of contact inhibition through epithelial-mesenchymal transition in human hepatocarcinoma. J Clin Invest. 2008;118(4):1354–1366. | ||
Wu YB, Huang YS, Xu YP, et al. A high level of TM4SF5 is associated with human esophageal cancer progression and poor patient survival. Dig Dis Sci. 2013;58(9):2623–2633. | ||
Lee SA, Kim TY, Kwak TK, et al. Transmembrane 4 L six family member 5 (TM4SF5) enhances migration and invasion of hepatocytes for effective metastasis. J Cell Biochem. 2010;111(1):59–66. | ||
Kang M, Ryu J, Lee D, et al. Correlations between transmembrane 4 L6 family member 5 (TM4SF5), CD151, and CD63 in liver fibrotic phenotypes and hepatic migration and invasive capacities. PLoS One. 2014;9(7):e102817. | ||
Choi S, Lee SA, Kwak TK, et al. Cooperation between integrin alpha5 and tetraspan TM4SF5 regulates VEGF-mediated angiogenic activity. Blood. 2009;113(8):1845–1855. | ||
Kwon S, Choi KC, Kim YE, et al. Monoclonal antibody targeting of the cell surface molecule TM4SF5 inhibits the growth of hepatocellular carcinoma. Cancer Res. 2014;74(14):3844–3856. | ||
Jung O, Choi S, Jang SB, et al. Tetraspan TM4SF5-dependent direct activation of FAK and metastatic potential of hepatocarcinoma cells. J Cell Sci. 2012;125(Pt 24):5960–5973. | ||
Lee JW. TM4SF5-mediated protein-protein networks and tumorigenic roles. BMB Rep. 2014;47(9):483–487. | ||
Kim H, Kang M, Lee SA, et al. TM4SF5 accelerates G1/S phase progression via cytosolic p27Kip1 expression and RhoA activity. Biochim Biophys Acta. 2010;1803(8):975–982. | ||
Ryu J, Kang M, Lee MS, et al. Cross talk between the TM4SF5/focal adhesion kinase and the interleukin-6/STAT3 pathways promotes immune escape of human liver cancer cells. Mol Cell Biol. 2014;34(16):2946–2960. | ||
Lee SA, Ryu HW, Kim YM, et al. Blockade of four-transmembrane L6 family member 5 (TM4SF5)-mediated tumorigenicity in hepatocytes by a synthetic chalcone derivative. Hepatology. 2009;49(4):1316–1325. | ||
Kwon S, Kim D, Park BK, et al. Prevention and therapy of hepatocellular carcinoma by vaccination with TM4SF5 epitope-CpG-DNA-liposome complex without carriers. PLoS One. 2012;7(3):e33121. | ||
Kwon S, Kim D, Park BK, et al. Induction of immunological memory response by vaccination with TM4SF5 epitope-CpG-DNA-liposome complex in a mouse hepatocellular carcinoma model. Oncol Rep. 2013;29(2):735–740. | ||
Kwon S, Kim YE, Kim D, et al. Prophylactic effect of a peptide vaccine targeting TM4SF5 against colon cancer in a mouse model. Biochem Biophys Res Commun. 2013;435(1):134–139. | ||
Kim YE, Kwon S, Wu G, et al. Therapeutic effect of a TM4SF5-specific monoclonal antibody against colon cancer in a mouse model. Oncotarget. 2014;5(18):8402–8415. | ||
Wu G, Kim D, Park BK, et al. Anti-metastatic effect of the TM4SF5-specific peptide vaccine and humanized monoclonal antibody on colon cancer in a mouse lung metastasis model. Oncotarget. 2016;7(48):79170–79186. | ||
Song KS, Jing K, Kim JS, et al. Omega-3-polyunsaturated fatty acids suppress pancreatic cancer cell growth in vitro and in vivo via downregulation of Wnt/Beta-catenin signaling. Pancreatology. 2011;11(6):574–584. | ||
Park BK, Park JY, Kim TH, et al. Production of an anti-TM4SF5 monoclonal antibody and its application in the detection of TM4SF5 as a possible marker of a poor prognosis in colorectal cancer. Int J Oncol. 2018;53(1):275–285. | ||
Cubas R, Zhang S, Li M, Chen C, Yao Q. Trop2 expression contributes to tumor pathogenesis by activating the ERK MAPK pathway. Mol Cancer. 2010;9:253. | ||
Yagublu V, Caliskan N, Lewis AL, et al. Treatment of experimental pancreatic cancer by doxorubicin-, mitoxantrone-, and irinotecan-drug eluting beads. Pancreatology. 2013;13(1):79–87. | ||
Subramaniam D, Ramalingam S, Linehan DC, et al. RNA binding protein CUGBP2/CELF2 mediates curcumin-induced mitotic catastrophe of pancreatic cancer cells. PLoS One. 2011;6(2):e16958. | ||
Schmidt T, Ziske C, Märten A, et al. Intratumoral immunization with tumor RNA-pulsed dendritic cells confers antitumor immunity in a C57BL/6 pancreatic murine tumor model. Cancer Res. 2003;63(24):8962–8967. | ||
Priebe TS, Atkinson EN, Pan BF, Nelson JA. Intrinsic resistance to anticancer agents in the murine pancreatic adenocarcinoma PANC02. Cancer Chemother Pharmacol. 1992;29(6):485–489. | ||
Vences-Catalán F, Rajapaksa R, Srivastava MK, et al. Tetraspanin CD81 promotes tumor growth and metastasis by modulating the functions of T regulatory and myeloid-derived suppressor cells. Cancer Res. 2015;75(21):4517–4526. | ||
Detchokul S, Williams ED, Parker MW, Frauman AG. Tetraspanins as regulators of the tumour microenvironment: implications for metastasis and therapeutic strategies. Br J Pharmacol. 2014;171(24):5462–5490. | ||
Zijlstra A, Lewis J, Degryse B, Stuhlmann H, Quigley JP. The inhibition of tumor cell intravasation and subsequent metastasis via regulation of in vivo tumor cell motility by the tetraspanin CD151. Cancer Cell. 2008;13(3):221–234. | ||
Liu CY, Lin HH, Tang MJ, Wang YK. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget. 2015;6(18):15966–15983. | ||
Tian X, Liu Z, Niu B, et al. E-Cadherin/β-Catenin Complex and the Epithelial Barrier. J Biomed Biotechnol. 2011;2011:567305. | ||
Horton DA, Bourne GT, Smythe ML. Exploring privileged structures: the combinatorial synthesis of cyclic peptides. J Comput Aided Mol Des. 2002;16(5–6):415–430. | ||
Colombo G, Curnis F, De Mori GM, et al. Structure-activity relationships of linear and cyclic peptides containing the NGR tumor-homing motif. J Biol Chem. 2002;277(49):47891–47897. | ||
Joo SH. Cyclic peptides as therapeutic agents and biochemical tools. Biomol Ther (Seoul). 2012;20(1):19–26. | ||
Gurrath M, Müller G, Kessler H, Aumailley M, Timpl R. Conformation/activity studies of rationally designed potent anti-adhesive RGD peptides. Eur J Biochem. 1992;210(3):911–921. | ||
Humphries MJ, Olden K, Yamada KM. A synthetic peptide from fibronectin inhibits experimental metastasis of murine melanoma cells. Science. 1986;233(4762):467–470. | ||
Gehlsen KR, Argraves WS, Pierschbacher MD, Ruoslahti E. Inhibition of in vitro tumor cell invasion by Arg-Gly-Asp-containing synthetic peptides. J Cell Biol. 1988;106(3):925–930. | ||
Chen Z, Deng J, Zhao Y, Tao T. Cyclic RGD peptide-modified liposomal drug delivery system: enhanced cellular uptake in vitro and improved pharmacokinetics in rats. Int J Nanomedicine. 2012;7:3803–3811. | ||
Schulz-Utermoehl T, Edwards RJ, Boobis AR. Affinity and potency of proinhibitory antipeptide antibodies against CYP2D6 is enhanced using cyclic peptides as immunogens. Drug Metab Dispos. 2000;28(5):544–551. | ||
Teague A, Lim KH, Wang-Gillam A. Advanced pancreatic adenocarcinoma: a review of current treatment strategies and developing therapies. Ther Adv Med Oncol. 2015;7(2):68–84. | ||
Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–2413. | ||
Amrutkar M, Gladhaug I. Pancreatic cancer chemoresistance to gemcitabine. Cancers (Basel). 2017;9(12):E157. | ||
Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2009;27(33):5513–5518. | ||
Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet. 2017;389(10073):1011–1024. | ||
Louvet C, Labianca R, Hammel P, et al; GERCOR; GISCAD. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol. 2005;23(15):3509–3516. | ||
Demols A, Peeters M, Polus M, et al. Gemcitabine and oxaliplatin (GEMOX) in gemcitabine refractory advanced pancreatic adenocarcinoma: a phase II study. Br J Cancer. 2006;94(4):481–485. | ||
Moore MJ, Goldstein D, Hamm J, et al; National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–1966. | ||
Barati Bagherabad M, Afzaljavan F, Shahidsales S, Mahdi Hassanian S, Avan A. Targeted therapies in pancreatic cancer: promises and failures. J Cell Biochem. Epub 2017 Jul 13. | ||
Philip PA. Targeted therapies for pancreatic cancer. Gastrointest Cancer Res. 2008;2(4 Suppl 2):S16–S19. | ||
Wu Z, Gabrielson A, Hwang JJ, et al. Phase II study of lapatinib and capecitabine in second-line treatment for metastatic pancreatic cancer. Cancer Chemother Pharmacol. 2015;76(6):1309–1314. | ||
Su D, Jiao SC, Wang LJ, et al. Efficacy of nimotuzumab plus gemcitabine usage as first-line treatment in patients with advanced pancreatic cancer. Tumour Biol. 2014;35(3):2313–2318. | ||
Rougier P, Riess H, Manges R, et al. Randomised, placebo-controlled, double-blind, parallel-group phase III study evaluating aflibercept in patients receiving first-line treatment with gemcitabine for metastatic pancreatic cancer. Eur J Cancer. 2013;49(12):2633–2642. | ||
Kindler HL, Wroblewski K, Wallace JA, et al. Gemcitabine plus sorafenib in patients with advanced pancreatic cancer: a phase II trial of the University of Chicago Phase II Consortium. Invest New Drugs. 2012;30(1):382–386. | ||
Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. | ||
Yamamoto S, Tomita Y, Hoshida Y, et al. Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2004;10(8):2846–2850. | ||
Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;2(11):1093–1103. | ||
Massihnia D, Avan A, Funel N, et al. Phospho-Akt overexpression is prognostic and can be used to tailor the synergistic interaction of Akt inhibitors with gemcitabine in pancreatic cancer. J Hematol Oncol. 2017;10(1):9. |
Supplementary material
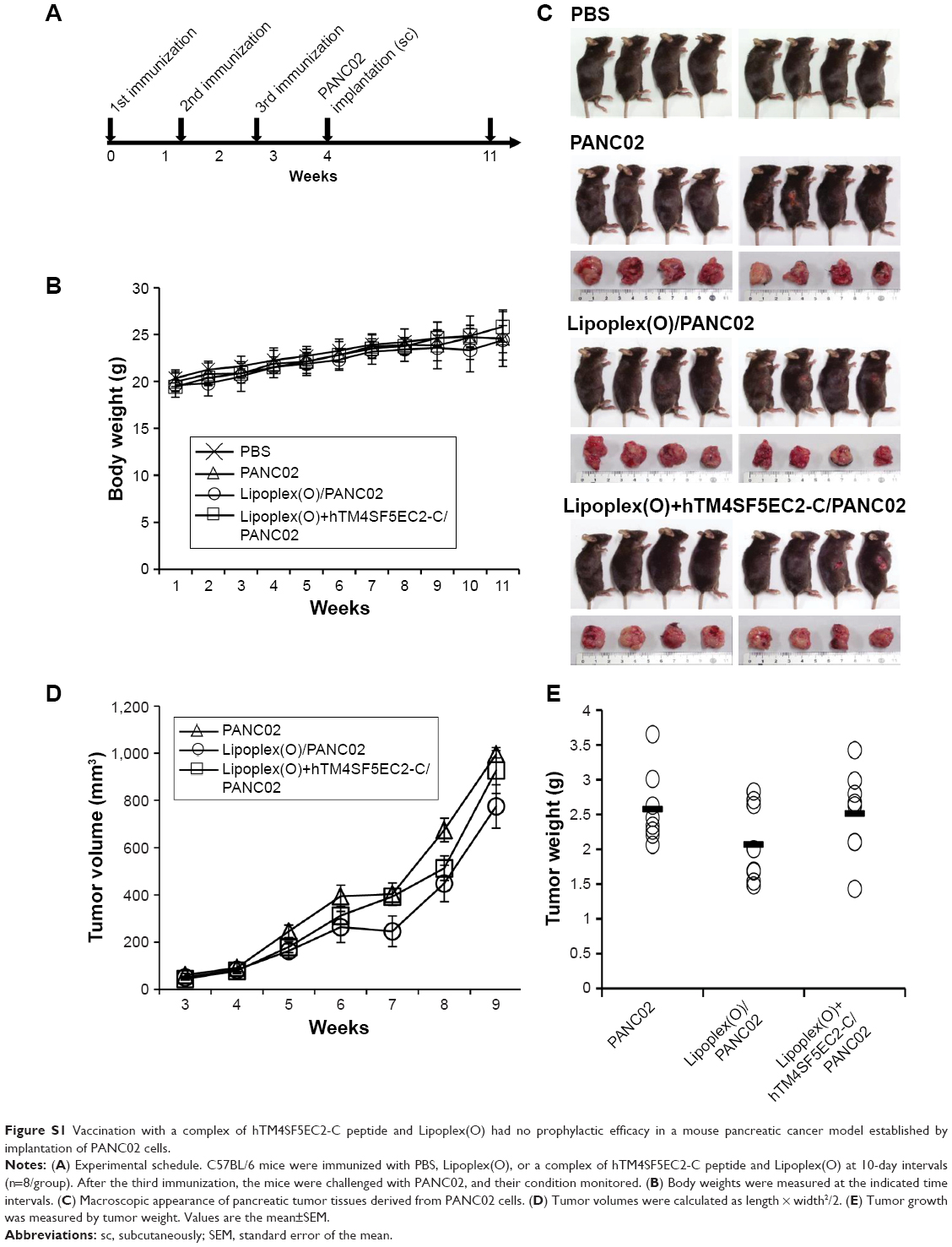 | Figure S1 Vaccination with a complex of hTM4SF5EC2-C peptide and Lipoplex(O) had no prophylactic efficacy in a mouse pancreatic cancer model established by implantation of PANC02 cells. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.